

Abstract
Most cancers rely disproportionately on glycolysis for energy even in the presence of adequate oxygen supply, a condition known as “aerobic glycolysis”, or the Warburg effect. Pharmacological reversal of the Warburg effect has been shown to cause selective apoptosis of tumor cells, presumably by stimulating mitochondrial respiratory chain activity and production of reactive oxygen species that, in turn, induce a caspase-mediated series of reactions leading to cell death. We reasoned that a similar effect on tumor cells might result from up-regulation of the E1α subunit gene (pda1) of the pyruvate dehydrogenase complex (PDC) that catalyzes the rate-limiting step in aerobic glucose oxidation and thus plays a major role in the control of oxidative phosphorylation. To test this postulate, we employed a self-complementary adeno-associated virus (scAAV)-based delivery and expression system for targeting pda1 to the mitochondria of primary cultures of human hepatoblastoma (HB) and hepatocellular carcinoma (HCC) cells. Serotypes 1-10 scAAV vectors that included enhanced green fluorescent (egfp) reporter gene driven by either cytomegalovirus (CMV) or chicken beta-actin (CBA) promoters were analyzed for transduction ability of HB (Huh-6) and HCC (Huh-7 and HepG2) cell lines and primary cultures of normal human hepatocytes. Serotype 3 scAAV-egfp (scAAV3-egfp) vector was the most efficient and transduced up to 90% of cells. We limited the transgene expression primarily to liver cancer cells by generating scAAV3 vectors that contained the human alpha-fetoprotein promoter (AFP)-driven reporter gene (scAAV3.AFP-egfp) and the potentially therapeutic gene scAAV3.AFP-pda1. Infection of Huh-6 cells by the scAAV3.AFP-pda1 vector increased protein expression of E1α, PDC catalytic activity, and late-stage apoptotic cell death. Apoptosis was also associated with increased protein expression of Bcl-X/S, an early marker of apoptosis, and release of cytochrome c into the cytosol of infected HB cells. These data indicate that molecular targeting of mitochondrial oxidative metabolism in liver cancer cells by AAV3-mediated delivery of pda1 holds promise as a novel and effective therapeutic approach for human hepatic tumors.
Keywords: Adeno-associated viral vectors, apoptosis, hepatoblastoma, hepatocellular carcinoma, gene therapy, pyruvate dehydrogenase complex
1. Introduction
The mitochondrial pyruvate dehydrogenase complex (PDC) catalyzes the irreversible oxidation of glucose-derived pyruvate to acetyl CoA. Decarboxylation of pyruvate is mediated by the E1 (pyruvate dehydrogenase; E.C. 1.2.4.1) component of PDC, a multimeric collection of nuclear-encoded enzymes situated in the mitochondrial matrix [1]. E1 is a heterotetramer of two alpha and two beta subunits that requires thiamine pyrophosphate as a cofactor for the oxidation of pyruvate. Rapid regulation of PDC activity occurs primarily by reversible phosphorylation of the E1α subunit [2]. Multiple copies of the PDC complex are distributed within each mitochondrion, although the cellular distribution and individual kinetic properties of the four known PDKs and two known PDPs in humans may account for the tissue-specific regulation of the complex. The PDK2 isoform is expressed ubiquitously and abundantly in tissues and is the most sensitive to activation or inactivation by endogenous regulatory molecules or xenobiotics, such as dichloroacetate (DCA). This investigational drug is a highly effective lactate-lowering agent in healthy organisms as well as in a number of acquired and congenital causes of lactic acidosis by virtue of its ability to inhibit PDK, thereby maintaining PDC in its unphosphorylated, catalytically active, form [3].
Transactivation of PDK in tumor cells causes phosphorylation and inactivation of PDC. Consequently, tumors rely on the much less efficient process of glycolysis for energy, although this is at least partly compensated by the transactivation of glucose transporters and glycolytic enzymes [4, 5]. The pivotal role of PDC in cellular energetics and lactate removal has focused interest on this complex as a therapeutic target for cancer. Several reports have documented the ability of DCA to stimulate aerobic metabolism in human tumor cells studied in vitro or implanted into immunocompromised rodents and to cause selective apoptosis in cancer, but not in normal, cells [6-10]. A mechanism to explain the drug’s efficacy as an antitumor agent holds that stimulation of PDC activity in tumor cells increases carbon flux through the tricarboxylic (TCA) cycle and electron flow through the respiratory chain. Enhanced respiratory chain activity increases production of reactive oxygen species (ROS) that, in turn, induce repolarization in the mitochondrial membrane potential, resulting in release of cytochrome c and stimulation of a caspase-mediated process of cell death [11].
We reasoned that the selective delivery and expression of the PDC E1α gene (pda1) to tumors could act in ways similar to drugs, such as DCA, by augmenting the oxidative capacity of tumor cells, thereby increasing their susceptibility to undergo of apoptosis. We tested this postulate using primary cultures of healthy human hepatocytes and hepatoblastoma (HB) and hepatocellular carcinoma (HCC) cells. HB and HCC are two of the most frequent and aggressive tumors worldwide in children and adults, respectively, and have a high mortality rate despite current treatments [12-14]. We observed that of the 10 available adeno-associated virus (AAV) serotype vectors, a self-complementary (sc) AAV3 serotype vector was the most efficient in transducing HB and HCC cell lines and primary human hepatocytes in vitro. The delivery of the pda1 transgene by scAAV3 resulted in significant stimulation of both E1α exoression and PDC activity as well as selective apoptosis of tumor cells.
