

Abstract
Synaptic plasticity is dependent upon the differential sorting, delivery and retention of neurotransmitter receptors, yet the mechanisms underlying these processes are poorly understood. In the present study, we have found that differential sorting of glutamate receptor subtypes begins within the endoplasmic reticulum (ER) of rat hippocampal neurons. While AMPARs are trafficked to the plasma membrane via the conventional somatic Golgi network, NMDARs are diverted from the somatic ER into a specialized ER sub-compartment that bypasses somatic Golgi, merging instead with dendritic Golgi outposts. Intriguingly, this ER sub-compartment is composed of highly mobile vesicles containing the NMDAR subunits NR1 and NR2B, the microtubule-dependent motor protein KIF17, and the postsynaptic adaptor proteins CASK and SAP97. Furthermore, our data demonstrate that the retention and trafficking of NMDARs within this ER sub-compartment requires both CASK and SAP97. These data indicate that NMDARs are sorted away from AMPARs via a non-conventional secretory pathway that utilizes dendritic Golgi outposts.
Keywords: Glutamate receptors, MAGUK proteins, NMDA receptors, protein trafficking, SAP97, CASK, shRNA, Endoplamic reticulum, Dendritic Golgi
N-methyl-D-Aspartate-type glutamate receptors (NMDARs) regulate synaptic plasticity by functioning as coincidence detectors that integrate synapse-specific information with the overall excitability of neuronal cells1. NMDAR activation is essential for eliciting changes in synaptic strength, primarily by regulating the levels of postsynaptic (AMPA)-type glutamate receptors (AMPARs)2,3. Numerous studies demonstrate that NMDARs and AMPARs are independently delivered to nascent synapses during synaptogenesis4,5,6, and to mature synapses during synaptic plasticity2,3. Mechanistically, sorting of these receptors into distinct vesicles4,6, and/or their differential retention at the postsynaptic density (PSD)7 may underlie these differences, though the relative contribution of each mechanism remains unclear.
Studies on AMPAR biogenesis, transport, and synaptic delivery suggest a three step mechanism, wherein receptor subunits are synthesized within somatic endoplasmic reticulum (ER) and Golgi, transported to the plasma membrane by constitutive membrane flow8,9, and subsequently internalized into recycling endosomes10 prior to their activity-dependent re-insertion at extrasynaptic sites11,12 and capture by scaffold proteins within the PSD7,13. It is unclear whether NMDARs follow a similar biosynthetic pathway, and/or at what stage they are sorted from AMPARs. Studies suggest that members of the membrane-associated guanylate kinase (MAGUK) family of synaptic scaffold proteins (e.g., SAP97, PSD-95, SAP102 and CASK; see7), the protein GRIP/ABP, and subclasses of microtubule and actin-dependent motor proteins contribute to the differential sorting and trafficking of NMDA and AMPARs14,15,16,17,18. For example, SAP97 associates with AMPAR subunits during their biosynthesis and transport to the plasma membrane15, and GRIP1 and KIF5 participate in the synaptic delivery of these receptors19. Similarly, subunits of the NMDAR form complexes with CASK, Velis/MALS, Mint, and KIF17 on vesicles that move rapidly along dendritic microtubules14,19 at rates distinct from vesicles carrying AMPARs6,20. Additional reports indicate that NMDAR subunits also form complexes with SAP102, sec8, and mPins21,22, as well as SAP9723,24,25. However, it remains unclear when and where each complex forms or how each contributes to individual steps in the biogenesis, transport, and recycling of NMDARs.
Here, we have explored the mechanisms that underlie sorting of AMPARs versus NMDARs. We show that NMDARs are trafficked via a SAP97 and CASK-dependent pathway from somatic ER to a dendritic ER sub-compartment, and subsequently to Golgi outposts. In contrast, AMPARs follow the conventional route from somatic ER and Golgi to reach the plasma membrane. These data not only provide novel insights into the cellular mechanisms underlying glutamate receptor sorting, but also indicate that dendritic Golgi outposts26 are part of a functionally distinct secretory pathway.
Results
NMDARs bypass somatic Golgi
Sorting of membrane proteins typically occurs at the trans-Golgi network (TGN)27. To determine whether glutamate receptors traffic through somatic TGN, we co-transfected EGFP-tagged AMPAR (GFP-GluR1) or NMDAR (NR1-GFP) subunits with a constitutively active mutant of the GTPase ARF1 (ARF1-Q71I), which causes the accumulation of cargo within the Golgi by preventing COPI-coated vesicle formation28.
When GFP-GluR1 was co-transfected into cultured hippocampal neurons with HA-tagged ARF1-Q71I (ARF1-Q71I-HA), both proteins accumulated in the cell soma and co-localized with antibodies against the Golgi marker GM130 (Fig. 1A). Quantifying the percent overlap with GM130 revealed a 45% increase in the levels of Golgi-localized GFP-GluR1 in neurons co-expressing ARF1-Q71I-HA (p=0.02, t-test). In contrast, we saw no accumulation of NR1-GFP in neurons co-expressing ARF1-Q71I-HA (Fig. 1A), indicating that NMDARs may bypass somatic Golgi. Similar results were obtained with two other conditions that block protein trafficking through the Golgi (Fig. S1).
Figure 1.


NMDARs exit the soma via a somatic Golgi-independent pathway and insert into dendritic Golgi outposts. A,B, Hippocampal neurons were transfected with GFP-GluR1 or NR1-GFP alone, or either subunit along with ARF-Q71I-HA. Cultures were fixed at 12–15h post-transfection. A) Co-expression with ARF-Q71I-HA significantly increased the Golgi localization of GFP-GluR1 (arrows, second row) but had no effect on NR1-GFP distribution, or golgi localization (fourth row). B) In dendrites, co-expression with ARF-Q71I-HA resulted in a significant accumulation of NR1-GFP at Golgi outposts (bottom panel), as compared to the subunit expressed alone (top panel). C–E, Accumulation of endogenous NMDARs at Golgi outposts following mutant ARF1 over-expression. C) Expression of the ARF1 mutant had no effect on NR1 localization in somatic Golgi (arrows, bottom row). D,E) Cultures transfected with the Golgi marker, Galtase-GFP, alone (top rows), or with ARF-Q71I-HA (bottom rows). Endogenous NR1 (D) and NR2B (E) exhibited minimal co-localization with outposts in neurons only expressing Galtase-GFP (arrows in top rows, merged panels). Co-expression of the ARF1 mutant resulted in an increase in the co-localization of both NR1 and NR2B with Golgi outposts (arrows in bottom rows, merged panels), as well as in the fluorescence intensity at these structures (pseudo-color intensity panels; increasing intensity is reflected in progression from blue-to-red-to-yellow). Scale bars, 10 μm.
NMDARs traffic through dendritic Golgi outposts
The absence of NR1-GFP in somatic Golgi following co-expression with ARF1-Q71I was puzzling because membrane proteins are usually post-translationally processed within the Golgi before their insertion at the plasma membrane. We thus tested whether NMDARs instead use dendritic Golgi membranes. These “Golgi outposts” are considered an extension of the somatic Golgi29, or a site of biosynthesis for integral membrane proteins translated from dendritically localized mRNAs26,30. To assess a role for Golgi outposts in NMDAR trafficking and processing, we again used ARF1-Q71I-HA to block Golgi trafficking. As with somatic Golgi, GM130 immunoreactivity identified dendritic Golgi outposts (Fig. 1B). In neurons expressing NR1-GFP alone, we observed modest co-localization with GM130-positive puncta. However, when ARF1-Q71I-HA was co-transfected with NR1-GFP, we detected a significant increase in Golgi outpost size and the intensity of co-localizing NR1-GFP puncta (~3–fold, p=0.029), consistent with NMDARs trafficking through dendritic rather than somatic Golgi. No accumulation of AMPAR subunits was observed in Golgi outposts following ARF1-Q71I block, as almost all GFP-GluR1 remained in the soma and failed to traffic beyond proximal dendritic segments (Fig. S2).
To rule out the possibility that NR1-GFP overexpression was responsible for these findings, we examined the effects of ARF1-Q71I-HA on endogenous NR1 and NR2B subunits. Again, ARF1-Q71I-HA did not trigger the accumulation of NR1 within somatic Golgi (Fig. 1C), but did significantly increase the percent overlap between dendritic NR1- and NR2B- immunoreactive puncta (60% and 43%, p=0.016 and 0.007, respectively) and Golgi outposts (labeled with the EGFP-tagged Golgi marker galactosyltransferase, Galtase-GFP). This accumulation was not due to alterations in synapse stability or formation by ARF1-Q71I-HA expression (Fig. S3). These data demonstrate that NMDARs traffic from the soma via a pathway that bypasses somatic Golgi but utilizes dendritic Golgi, in contrast to the conventional sorting pathway of AMPARs.
NMDAR transport in dendrites occurs via a mobile ER sub-compartment
If NMDARs bypass somatic Golgi by budding directly from the ER, then NMDAR-containing transport vesicles in dendrites may have features in common with the ER. Recent studies have demonstrated the existence of an ER sub-compartment throughout dendrites of hippocampal neurons29. The ER sub-compartment contains ER-resident proteins, proteins involved in Ca+2 storage and release31,32, and associated vesicles that move via kinesin motors in both the anterograde and retrograde direction at velocities of 0.2 – 0.3 μm/sec at 36–37°C31,32. To assess whether NMDAR-containing vesicles are part of this compartment, we utilized three different ER markers: 1) anti-KDEL Abs, 2) anti-IP3 receptor Abs, and 3) DsRed-ER (a DsRed fusion protein containing ER-retention signals from both calreticulin and KDEL). All three markers co-localize with each other and with newly synthesized NR1-GFP in small punctate structures within dendrites (Fig. 2A).
Figure 2.


NMDARs traffic in a mobile, vesicular, ER sub-compartment. Hippocampal neurons were transfected with DsRed-ER and/or NR1-GFP, fixed at 1day post-transfection, and stained with antibodies against KDEL, NR1, or IP3R. A) Co-distribution of previously characterized markers for the mobile ER sub-compartment in dendrites (top row). Newly synthesized, trafficking puncta of NR1-GFP co-localize with KDEL, IP3R, and DsRed-ER (arrows; second, third, and fifth row, respectively). Dendritic DsRed-ER puncta co-localize with endogenous NR1 subunits (arrows, fourth row). Scale bars, 5 μm. B) Inverted monochrome images from time-lapse imaging of neurons co-expressing NR1-GFP (top row) and DsRed-ER (bottom row). NR1-GFP and DsRed-ER-containing puncta (arrows) were highly mobile (arrowhead indicates position at time = 0 s). Scale bar, 5 μm. C) Cultures were transfected with DsRed-ER, treated with BFA at 2h post-transfection for a period of 10h, then fixed and stained with antibodies against NR2B and GluR1. DsRed-ER-containing puncta that trafficked out of the soma despite Golgi membrane disruption co-localized highly with NR2B (arrows), but not GluR1 (arrows), in both proximal (top panels) and distal (bottom panels) dendritic processes. Scale bar, 10 μm.