2. Materials and methods
2.1 Cell culture lines
Human hepatoblastoma (Huh-6), hepatocellular carcinoma (Huh-7, HepG2), and adenovirus-transformed human embryonic kidney (HEK293) cell lines were maintained in complete DMEM medium with 4.5 g/L glucose, L-glutamine and sodium pyruvate (Mediatech Inc, Manassas, VA, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Lonza, Walkersville, MD), 100 units/ml penicillin and 100 mg/ml streptomycin (PS, Lonza). Primary human hepatocytes were obtained from CellzDirect Inc. (Durham, NC, USA). The cell isolation and culture procedure were performed according to our published protocol [15]. Cells were grown as adherent culture in a humidified atmosphere at 37°C in 5% CO2 and were sub-cultured after trypsinisation by a trypsin-versene mixture (Lonza, Walkersville, MD) for 2-5 minute at room temperature, washed and re-suspended in complete DMEM medium.
2.2 Recombinant AAV vectors
Table 1 lists the properties of the vectors used in this research. Two pscAAV constructs were made, each containing an alpha-fetoprotein promoter region (AFP) - SV40 enhancer sequence upstream of pda1-therapeutic gene or egfp-reporter gene. The pscAAV- CBA-pdh backbone [16] was used to generate pscAAV-AFP-egfp and pscAAV-AFP-pda1 constructs. The 515-bp fragment of the AFP promoter-SV40 enhancer region was amplified from a pDrive-SV40-hAFP (InvivoGene, San Diego, CA) template (region from 7 bp till 522 bp) by a set of primers including the Kpn1 site from the 5′- terminus and the Bam H1 site from the 3′- terminus of an amplicon (underlined). The following primer-pair was used: forward, 5′-GCGGTACCGGGCCTGAAATAACCTCTG-3′ and reverse, 5′-CGGATTCGTTATTGGCAGTGGTGGAAGC -3′. PCR was performed (Expand High Fidelity PCR System; Roche Diagnostics Corp., Indianapolis, IN) under the following cycling conditions: initial denaturation at 94°C for 3 minutes, then 30 cycles at 94°C for 30 seconds, 58°C for 30 seconds, 72°C for 30 seconds and a final extension at 72°C for 7 minutes. The integrity of the PCR product was verified by agarose gel electrophoresis with ethidium bromide staining and then it was directionally cloned into Kpn1-BamH1 sites of pscAAV-CBA-pdh plasmid [16] replacing a CBA promoter. The sequence analysis of both strands confirmed the identity and the orientation of the SV40 enhancer-AFP promoter fragment in the constructs. Self-complementary AAV.CMV-egfp vector plasmid was generously provided by Dr. D. M. McCarty, Ohio State University, Columbus, OH. ScAAV-CMV-egfp vector serotypes 1-10 were packaged by transient co-transfection of HEK 293 cells with the vector plasmid pscAAV.CMV-egfp and helper-packaging plasmids that supply all the necessary adenovirus helper functions as well as AAV rep and cap proteins in trans. ScAAV3.AFP-egfp, scAAV3.AFP-pda1 and pscAAV3.CBA-egfp vectors were also packaged in AAV3 capsids as described above. Vector particle titers were determined by dot-blot hybridization and quantitative PCR. The titers of scAAV.AFP-egfp and scAAV.AFP -pda1 vectors were 2.38 × 1012 and 3.26 × 1012 viral genomes/ml, respectively.
Table 1.
Vectors used in this study.
| Vector | Serotype | Transgene | Transgene promoter | Titer (viral particles /ml) |
|---|---|---|---|---|
| scAAV.CMV.egfp | 1 - 10 | egfp (enhance green fluorescent protein) |
CMV (cytomegalovirus) |
2× 1011 -7× 1012 |
| scAAV.CBA.egfp | 3 | Egfp | CBA(chicken beta- actin) |
7.2× 1011 |
| scAAV.AFP.egfp | 3 | Egfp | AFP (alpha feto- protein) |
2.38 × 1012 |
| scAAV.AFP.pda1 | 3 | pda1 (E1α subunit) | AFP | 3.26 × 1012 |
2.3 Antibodies
A primary monoclonal antibody against the PDH E1 alpha subunit (MSP07) (MitoSciences, Inc., Eugene, OR) was used for Western blotting at a final concentration of 0.5 μg/ml and a secondary was goat anti-mouse antibody (IgG HRP, sc 2005; Santa Cruz Biotechnology, Inc; Santa Cruz, CA; 1:2000). Other antibodies and dilutions were: rabbit polyclonal primary antibody (L-19, Santa Cruz, CA; 1:5000) and goat anti-rabbit secondary antibody (IgG HRP, sc 2004; Santa Cruz, CA; 1:5000) to visualize of Bcl-X/L and Bcl-X/S splice-isoforms; rabbit alpha-tubulin antibody (1H10; Cell Signaling Technology, Inc, Beverly, MA; 1:2000); goat polyclonal cytochrome c antibody (C-20; Santa Cruz, CA; 1:100) for immunocytochemistry; and secondary donkey anti-goat antibody conjugated with Texas Red (TR) (IgG-TR, sc 2783; Santa Cruz, CA; 1:1000). All antibodies were diluted in PBS containing 1% of non-fat milk for Western blotting and in PBS containing 1% of bovine serum albumin (BSA), for immunocytochemistry.