To further characterize these NMDAR-containing vesicles, we performed live imaging on 13 day old neurons co-expressing newly-synthesized NR1-GFP and DsRed-ER. We routinely observed small, discrete, highly mobile NR1-GFP puncta that also contained DsRed-ER (Fig. 2B). These puncta moved rapidly along dendrites in both anterograde and retrograde directions at a mean rate of 0.26 ±0.03 μm per second at 36–37°C (for 11 measured events). This rate is similar to that reported previously for ER-like vesicles in dendrites31,32, as well as for NMDAR-containing vesicles6,17.
We next tested whether other NMDAR or AMPAR subunits were also present on ER sub-compartment vesicles. Brefeldin A (BFA) was used to enrich for ER-vesicles trafficking from the soma. Cultures were transfected with DsRed-ER, treated with BFA (2h post-transfection), and assayed for endogenous NR2B and GluR1 subunits on ER sub-compartment puncta (Fig. 2C). We observed a high degree of co-localization between DsRed-ER and NR2B (60%), but not GluR1 (19%), consistent with NR1 and NR2B-containing NMDARs, but not GluR1-containing AMPARs, in ER-derived vesicles that traffic to dendrites.
It was previously shown that dendritic transport of NMDARs is mediated by a complex composed of three multi-domain scaffold proteins, CASK (Lin2), Velis/MALS (Lin7), and Mint (Lin10), that tether NMDARs to the kinesin KIF1714,33. We used immunostaining to evaluate whether the observed NMDAR-containing vesicles contained CASK and KIF17 (Fig. S4). We found that clusters of CASK and KIF17 exhibited a high degree of overlap with NR1-GFP (Fig. S4A,B) and DsRed-ER puncta (Fig. S4C,D), suggesting that mobile NMDAR/ER vesicles share features with previously characterized NMDAR transport vesicles.
SAP97 associates with NMDARs and CASK and alters NMDAR trafficking
CASK not only forms complexes with Velis/MALS and Mints33,34, but also with another MAGUK, SAP9735,36. SAP97 interacts via its N-terminal PDZ domains with the C-terminal tails of NR2A or NR2B subunits of the NMDAR23, and is known to do so within the ER of transfected cells25,37. Taken together, these features led us to hypothesize that a complex containing SAP97 and CASK may traffic NMDARs into the dendritic ER sub-compartment.
This concept was initially tested in HEK293 cells by co-expressing HA-NR1 (HA-tagged on its extracellular domain) and NR2B subunits. We detected NMDARs at the cell surface, consistent with assembled NMDARs having transited through the ER and Golgi (Fig. 3A,E). However, when co-transfected with EGFP-tagged SAP97, surface NMDAR expression was lost (Fig. 3B,E), and nearly all NMDARs were sequestered in large structures co-localizing precisely with EGFP-SAP97 fluorescence (Fig. 3B). These clusters also co-localized with ERp57 (Fig. 3B), a resident ER protein, suggesting that SAP97 sequestered NMDARs within an ER-derived compartment. While SAP97 can also bind AMPAR GluR1 subunits38, it had no effect on the surface expression or ER distribution of these receptors when a HA-tagged version of GluR1 (HA-GluR1) was co-expressed with EGFP-SAP97 (Fig. 3A).
Figure 3.


SAP97 and CASK alter NMDAR trafficking in HEK293 cells. A) Top row, transient transfection with: 1) GFP-SAP97, 2) HA-tagged-NMDARs containing NR1 and HA-NR2B, and 3) HA-GluR1, respectively. NR2B and GluR1 subunits contain N-terminal, hemaglutinnin (HA) epitope tags to permit surface HA staining. Bottom row, GFP-SAP97 and HA-GluR1 co-localize at the cell surface. B) Loss of GFP-SAP97 and HA-tagged NMDAR trafficking to the plasma membrane. Second row, permeabilization revealed the intracellular retention of both proteins. Bottom row, intracellular clusters were specifically labeled with antibodies against ERP57. C–D) Cells expressing GFP-SAP97, HA-NR1/wt-NR2B, and Myc-CASK (C) or Myc-CASK-ΔL27N (D). Single Z-planes of intact cells demonstrating that co-expression of Myc-CASK (C), and not Myc-CASK-ΔL27N (D), restores surface expression of SAP97 and NMDARs (surface panels). Permeabilized cells expressing Myc-CAS K (C) lack intracellular clusters of NMDAR/SAP97 complexes. Expression of Myc-CASK-ΔL27N (D) does not relieve the clustering of NMDA receptors with SAP97. E). Live cells were labeled with anti-HA antibodies, and flow cytometry was used to quantitatively assay for GFP and surface HA-expression. Surface NMDAR expression of GFP-expressing cell populations is shown on the x-axis (arbitrary fluorescence units) and cell granularity (side scatter) on the y-axis. Total fluorescence intensities of surface NMDARs were quantified (right panel). Co-expression with GFP-SAP97 caused a reduction in surface NMDARs (54.93% ± 6.80%, p=0.000882). This reduction was rescued by Myc-CASK (93.34% ± 15.94%) but not Myc-CASK-ΔL27N (69.03% ± 7.83%, p=0.006717). Data are expressed as mean ± SEM, n=3 independent experiments assaying 50000 cells/experiment. Scale bars, 5 μm.
We also examined whether CASK affected the distribution of NMDARs in HEK293 cells. In the presence of myc-tagged CASK and EGFP-SAP97, NMDARs trafficked to the cell surface and were no longer retained in the ER (Fig. 3C,E). This effect required a direct interaction between CASK and SAP97, as a CASK deletion mutant lacking its N-terminal L27 domain (L27N), which binds the N-terminal L27 domain of SAP9735, had no effect when transfected with GFP-SAP97 (Fig. 3D,E). These data demonstrate that SAP97 can mediate the ER retention of NMDARs, and together with CASK may regulate the sorting and trafficking of NMDARs through the ER and Golgi.
We next examined whether SAP97 and CASK formed complexes with NMDARs in hippocampal neurons. This was initially explored by immunostaining cultured hippocampal neurons with antibodies against NR1, SAP97, and/or CASK. We observed strong co-localization of NR1 with SAP97 or CASK, in somata and punctate structures along the dendrites of these neurons (Fig. 4A). To distinguish between synaptic puncta and NMDAR transport vesicles, we first immunostained cultures with SAP97 and synapsin antibodies. Endogenous SAP97 was present not only at synapses, as defined by the co-localization of SAP97 and synapsin puncta along dendrites (Fig. 4A), but also in a distinct population of smaller puncta lacking synapsin (Fig. 4A, arrows). These latter data suggested that non-synaptic SAP97 might be associated with dendritic vesicles containing NMDARs. As another test, neurons were transfected with YFP-tagged SAP97 and GFP-tagged NR1, and then immunostained for synapsin and CASK. As shown in Figure 4B, using a filter set that separated YFP and GFP spectra, we saw two populations of YFP-SAP97 puncta: larger synaptic puncta that co-localized with NR1-GFP, CASK and synapsin, and smaller puncta that co-localized only with CASK and NR1-GFP.
Figure 4.

SAP97 co-distributes with NR1 and CASK in non-synaptic, mobile puncta. A) Hippocampal neurons were fixed at DIV 14, permeabilized, and double-labeled with combinations of antibodies against SAP97, NR1, CASK, and synapsin. SAP97 (top row, scale bar 10 μm) and CASK (second row, scale bar 5 μm) co-localized with NR1 in the soma and in dendritic puncta. Bottom row, SAP97 is found at synaptic and non-synaptic dendritic puncta (arrows, scale bar 10 μm). B) Cultures were transfected for 1day with NR1-GFP and YFP-SAP97. Neurons were then fixed, permeabilized, and double-labeled with antibodies against CASK, and synapsin. NR1-GFP, YFP-SAP97, and CASK co-localize in synaptic (arrowheads), and non-synaptic (arrows), dendritic puncta (scale bar 10 μm). C) Time-lapse imaging of neurons transfected for 1day with NR1-GFP and RFP-SAP97 (top panels, scale bar 5 μm), or GFP-SAP97 and DsRed-ER (bottom panels, scale bar 5μm). Co-migrating puncta for both transfection combinations trafficked down dendrites at similar velocities (0.13 ± 0.07 μm/s, n=13; 0.34 ± 0.05 n=15, respectively).
To test whether SAP97 and NMDARs were found together in mobile ER vesicles, cells were co-transfected with RFP-SAP97 and NR1-GFP, or EGFP-SAP97 and DsRed-ER prior to time-lapse imaging (Fig. 4C). We detected highly mobile RFP-SAP97 and EGFP-SAP97 puncta that co-migrated along dendrites with NR1-GFP (average velocity in both the retrograde and anterograde directions of 0.13 ± 0.07 μm/sec; mean ± SD; n=13; see Supplementary Video 1) or DsRed-ER puncta (average velocity in both the retrograde and anterograde directions of 0.34 + 0.05 μm/sec; mean ± SD; n=15), respectively. These data demonstrate that SAP97 and NMDARs together associate with motile, ER-like vesicles in dendrites.
We next assessed whether SAP97 exists in a complex with NMDARs and CASK by immunoprecipitating SAP97 from post-natal day 2 (P2) rat brain microsomal membranes. Using Western blot analysis, we found that CASK and NR2B subunits of the NMDAR co-immunoprecipitated with SAP97, consistent with a complex containing SAP97, CASK and NMDARs (Fig. 5A). The formation of this complex was confirmed in HEK293 cells. HA antibodies immunoprecipitated HA-tagged NMDAR subunits as well as co-transfected GFP-SAP97 and Myc-CASK (Fig. 5B). We failed to detect any myc-CASK in this complex when the L27N CASK deletion was used (Fig. 5B), indicating that its association with the SAP97/NMDAR complex requires a L27 domain-mediated interaction.
Figure 5.