2.4 Immunocytochemistry
Huh-6 cells were seeded in Poly-D-Lysine Cellware 8-well culture slides (BD Biosciences; San Jose, CA) at a concentration of about 1.5 ×104 cells per well in complete DMEM medium. On the next day cells were infected by scAAV3.AFP-pda1 or scAVV3.AFP-egfp vectors with the multiplicity of 2 × 103 or 5 × 103 viral genomes/cell in serum and antibiotic-free DMEM medium. After four hours the medium was switched to complete DMEM medium supplemented by 10% of FBS and 1% of PS solution (Mediatech, Inc. Hemdon, VA). The medium was discarded seven days after infection and cells were rinsed twice with PBS and fixed in 4% paraformaldehyde in PBS for 10 minutes. Slides were rinsed with PBS and blocked with blocking buffer (1 X PBS, 5% BSA, 0.05% Tween-20) for 1hour at room temperature or overnight at 4° C. Fixed cells were probed by a goat cytochrome C primary antibody. After washing, an anti-goat secondary antibody conjugated with Texas Red (TR) was applied for 1 hour. Slides were mounted under cover slips in Vectashield mounting medium for fluorescence (Vector Laboratories, Burlingame, CA). Images were taken with an inverted Zeiss Axiovert 25 fluorescent microscope (Carl Zeiss, Inc., Thornwood, NY) and were processed using PhotoShop Version 7 image analysis software (Adobe, San Jose, CA).
2.5 PscAAV.AFP-pda1 transfection HepG2 cells and Annexin V assay
PscAAV.AFP-egfp and pscAAV.AFP-pda1 DNAs were transfected into HepG2 cells using Lipofectamine-2000 reagent (Invitrogen) according to the manufacturer’s protocol. Cells were grown in 6-well plates and were 90-95% confluent at the time of transfection. Each procedure was performed with 4 μg of plasmid DNA and 8 μl of lipofectamine-2000 reagent in 2 ml of antibiotic-free growth medium. After 6 hours the transfection medium was changed to complete DMEM medium. The next day the cells were passaged into 8-well culture slides (BD Biosciences, San Jose, CA) at a concentration of 1.5 × 104 cells per well. Cells were double-labeled with Annexin V-FITC and PI (Trevigen, Inc., Gaithersburg, MD) 3-4 days later. Slides were mounted under cover slips in Vectashield mounting medium for fluorescence. Representative images were taken with an inverted Zeiss Axiovert 25 fluorescent microscope (Carl Zeiss, Inc., Thornwood, NY). Images were processed using PhotoShop Version 7 image analysis software (Adobe).
2.6 AAV vector transduction
Huh-6, Huh-7, HepG2, HEK293, or normal human hepatocytes were seeded in 96 well plates at a concentration of 5 × 103 cells per well in complete DMEM medium. The infections were performed with the multiplicity of 1 × 103 or 1 × 104 viral genomes/cell in serum- and antibiotic-free DMEM medium. The expression of EGFP was analyzed in infected cells by direct fluorescence imaging using a Zeiss Axiovert 25 fluorescent microscope. Representative images were taken with an X20 objective. The viral transduction efficiency was quantitated by ImageJ (NIH, Bethesda, MD). Three visual fields were analyzed for each experiment.
2.7 AAV transduction efficiency
Huh-6 or HEK293 cells infected with scAAV3.AFP-egfp virus and non-infected cells were grown in 6 well plates to about 70% confluence at the time of infection. Five-9 days after infection the cells were trypsinized, washed twice with cold PBS, and then counted. 1 × 106 cells were re-suspended in PBS. FACS data were acquired using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) and were analyzed using Cell Quest software (BD Biosciences, San Jose, CA). An FITC filter was applied to measure GFP fluorescence in scAAV3.AFP-egfp infected cells. 2 × 104cells were analyzed for each sample. Results are presented as the percentage of EGFP-positive cells per sample.
2.8 Annexin V-ACP/ 7-AAD
Huh-6 cells were infected with scAAV3.AFP-pda1 vector or its control counterpart, scAAV3.AFP-egfp vector as described. Cells were washed twice with cold PBS and resuspended in 1X binding buffer (BD Pharmingen Biosciences, Bedford, MA) 5-7 days after infection. 100 μl of a suspension containing 1 × 106 cells were transferred to a 5 ml culture tube. 5 μl of AnnexinV-Allophycocyanin conjugate (AnnexinV-APC) and 5 μl of 7-Amino-Actinomycin D (7-AAD) (BD Biosciences) solutions were added to each tube. Cells were gently vortexed and incubated for 15 minutes in the dark at room temperature. 400 μl of binding buffer was added to each tube. FACS data were acquired and analyzed as described above. 3 × 104 cells were analyzed per sample.
2.9 Western Blotting
Huh-6 cells were infected with scAAV3.AFP-egfp and scAAV3.AFP-pda1 viruses as described above. Cells were passaged into 10 cm plates after 2 days. 5-9 days after infection cells were washed twice with PBS. 600 μl of 1 X RIPA buffer (150 mM NaCl, 10 mM Tris [pH 7.2], 0.1% SDS, 1% Triton X-100, 1% deoxycholate, 5 mM EDTA) that contained Inhibitor Cocktail Set III (EMD Biosciences Inc., La Jolla, CA) were added to the cells for 20-30 minutes on ice. Lysates were collected, passed through 20 gauge needles, and clarified by centrifugation for 10 minutes at 13,000 rpm/min (4°C). Pellets were sonicated in 100 μl of 1 X RIPA buffer, clarified by centrifugation, and recombined with their supernatants.
Samples were normalized according to protein concentration (Bio-Rad Protein Assay, Bio-Rad Laboratories, Hercules, CA), separated by 4-20% SDS-PAGE and transferred to a PVDF membrane (Bio-Rad Laboratories, Hercules, CA). The membrane was blocked with a blocking buffer (1 X PBS, 5% non-fat milk, Bio-Rad Laboratories, Hercules, CA) for 1 hour with gentle agitation at room temperature and then probed with primary antibody for 1 hour at room temperature or overnight at 4°C. After three 10 minutes washes (1 X PBS, 0.05% Tween-20) the membrane was incubated with secondary antibody for 1 hour at room temperature. After three 5-minutes washes enhanced chemiluminescence substrate (Pierce, Rockford, IL) was applied to the membrane for 5 minutes and the proteins were visualized by exposure to x-ray film (Kodak, Rochester, NY).