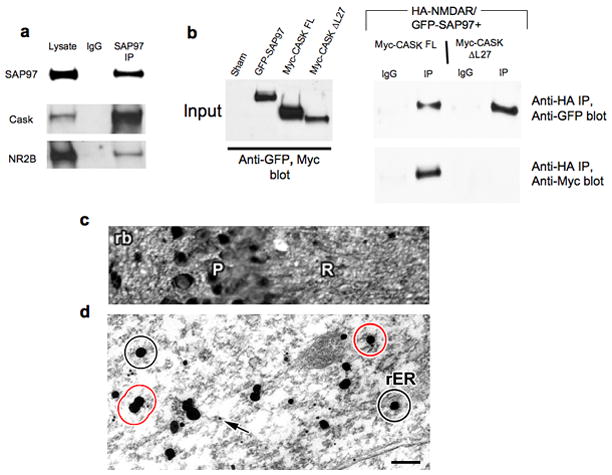
SAP97, NMDARs, and CASK form a complex in brain. A) Co-immunoprecipitation of SAP97, CASK, and NR2B from P2 rat brain lysates. B) HEK293 cells were transfected with cDNAs encoding GFP-SAP97, HA-NR1/wt-NR2B, and Myc-CASK or Myc-CASK-ΔL27. Lysates were subjected to anti-HA immunoprecipitation and precipitated proteins were immunoblotted with GFP and c-Myc Abs. C, D) Ultrastructural co-localization of SAP97 and NMDARs. C) Photograph showing pyramidal cell (P) and radiatum (R) layers of CA1 in P7 rat hippocampus. The tissue was labeled for SAP97 by silver intensified colloidal gold (SIG) using a polyclonal rabbit antibody. The micrograph shows SAP97 labeling in the somata as well as in the neuropil. D) Electron micrograph: A high-magnification image of the dendrites of a CA1 pyramidal neuron. SAP97 is immuno-labeled with SIG, the irregularly-shaped and -sized black spots. They were observed in the cytoplasm as well as in association with membraneous organelles, such as the rough endoplasmic reticulum (rER). NR2B subunits were immuno-labeled with 10 nm colloidal gold, examples of which are indicated by arrows. The circles represent the 60 nm-radius area around SAP97 labeling; if NR2B labeling falls within this area, the two were considered to be co-localized. Black circles are examples of SAP97 labeling without any NR2B co-localization, and red circles are those with NR2B co-localization. Scale bar: 25μm for the light micrograph, 200nm for the electron micrograph.
To test whether SAP97 and NMDARs co-localized on cellular membranes, we also performed dual label immunogold electron microscopy on dendrites of CA1 pyramidal cells. SAP97 immunoreactivity was detected at the pre-embed stage and enhanced by silver-intensified gold (SIG), and NR2B immunoreactivity at the post-embed stage with 10 nm colloidal gold. As shown in Fig. 5D, 10nm gold particles (arrows) were readily detected in the vicinity of larger SIG particles (Fig. 5D; circles in black), many of which associated with dendritic membranous structures. Our limit of resolution for co-localization was 60 nm, the estimated maximal distance that could be traversed by four IgG molecules linked to a single SAP97-NR2B complex. Altogether, our findings of a SAP97-CASK-NMDAR complex in mobile ER-like vesicles are consistent with the concept that SAP97 and CASK participate in the sorting of NMDARs into the ER sub-compartment found in dendrites.
SAP97 and CASK regulate NMDAR sorting through Golgi outposts
To further examine the roles of SAP97 and CASK in NMDAR trafficking, we generated short-hairpin RNAs (shRNAs) against SAP97 or CASK transcripts. Based on Western blot analysis, these shRNAs dramatically suppressed the expression of SAP97 (>90%) and CASK (>70%), while a scrambled shRNA had no effect (Fig. S5). To determine whether SAP97 or CASK knockdown affected NMDAR trafficking, we examined the co-localization of NR1 subunits with the Golgi marker GM130 in the somas of shRNA-expressing neurons (Fig. 6A). We observed a significant increase in the perinuclear distribution of NR1 in neurons expressing shRNAs for SAP97 or CASK versus control cells (Fig. 6A), and significant colocalization with GM130, suggesting that NR1 now trafficked via the somatic Golgi. Quantitatively, SAP97 knockdown led to a 33% increase in NR1 at the somatic Golgi, and CASK knockdown to a 48% increase compared to neurons expressing scrambled shRNAs (Fig. 6B). These results suggest that SAP97 and CASK are required for the selective sorting of NMDARs away from somatic Golgi.
Figure 6.


Knockdown of SAP97 or CASK reroutes NMDARs to the somatic Golgi and away from dendritic Golgi outposts. Cultures were infected at DIV0 with lentivirus encoding hairpin sequences against SAP97, CASK, or a randomized shRNA sequence (scrambled). Cultures were fixed at DIV16 and stained with antibodies against NR1 and GM130 (A), or GluR1 (C) or synaptophysin (E) and TGN38. shRNA knockdown of SAP97 (A) or CASK (B) resulted in a significant buildup of NR1 in the Golgi versus control cultures. B) Quantitative analysis demonstrated increases of 33% and 48% (mean ± S.E.M. from 10 neurons per group; p = 0.004 and p = 0.019 respectively). C) While SAP97 knockdown yielded a significant accumulation of GluR1 in the Golgi (D, mean ± S.E.M. from 10 neurons per group; p = 0.008), CASK knockdown had no effect on its distribution. E) Knockdown of SAP97 and CASK had no qualitative or quantitative (F) effect on the Golgi localization of synaptophysin, another integral membrane protein. G) Over-expression of ARF1-Q71I-HA had no effect on the somatic distribution of NR1 in uninfected (top row) and scrambled control (second row) cultures. Neurons infected with SAP97 shRNA displayed an increased somatic Golgi concentration of NR1 (bottom row). H) Quantitative analysis of the effect of ARF1-Q71I-HA over-expression in somatic Golgi membranes (mean ± S.E.M. from 10 neurons per group, p = 0.03). I) Knockdown of SAP97 yielded a loss of NR1 at ARF1-Q71I-HA puncta in the dendrites of transfected neurons. Scale bars, 10 μm.
We also tested the effects of SAP97 and CASK knockdown on AMPAR trafficking. We observed an accumulation of endogenous GluR1 within the somatic Golgi of neurons infected with the SAP97 shRNA. No accumulation was seen in uninfected neurons, neurons infected with a scrambled shRNA, or neurons infected with shRNA against CASK (Fig. 6C,D). Reducing the levels of SAP97 or CASK had no effect on the trafficking of another synaptic protein, synaptophysin, through somatic Golgi (Fig. 6E,F). Together, these observations indicate that SAP97, but not CASK, performs a specific role in the forward trafficking of GluR1 through somatic Golgi. This result is consistent with previous studies demonstrating that a SAP97-GluR1 interaction has a role in forward trafficking of AMPARs to the plasma membrane15. No such involvement of CASK in AMPAR trafficking has been described.