2.10 PDH activity
PDH complex specific activity was determined as the rate of 14CO2 formed from [1-14 C]-pyruvate/mg protein/min as described previously [17, 18].
2.11 Statistical analysis
Results are presented as mean ± standard deviation (SD). Differences between groups were identified using a grouped-unpaired two-tailed distribution of Student’s T test. P values ≤ 0.05 were considered statistically significant.
3. Results
3.1 Transduction efficiency of AAV serotype vectors in human HB and HCC cells
The recombinant AAV serotype 8 vector has shown strong tropism and high transduction efficiency for mouse hepatocytes [19, 20], while the relative capability of AAV serotypes to transduce human HB and HCC is unknown. Therefore, we transduced Huh-6, Huh-7 and HepG2 cells with recombinant scAAV serotype vectors 1 through 10 containing a CMV promoter-driven enhanced green-fluorescent protein gene (egfp). CMV is known to be a strong promoter capable of transgene expression within a wide variety of cell types. Cells were either mock-infected or infected with ∼2,000 AAV vector particles per cell under identical conditions and were visualized under fluorescent microscopy 96 hours after infection. Fig. 1 demonstrates that AAV3 is by far the most efficient vector in transducing HB and HCC cell lines in vitro such that over 60% of cells expressed EGFP.
Fig. 1.



Relative efficiency of AAV serotype vectors for transduction of HB (A,) and HCC (B, C) cells in vitro. Representative direct fluorescent images of Huh-6 (A), Huh-7 (B) and HepG2 (C) cell lines infected with scAAV.CMV-egfp serotype vectors 1 through 10 are presented (2,000 viral genomes per cell). Images were taken on the fourth day after infection. Histograms are quantified for transduction efficiency of each cell line infected. P<0.001 for scAAV3 vs other serotypes.
We then investigated the ability of a CBA promoter to drive the reporter expression from scAAV3 vector and infected Huh-6 cells with multiplicities of either 1,000 or 5,000 vector genomes per cell (Fig. 2). The transduction efficiency was vector-dose dependent, from about 60% to almost 90% transduction at the highest vector dose used.
Fig. 2.

Transduction of Huh-6 cells by scAAV3 vectors is dose-dependent. Panel A: Fluorescent micrographs of cells infected by scAAV3.CBA-egfp vector with either 1,000 or 5,000 viral genomes per cell vs. mock infection; phase contrast (PC) images are also shown. Panel B: Quantification of transduction by FACS analysis with FITC filter. Vg/cell, viral genomes per cell.
HEK 293 cells and human primary hepatocytes were also tested for their susceptibility to transduction by a scAAV3.CMV-egfp vector (Fig. 3). At a dose of ∼2,000 virus particles per cell, HEK293 cells were transduced poorly whereas human primary hepatocytes were transduced efficiently.
Fig. 3.

Transduction of HEK 293 cells (A) and primary human hepatocytes (B). Representative direct fluorescent micrographs are from mock infected cells or from cells 72 hours after infection with scAAV3.CMV-egfp vectors (2,000 viral genomes per cell).
3.2 HB- and HCC- specific transgene expression can be achieved by AAV3 serotype vectors
HB, HCC, HEK 293 cells and primary human hepatocytes were infected with∼2,000 particles per cell of scAAV3 vectors containing the egfp gene under the control of the human alpha-fetoprotein (AFP) promoter. AFP is a major plasma protein produced by the yolk sac and the liver during fetal life and is considered to be the fetal counterpart of albumin [21]. AFP levels decrease gradually after birth and serve as a biomarker for a subset of tumors, principally HB and HCC [22-25].
ScAAV3.AFP-egfp vector-mediated transgene expression indeed occurred in HB and HCC, but not HEK 293 cells and normal human hepatocytes (Fig. 4A). About 64% of HB cells could be transduced with a dose of 2,000 vector particles/cell and this transduction efficiency was sustained for at least 9 days, when infected cells were passaged and propagated further (Fig. 4B).
Fig. 4.
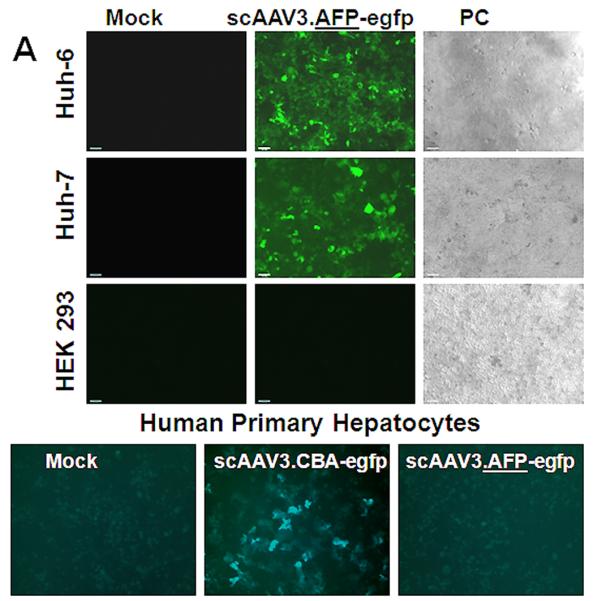

HB- and HCC- specific transgene expression directed by scAAV3.AFP-egfp vector. Panel A: Representative direct fluorescent images were taken on the fifth day after infection with a multiplicity of 2,000 viral genomes per cell. Panel B: Quantification of transduction by FACS analysis with FITC filter.
We next tested pda1 transgene expression and activity in Huh-6 cells after infection with the scAAV3.AFP-pda1 vector (Fig. 5). To discriminate a pda1 transgene expression from its genomic counterpart expression, three groups of samples were compared by Western blotting: lysates of mock-infected cells, lysates of cells infected with control, scAAV3.AFP-egfp and lysates infected with the scAAV3.AFP-pda1 vector. Only cells infected with the vector containing the pda1 transgene inc with an increase in PDH E1α expression (Fig. 5A), which correlated well with an increase in PDH complex enzymatic activity (Fig. 5B).
Fig. 5.
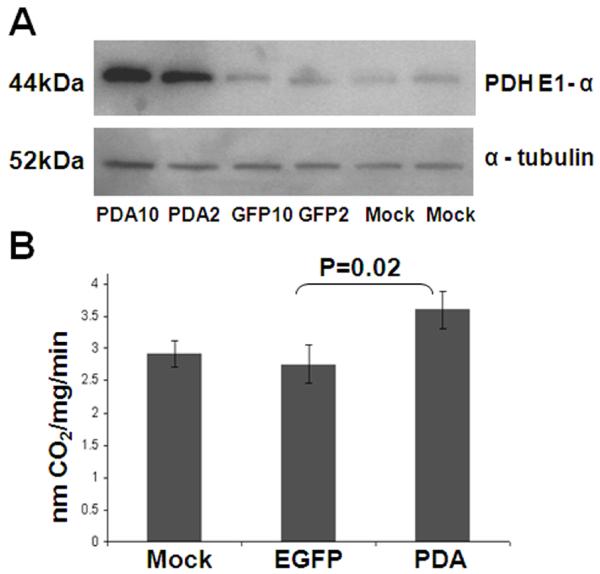
ScAAV.AFP-pda1 vector induces PDH E1 alpha subunit expression and increases PDC activity in Huh-6 cells. Panel A: Lysates of uninfected (mock) Huh-6 cells and cells infected with scAAV3.AFP-egfp (EGFP, control) and scAAV3.AFP-pda1 (PDA) viruses with 2,000 (EGFP 2, PDA 2) or 10,000 (EGFP 10, PDA 10) viral genomes per cell were analyzed by Western blotting with antibodies that recognized PDH E1αsubunit 44k Da protein. The loading control was α-tubulin. Panel B: PDH activity was measured and expressed as nmoles 14CO2 per mg protein·min. Samples were collected on the eighth day after infection.
3.3 Apoptosis of scAAV3.AFP-pda1-infected Huh-6 cells
Early in apoptosis, phosphatidylserine becomes exposed on the cell surface by flipping from the inner to outer leaflet of the cytoplasmic membrane. Conjugated Annexin V binds to the exposed phosphatidylserine while 7-AAD subsequently enters cells and binds to DNA. Virus-infected and mock-infected cells were doubly labeled by Annexin V-APC conjugate and 7-AAD and were then processed by FACS analysis (Fig.6). Transduction by the scAAV.AFP-pda1 vector induced a significant (p<0.005) increase in late-stage apoptosis within five days of infections, compared to scAAV.AFP-egfp-infected or mock-infected cells. The population of cells treated with scAAV.AFP-pda1 entering late-stage apoptosis increased progressively and was greater than that measured in control cells seven days after transduction (p<0.001; Fig. 6A). In contrast, neither scAAV.AFP-pda1 nor scAAV.AFP-egfp vectors caused significant cell death in either HEK 293 cells eight days after infection (Fig.6B).
Fig. 6.

Induction of apoptosis in pda1-transduced Huh-6 cells. Huh-6 cells and normal HEK 273 cells were infected with scAAV3.AFP-pda1 and control scAAV.AFP-egfp vectors (2,000 viral genoms per cell) or were mock-infected. Cells were double-labeled with Annexin V-APC and 7-AAD and processed by FACS 7 days after infection (Panel A, B). Representative scatterplots from FACS analysis are shown. Data are expressed as DNA staining by 7-AAD relative to Annexin V binding to cell membrane. Bar graphs indicate mean ± SD of FACS data from 3 separate experiments and indicate the percentage of cells in each quadrant. No significant differences in apoptosis were detected between mock and scAAV3.AFP-egfp infected Huh-6 cells (Panel A) or HEK 293 cells (Panel B). In contrast, infection of Huh-6 cells with scAAV3.AFP-pda1 resulted in a significant increase in late-stage apoptosis (Panel A; p<0.001) but not in HEK 293 cell (Panel B, p>0.5). live cells
(Panel B, p>0.5). live cells ower left panel); early-stage apoptosis (lower right panel); □ late-stage apoptosis (upper right panel);
ower left panel); early-stage apoptosis (lower right panel); □ late-stage apoptosis (upper right panel);  dead cells (upper left panel).
dead cells (upper left panel).
3.4 Apoptosis of HepG2 cells transfected with pscAAV.AFP-pda1
Hep G2 cells were transfected with either pscAAV.AFP-pda1 proviral plasmid, including pda1 gene under AFP promoter and SV40 enhancer regulation or control plasmid pDrive-SV40-hAFP that contained the same promoter-enhancer driven lacZ gene (Fig.7). The quality of the transfection was determined by a control transfection with a pscAAV.AFP-egfp plasmid (Fig. 7A). Adherent cells were labeled in situ with AnnexinV-FITC conjugate and PI and visualized by fluorescence microscopy (Fig. 7B). Only the transfection with pscAAV.AFP-pda1 plasmid resulted in the appearance of apoptotic AnnexinV-FITC positive cell clusters that co-localized well with PI staining. A background level of AnnexinV positive cells was seen using the control plasmid (Fig. 7C).