The accumulation of NR1 in somatic Golgi with knockdown of SAP97 or CASK suggests that these MAGUKs are required for the forward trafficking of NMDARs through Golgi membranes. Alternatively, NMDARs may still have the capacity to transit through somatic Golgi in the absence of SAP97 or CASK, but accumulate due to a slowed transport rate. To distinguish between these possibilities, we examined whether ARF1-Q71I Golgi block further enhanced NMDAR levels in the somatic Golgi of neurons lacking SAP97. Neurons expressing both SAP97 shRNAs and ARF1-Q71I-HA showed a marked increase in NR1 immunofluorescence in somatic Golgi (Fig 6G; bottom row, H) compared to neurons with SAP97 shRNAs alone (Fig. 6A; middle row). The average overlap between NR1 and GM130 intensities in somata was increased by 80% in neurons with ARF1-Q71I-HA and SAP97 knockdown (Fig. 6H), compared to 33% in neurons without ARF1-Q71I-HA (Fig. 6B).
Finally, we examined whether NMDARs still accumulate in Golgi outposts during an ARF1-Q71I block in neurons lacking SAP97. Endogenous NR1 accumulated at ARF1-Q71I-HA positive puncta in dendrites of neurons expressing a scrambled shRNA, but exhibited a 65% (p=0.00004) reduction at these same sites in neurons expressing the SAP97 shRNA (Fig. 6I).
SAP97 and CASK influence the synaptic localization of NMDARs
To address whether the alternative secretory pathway taken by NMDARs contributes to their synaptic delivery, we assessed whether SAP97 or CASK knockdown altered synaptic levels of NMDARs. This was tested by measuring the average intensity of NR1 immunoreactive puncta co-localizing with synaptophysin in neurons infected with the shRNAs (Fig. 7). The normalized ratio of NR1 to synaptophysin intensity was 30% less in neurons with SAP97 shRNA, and 46% less in those with CASK shRNA, when compared to uninfected neurons or neurons expressing a scrambled shRNA sequence (Fig. 7A–C). These results are consistent with the 45% decrease in NMDAR EPSCs observed previously with a SAP97 knockdown in hippocampal neurons39. Furthermore, they correlate with the 33% and 48% increases in NMDAR somatic Golgi accumulation observed with the same SAP97 and CASK knockdowns (Fig. 6A,B). These effects were not due to alterations in synapse formation or stability, as neither SAP97 nor CASK knockdown yielded a change in synaptic density versus scrambled shRNA controls (Fig. S6). Taken together, these data indicate that NMDARs traffic to synapses via the conventional secretory pathway in the absence of SAP97 or CASK, but are more efficiently delivered to synapses when they utilize the atypical pathway through the ER sub-compartment and dendritic Golgi outposts.
Figure 7.

Knockdown of SAP97 or CASK reduces synaptic NMDARs. Hippocampal neurons were infected shortly after plating, at DIV0, with lentivirus encoding shRNA against SAP97 or CASK, respectively, or a lentivirus with a randomized shRNA sequence (scrambled). A) Representative images from an uninfected sister culture (top row), and those infected with a high titer of SAP97 shRNA lentivirus (bottom row). Cultures were fixed at DIV16 and stained with antibodies against NR1 and synaptophysin. The relative intensities of the synaptic NR1 puncta in each field are shown in the third panels (NR1:synaptophysin, pseudo-color intensity image). These were obtained by thresholding and dividing the synaptic NR1 image by the synaptophysin image (see Methods). The resultant pixels were displayed in a pseudo-color channel (progression from blue to red to yellow pixels reflects increasing intensities). B) Representative images from an uninfected sister culture (top row), and those infected with a high titer of CASK shRNA lentivirus (bottom row). C) Quantification of the decrease in synaptic NMDAR levels following SAP97 or CASK knockdown. Fluorescence intensities of overlapping NR1 and synaptophysin puncta were measured. Shown are the normalized ratios of NR1 to synaptophysin average intensities (mean ± S.E.M. from 10 fields per group; n = 1103, 1108, and 881 puncta from uninfected, scrambled, and SAP97 shRNA cultures, respectively; p = 0.001; n = 1524, 1294, and 840 puncta from uninfected, scrambled, and CASK shRNA cultures, respectively; p = 0.0000003). Scale bar, 10 μm.
Discussion
NMDARs utilize an atypical secretory pathway
With their long processes and polarized structures, neurons face unparalleled demands to correctly sort and target proteins to the appropriate destinations. Like other cells, neurons have partially solved this problem by sorting proteins into vesicles at the TGN prior to their transport to axons or dendrites40,41,42. Sorting of postsynaptic glutamate receptors was initially thought to only occur at the TGN. However, recent studies have demonstrated that sorting of AMPARs utilizes an endosomal pathway after their insertion at or near the PSD10.
In this study, we have uncovered yet another pathway for glutamate receptor trafficking, that involves a dendritic ER sub-compartment and Golgi outposts and is utilized by NMDARs but not AMPARs. This trafficking pathway requires SAP97 and CASK, and appears essential for the efficient synaptic delivery of NMDARs. Our data reveal three novel aspects of NMDAR trafficking. First, the majority of NMDARs use this alternative pathway, as no increase in NMDAR immunoreactivity or NR1-GFP fluorescence was seen in somatic Golgi following an ARF1-Q71I block. Second, NMDAR trafficking from the soma to dendrites is independent of COPI vesicle formation, and NMDARs remain in an ER-like compartment during their trafficking into dendrites. Finally, NMDAR transit through dendritic Golgi outposts is dependent on COPI vesicle formation, suggesting that Golgi outposts for NMDARs much like somatic TGN functions for other proteins, i.e. as a transit and processing station. This may enable neurons to more tightly and locally regulate the delivery of synaptic NMDARs.
SAP97 and CASK direct NMDARs to Golgi outposts
Our data indicate that NMDARs exit the conventional secretory pathway just after their exit from the somatic ER compartment. This prediction is supported by our 15°C temperature shift experiments that led to the somatic accumulation of newly synthesized NMDARs (Fig. S1). Furthermore, we found that a signification fraction of dendritically localized, extra-synaptic NMDARs co-localized with ER markers, including KDEL, IP3R, and DsRed-ER (Fig. 2). Importantly, these NMDAR-associated, ER-derived vesicles were found to be highly dynamic, moving at speeds consistent with their transport along dendritic microtubules31, and to share features with previously characterized transport vesicles14 containing NMDARs, CASK, and KIF17.
By further characterizing ER-derived NMDAR transport vesicles, we detected SAP97 associated with NMDAR/CASK/KIF17 complexes. SAP97 directly binds CASK and NMDARs35,36,23 and causes ER retention of voltage-gated ion channels in HEK293 cells37. These data suggest that SAP97 is necessary for sorting NMDARs into the alternative secretory pathway. Three lines of evidence support this hypothesis: 1) SAP97 caused the ER retention of NMDARs when co-transfected with NMDAR subunits into HEK293 cells (Fig. 3); 2) SAP97 knockdown in neurons caused NMDAR accumulation in somatic Golgi (Fig. 6); and 3) endogenous NMDAR localization in dendritic Golgi outposts was lost with SAP97 knockdown and ARF1-Q71I-HA block of Golgi trafficking (Fig. 6). These data suggest that SAP97 associates with NMDARs during/after their assembly in the ER, and that this interaction prevents their ability to transit from ER exit sites to the somatic Golgi (See Model Fig. S7).
Our data also indicate that SAP97 does not act alone in diverting NMDARs from somatic Golgi, although it has the ability to directly bind NR2 subunits of the NMDAR23. CASK, a second component of the NMDAR transport complex, is also required for this process, though it does not directly bind NMDAR subunits. Two sets of experiments support this conclusion. First, CASK co-expression enabled NMDAR transport to the cell surface and prevented SAP97-dependent ER retention of NMDARs (Fig. 3). This activity required the L27N domain of CASK, necessary for complex formation with SAP9735. Second, CASK knockdown in cultured hippocampal neurons re-routed NMDARs through somatic Golgi (Fig. 6). These data indicate that a CASK-SAP97 complex functions in concert to sort NMDARs through this atypical secretory pathway.
Functional significance of the alternative secretory pathway
We evaluated whether levels of synaptic NMDARs were altered by knockdown of SAP97 or CASK, and found that either reduced synaptic NMDARs by 30–40% (Fig. 7). These values are consistent with the 45% decrease in NMDAR EPSCs observed previously with SAP97 knockdown39. A similar reduction in synaptic NMDARs was reported for neurons lacking KIF17, the microtubule-dependent motor thought to be responsible for the anterograde transport of vesicles carrying NMDARs/CASK17. When this alternative pathway is unavailable, NMDARs can traffic to synapses via the conventional secretory pathway, as evidenced by the accumulation of NMDARs in somatic Golgi during the knockdown of SAP97 or CASK (33% and 48%, respectively) (Fig. 6), as well as by the 80% increase in NMDARs on somatic Golgi during a SAP97 knockdown/ARF1-Q71I block (Fig. 6). We also detected a reciprocal relationship between the increase in NMDARs at the Golgi (33% and 48%; Fig. 6) and their reduction at synapses (30% and 46%; Fig. 7) during the SAP97 or CASK knockdown, respectively. Our data suggest that loss of synaptic NMDARs is caused by a reduced rate of trafficking as they move through the conventional secretory pathway, and not by reduced synaptic anchoring of the receptors by SAP97 or CASK.
Another question raised by our studies is whether this alternative secretory pathway is specific for NMDARs, or whether other integral membrane and/or secreted proteins traffic through this pathway. Clearly, proteins previously shown to reside within the dendritic ER sub-compartment, such as the integral membrane IP3 receptor and the calcium ATPase-2a are trafficked using this pathway31. Our studies on GluR1 subunits of the AMPAR indicate that this pathway is not available to all proteins and that there is some degree of specificity. Other studies have found that recombinant BDNF, a secreted protein, and VSVG, an integral membrane protein, traffic though dendritic Golgi outposts, though both proteins also appear to use somatic Golgi, perhaps due to over-expression29. Inwardly rectifying potassium channels (Kir2) also complex with SAP97, CASK, Mint and Velis/MALS in fibroblast and neuronal cells36. Although not explored here, the ability of SAP97 and CASK to form complexes with channels other than NMDARs suggest that this alternative sorting pathway may be used by multiple dendritically sorted ion channels and receptors.
In summary, we find that the differential sorting of NMDARs and AMPARs begins in the somatic ER, causing a majority of NMDARs to bypass somatic Golgi in favor of dendritic Golgi outposts. Our data also reveal that the MAGUKs SAP97 and CASK are required for the sorting of NMDARs from AMPARs. Finally, our data indicate that this alternative pathway allows NMDA receptors to be more efficiently delivered to synapses and may provide a platform for local control of NMDAR insertion near synapses.