Fig. 7.

Induction of apoptosis in HepG2 cells. Panel A: Representative micrographs of cells transfected with pscAAV3.AFP-egfp (Panel A), pscAAV3.AFP-pda1 (Panel B) or control plasmid (Panel C). PC, phase contrast.
3.5 Induction of Bcl-X/S with scAAV3.AFP-pda1 vector
Bcl-X is a member of the Bcl-2 family of proteins that regulates the permeability of the outer mitochondrial membrane [26]. Alternative splicing results in two distinct Bcl-X isoforms: Bcl-X/L and Bcl-X/S [27-29]. Bcl-X/L (∼31 kDa) is an anti-apoptotic protein that prevents release of cytochrome C from the mitochondrial intermembrane space into the cytosol. Bcl-X/S (∼21 kDa) is a proapoptotic member of the Bcl-2 protein family that is localized in the mitochondria and induces apoptosis in a caspase and BH3-dependent manner by a mechanism involving cytochrome c release [30]. We analyzed these two products of the Bcl-x gene in extracts of mock-infected Huh-6 cells or cells infected either with scAAV.AFP-pda1 or scAAV.AFP-egfp vectors (Fig. 8). Western blotting using Bcl-X specific antibody against both splice-isoforms revealed a 31 kDa- protein that corresponded to the Bcl-X/L isoform in extracts of all three cell samples. In contrast, the 21 kDa Bcl-X/S isoform was expressed only in extracts of scAAV.AFP-pda1-infected cells. A large Bcl-X positive signal (∼50 kDa) was also observed in cells transduced with the scAAV.AFP-pda1 vector and may represent a homo or hetero-dimer of Bcl-X/S [31].
Fig. 8.

Induction of Bsl-X/S in Huh-6 cells by scAAV3.AFP-pda1 vector. Panel A: Bcl-X/L and Bcl-X/S proteins are splice isoforms of Bcl-X: BH, Bcl-2 homology domains; TM transmembrane domain. Panel B: Lysates of Huh-6 cells infected with scAAV3.AFP-egfp (EGFP) or scAAV3.AFP-pda1 (PDA) viruses or mock infected were analyzed by Western blotting with antibody that recognize both isoforms of Bcl-X (L-19). The loading control was α-tubulin.
3.6 Release of cytochrome c with scAAV.AFP-pda1
Bcl-X/S can induce apoptosis via cytochrome c release [30] that, in turn, activates caspase 9, a cysteine protease, which then activates caspases 3 and 7 and precipitates apoptotic cell death.
Fig. 9 illustrates that a strong cytochrome c signal was found in the cytoplasm of Huh-6 cells infected by scAAV.AFP-pda1 but was not found in mock-infected or in cells infected with the control vector.
Fig. 9.

Cytochrome c release in Huh-6 cells infected with scAAV3.AFP-pda1 virus. Representative fluorescence micrographs were taken on the 7th day after infection. Cytochrome c is visualized with Texas Red conjugated secondary antibodies after mock infection or after infection with scAAV3.AFP-pda1 and scAAV3.AFP-egfp vectors. Scale bar, 100 pixels.
4. Discussion
Cancer cells are considered to be metabolic anomalies because they rely disproportionately on glycolysis for energy, even in the presence of adequate oxygen supply. First described in detail by Warburg [32], the “aerobic glycolysis” of tumors was initially considered a compensatory mechanism for survival by cells whose capacity for oxidative phosphorylation was intrinsically impaired. We now know that the transcriptional activation of specific genes involved in carbohydrate metabolism in tumors leads to both upregulation of glycolysis in the cytosol and downregulation of glucose oxidation by the mitochondria [5, 33-35]. The temporal coupling of these events results in the abnormal accumulation of lactate and protons that, in turn, may enhance the invasive potential of cancer cells [36].
The recognition of the molecular mechanisms underlying the metabolic remodeling of tumors has generated considerable interest in identifying potential therapeutic targets along the pathway of carbohydrate metabolism from the transport of glucose across the plasma membrane, to its cytoplasmic conversion to pyruvate and lactate and to the transport of pyruvate into mitochondria and its subsequent oxidation by PDC [33-35, 37].
Several key steps were required to successfully undertake this study. First, we employed self-complementary (sc) AAV vectors that contained double-stranded DNA. This eliminated the requirement for viral second-strand DNA synthesis and thus increased the efficiency of transgene expression. Next, we examined a panel of 10 AAV serotypes in which each vector contained a CMV promoter-driven egfp gene to identify the optimal serotype for high-efficiency transduction of HB and HCC cells. We discovered that scAAV3 vectors were extremely efficient in transducing HB and HCC cell line and primary cultures of normal human hepatocytes and were far superior to any other AAV serotype. This finding was unanticipated because AAV3 has not previously been shown to be particularly effective in transducing mammalian cells [38]. The mechanism accounting for the marked trophism of AAV3 vectors toward liver cells is unknown and warrants further investigation. Lastly, we incorporated the AFP gene promoter in scAAV3 vector to selectively drive the transgene expression in tumor, but not normal, liver cells.
Infection of HB and HCC cells with the scAAV3.AFP-pdal vector resulted in enhanced PDC catalytic activity. This was temporally associated with induction of Bcl-X/S, release of cytochrome c and apoptosis of infected cells. These findings are consistent with results obtained by several laboratories [6-10] showing that pharmacological stimulation of PDC by DCA also leads to selective apoptotic cell death of human cancer cells in vitro or in vivo. Thus, we interpret our results as demonstrating the potential of genetic upregulation of PDC to induce a state of metabolic remodeling of tumor cells, leading an increase in their capacity for oxidative metabolism, ROS accumulation and apoptosis.
HB is the most frequent liver cancer among children and HCC is a common cancer in adults worldwide [39, 40]. HCC accounts for approximately 600,000 deaths per year in the world [41]. Unfortunately, except for surgical resection or liver transplantation for early HCC (less than 10% of the cases), currently available chemotherapy and radiation therapy are not effective for adult HCC, although chemotherapy is effective for HM [42]. Thus, more effective therapies are urgently needed. If scAAV3 vector-mediated transduction can be documented to cause selective elimination of human HB and HCC cells in an animal xenograft model of human liver tumors in vivo, the gene delivery and expression strategy described here may be feasible for targeting liver cancer in humans.
Acknowledgements
We thank Candace Caputo for editorial assistance. This work was supported by R01 HL-07691, P01 DK-058327 (Project 1) and U54RR025208 from the National Institutes of Health.
Abbreviations
- PDC
pyruvate dehydrogenase complex
- DCA
dichloroacetate
- ROS
reactive oxygen species
- TCA
tricarboxylic
- HB
hepatoblastoma
- HCC
hepatocellular carcinoma
- AAV
adeno-associated virus
- EGFP
enhanced green fluorescent protein
- CMV
cytomegalovirus
- CBA
chicken beta-actin
- AFP
alpha-fetoprotein.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Patel MS, Roche TE. Molecular biology and biochemistry of pyruvate dehydrogenase complexes. Faseb J. 1990;4:3224–3233. doi: 10.1096/fasebj.4.14.2227213. [DOI] [PubMed] [Google Scholar]
- [2].Holness MJ, Sugden MC. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem Soc Trans. 2003;31:1143–1151. doi: 10.1042/bst0311143. [DOI] [PubMed] [Google Scholar]
- [3].Stacpoole PW. The pharmacology of dichloroacetate. Metabolism. 1989;38:1124–1144. doi: 10.1016/0026-0495(89)90051-6. [DOI] [PubMed] [Google Scholar]
- [4].Gillies RJ, Robey I, Gatenby RA. Causes and consequences of increased glucose metabolism of cancers. J Nucl Med. 2008;49(Suppl 2):24S–42S. doi: 10.2967/jnumed.107.047258. [DOI] [PubMed] [Google Scholar]
- [5].Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13:472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- [6].Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- [7].Cairns RA, Papandreou I, Sutphin PD, Denko NC. Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy. Proc Natl Acad Sci U S A. 2007;104:9445–9450. doi: 10.1073/pnas.0611662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate. 2008;68:1223–1231. doi: 10.1002/pros.20788. [DOI] [PubMed] [Google Scholar]
- [9].Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol. 2008;109:394–402. doi: 10.1016/j.ygyno.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sun W, Zhou S, Chang SS, McFate T, Verma A, Califano JA. Mitochondrial mutations contribute to HIF1alpha accumulation via increased reactive oxygen species and up-regulated pyruvate dehydrogenease kinase 2 in head and neck squamous cell carcinoma. Clin Cancer Res. 2009;15:476–484. doi: 10.1158/1078-0432.CCR-08-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer. 2008;99:989–994. doi: 10.1038/sj.bjc.6604554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Thomas MB, Zhu AX. Hepatocellular carcinoma: the need for progress. J Clin Oncol. 2005;23:2892–2899. doi: 10.1200/JCO.2005.03.196. [DOI] [PubMed] [Google Scholar]
- [13].Deuffic S, Poynard T, Buffat L, Valleron AJ. Trends in primary liver cancer. Lancet. 1998;351:214–215. doi: 10.1016/S0140-6736(05)78179-4. [DOI] [PubMed] [Google Scholar]
- [14].Wilson JF. Liver cancer on the rise. Ann Intern Med. 2005;142:1029–1032. doi: 10.7326/0003-4819-142-12_part_1-200506210-00024. [DOI] [PubMed] [Google Scholar]
- [15].Zhu H, Elyar J, Foss R, Hemming A, Hall E, Lecluyse EL, Liu C. Primary human hepatocyte culture for HCV study. Methods Mol Biol. 2009;510:373–382. doi: 10.1007/978-1-59745-394-3_28. [DOI] [PubMed] [Google Scholar]
- [16].Han Z, Berendzen K, Zhong L, Surolia I, Chouthai N, Zhao W, Maina N, Srivastava A, Stacpoole PW. A combined therapeutic approach for pyruvate dehydrogenase deficiency using self-complementary adeno-associated virus serotype-specific vectors and dichloroacetate. Mol Genet Metab. 2008;93:381–387. doi: 10.1016/j.ymgme.2007.10.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Berendzen K, Theriaque DW, Shuster J, Stacpoole PW. Therapeutic potential of dichloroacetate for pyruvate dehydrogenase complex deficiency. Mitochondrion. 2006;6:126–135. doi: 10.1016/j.mito.2006.04.001. [DOI] [PubMed] [Google Scholar]
- [18].Han Z, Gorbatyuk M, Thomas J, Jr., Lewin AS, Srivastava A, Stacpoole PW. Down-regulation of expression of rat pyruvate dehydrogenase E1alpha gene by self-complementary adeno-associated virus-mediated small interfering RNA delivery. Mitochondrion. 2007;7:253–259. doi: 10.1016/j.mito.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Thomas CE, Storm TA, Huang Z, Kay MA. Rapid uncoating of vector genomes is the key to efficient liver transduction with pseudotyped adeno-associated virus vectors. J Virol. 2004;78:3110–3122. doi: 10.1128/JVI.78.6.3110-3122.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shen X, Storm T, Kay MA. Characterization of the relationship of AAV capsid domain swapping to liver transduction efficiency. Mol Ther. 2007;15:1955–1962. doi: 10.1038/sj.mt.6300293. [DOI] [PubMed] [Google Scholar]
- [21].Harper ME, Dugaiczyk A. Linkage of the evolutionarily-related serum albumin and alpha-fetoprotein genes within q11-22 of human chromosome 4. Am J Hum Genet. 1983;35:565–72. [PMC free article] [PubMed] [Google Scholar]
- [22].Rebischung C, Pautier P, Morice P, Lhomme C, Duvillard P. Alpha-fetoprotein production by a malignant mixed Mullerian tumor of the ovary. Gynecol Oncol. 2000;77:203–205. doi: 10.1006/gyno.1999.5653. [DOI] [PubMed] [Google Scholar]
- [23].Crocoli A, Madafferi S, Jenkner A, Zaccara A, Inserra A. Elevated serum alpha-fetoprotein in Wilms tumor may follow the same pattern of other fetal neoplasms after treatment: evidence from three cases. Pediatr Surg Int. 2008;24:499–502. doi: 10.1007/s00383-007-2067-7. [DOI] [PubMed] [Google Scholar]
- [24].Paul SB, Gulati MS, Sreenivas V, Madan K, Gupta AK, Mukhopadhyay S, Acharya SK. Evaluating patients with cirrhosis for hepatocellular carcinoma: value of clinical symptomatology, imaging and alpha-fetoprotein. Oncology. 2007;72(Suppl 1):117–123. doi: 10.1159/000111717. [DOI] [PubMed] [Google Scholar]
- [25].Kim do Y., Paik YH, Ahn SH, Youn YJ, Choi JW, Kim JK, Lee KS, Chon CY, Han KH. PIVKA-II is a useful tumor marker for recurrent hepatocellular carcinoma after surgical resection. Oncology. 2007;72(Suppl 1):52–57. doi: 10.1159/000111707. [DOI] [PubMed] [Google Scholar]
- [26].Kirkin V, Joos S, Zornig M. The role of Bcl-2 family members in tumorigenesis. Biochim Biophys Acta. 2004;1644:229–249. doi: 10.1016/j.bbamcr.2003.08.009. [DOI] [PubMed] [Google Scholar]
- [27].Taylor JK, Zhang QQ, Wyatt JR, Dean NM. Induction of endogenous Bcl-xS through the control of Bcl-x pre-mRNA splicing by antisense oligonucleotides. Nat Biotechnol. 1999;17:1097–1100. doi: 10.1038/15079. [DOI] [PubMed] [Google Scholar]
- [28].Mercatante DR, Bortner CD, Cidlowski JA, Kole R. Modification of alternative splicing of Bcl-x pre-mRNA in prostate and breast cancer cells. Analysis of apoptosis and cell death. J Biol Chem. 2001;276:16411–16417. doi: 10.1074/jbc.M009256200. [DOI] [PubMed] [Google Scholar]
- [29].Wilhelm E, Pellay FX, Benecke A, Bell B. Determining the impact of alternative splicing events on transcriptome dynamics. BMC Res Notes. 2008;1:94. doi: 10.1186/1756-0500-1-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lindenboim L, Kringel S, Braun T, Borner C, Stein R. Bak but not Bax is essential for Bcl-xS-induced apoptosis. Cell Death Differ. 2005;12:713–723. doi: 10.1038/sj.cdd.4401638. [DOI] [PubMed] [Google Scholar]
- [31].Lindenboim L, Borner C, Stein R. Bcl-x(S) can form homodimers and heterodimers and its apoptotic activity requires localization of Bcl-x(S) to the mitochondria and its BH3 and loop domains. Cell Death Differ. 2001;8:933–942. doi: 10.1038/sj.cdd.4400888. [DOI] [PubMed] [Google Scholar]
- [32].Warburg O. The metabolism of tumor. Constable; London: 1930. [Google Scholar]
- [33].Kroemer G. Mitochondria in cancer. Oncogene. 2006;25:4630–4632. doi: 10.1038/sj.onc.1209589. [DOI] [PubMed] [Google Scholar]
- [34].Gatenby RA, Gillies RJ. Glycolysis in cancer: a potential target for therapy. Int J Biochem Cell Biol. 2007;39:1358–1366. doi: 10.1016/j.biocel.2007.03.021. [DOI] [PubMed] [Google Scholar]
- [35].Pan JG, Mak TW. Metabolic targeting as an anticancer strategy: dawn of a new era? Sci STKE. 2007;381:1–4. doi: 10.1126/stke.3812007pe14. [DOI] [PubMed] [Google Scholar]
- [36].Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev Cancer. 2008;8:56–61. doi: 10.1038/nrc2255. [DOI] [PubMed] [Google Scholar]
- [37].Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, Wahl ML, Feron O, Dewhirst MW. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930–3942. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- [39].Finegold MG, Egler RA, Goss JA, Guillerman RP, Karpen SJ, Krishnamurthy R, O’Mahony CA. Liver tumors: pediatric population. Liver Transpl. 2008;14:1545–56. doi: 10.1002/lt.21654. [DOI] [PubMed] [Google Scholar]
- [40].Williams R. Global challenges in liver disease. Hepatology. 2006;44:521–6. doi: 10.1002/hep.21347. [DOI] [PubMed] [Google Scholar]
- [41].El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–76. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- [42].El-Serag HB, Marrero JA, Rudolph L, Reddy KR. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology. 2008;134:1752–63. doi: 10.1053/j.gastro.2008.02.090. [DOI] [PubMed] [Google Scholar]