Methods
Antibodies
The following primary antibodies were used in immunofluorescence and Western blot experiments: Anti-HA mAb HA.11 (Covance), anti-cMyc 9E10 mAb and pAb (Santa Cruz), anti-GFP pAb (Invitrogen), anti-ERP57 pAb (StressGen), anti-NR2B mAb (BD Biosciences), chicken and rabbit anti-HA pAbs (Bethyl), anti-KDEL mAb (StressGen), anti-IP3R pAb (Affinity Bioreagents), anti-NR1 mAb363 (Chemicon), anti-synapsin mAb (Chemicon), anti-SAP97 pAb (Affinity Bioreagents), anti-CASK (pAb, Zymed; mAb, BD Biosciences), anti-KIF17 pAb (Sigma), anti-GluR1 pAb (Calbiochem), anti-tubulin mAb (Sigma), anti-TGN38 mAb (BD Biosciences), anti-GM130 pAb (Calbiochem), anti-MAP2 pAb (Sigma), anti-PSD95 mAb (Neuromab), and anti-synaptophysin pAb (Santa Cruz). The secondary antibodies were from Molecular Probes (goat anti-mouse and rabbit Alexa Fluors 488, 568, and 647; goat anti-chicken DSB-X biotin and Streptavidin-Marina Blue), Aurion (10 nm and 0.8 nm colloidal gold-conjugated goat anti-rabbit IgG and 0.8 nm colloidal gold-conjugated goat anti-mouse IgG).
Cell Culture and Transfection
HEK-293 cells (gift from Dr. J. Kyle, University of Chicago, Chicago, IL) were maintained in DMEM supplemented with 10% calf serum (Hyclone). Cells were transiently transfected with cDNA using a calcium phosphate protocol 43. Cells transfected with HA-GluR1 or HA-NMDAR were maintained in media containing 1mM Kynurenic acid (Sigma), or 100μM D-APV (Sigma) and 10μM MK-801 (RBI), respectively, to prevent excitotoxicity.
Hippocampal cultures were prepared using a modified Banker culture protocol44. Briefly, hippocampi from embryonic (E18–19) Sprague-Dawley rats were dissected out, dissociated in 0.05% trypsin (Invitrogen), and cells plated at a density of 165/mm2 on poly-L-lysine coated coverslips (Carolina Biological). One hour after plating, coverslips were transferred in pairs to 60 mm dishes containing a glial feeder layer, where they were inverted (to maximize neuronal contact with secreted glial factors) and maintained in Neurobasal medium containing B27 and GlutaMAX (all from Invitrogen).
Neuronal cultures were transfected at DIV12–14 with the Lipofectamine 2000 transfection reagent (Invitrogen) according to manufacturer’s recommendations, except that 1–2.5 μg of each cDNA in 62.5 μl Neurobasal media and 2.5 μl of Lipofectamine 2000 in 62.5 μl Neurobasal media were mixed and added to coverslips in 6 well plates.
cDNA Constructs
Rat NR1 and NR2B were obtained from J. Boulter (UCLA). All subunits were tagged at the NH2 terminus with the hemaglutinin (HA:YPYDVPDYA) epitope using the extension overlap PCR method45. These cDNAs were then subcloned into a pCB6 mammalian expression vector. Myc-CASK and Myc-CASK L27 were gifts from Ben Margolis (University of Michigan). GluR1-GFP was a gift from R. Malinow (Cold Spring Harbor). Construction of NR1-GFP and GFP-SAP97 has been previously described46,47. YFP-SAP97 and RFP-SAP97 were generated by excising SAP97 from pEGFP-C1 (Clontech) with EcoRI and KpnI, and subcloning into pEYFP-C1 and pDsRed1-C1 (Clontech), respectively. ARF1-Q71I-HA was a gift from A. El-Husseini (University of British Columbia). Galactosyltransferase-GFP (Galtase-GFP) was a gift from Jennifer Lippincott-Schwartz (NIH), and pDsRed-ER is from Clontech Laboratories.
Immunofluorescence Staining
HEK-293 cells (18–24h post-transfection), and neuronal cultures, were washed 2x in PBS at room temperature, fixed in 4% paraformaldehyde/sucrose (4° C; 15min.), and washed 3x in PBS (5–10min.). For permeabilization, cells were incubated in 0.1% Triton-X-TBS (10min.), incubated in blocking solution (BS) (2% Glycine, 2% BSA, 0.2% Gelatin, and 50mM NH4Cl in 1X PBS; 10min.), and then incubated with the indicated primary antibody diluted in BS (1h). Following primary antibody incubation, cells were washed 3x in BS (5–10min.) and overlaid with an appropriate secondary antibody diluted in BS (1h). Cells were then washed 3x in PBS (5–10min.) and the slips mounted in Vectashield (Vector laboratories) or Prolong Gold (Invitrogen). Fluorescence images were acquired using the Leica SP2 AOBS spectral laser scanning confocal microscope (LSCM, Leica Microsystems), or with a Yokogawa spinning disc confocal head (Perkin Elmer), fitted on a Zeiss Axiovert 200M microscope. Images were processed using Image J (NIH) and Adobe Photoshop software.
Image Analysis
Quantification of fluorescence data was performed using MetaMorph (Universal Imaging, Downing-town, PA) and Image J software. Somatic Golgi localization was assayed by setting the Z-plane limits for acquisition (~ 0.5–1.0 μm/slice, 5–10 slices) based on fluorescence signal for the Golgi marker (GM130 or TGN38). The signal was then thresholded and used to create a binary mask, allowing for the measurement of subunit pixel intensities in each plane. Averaged pixel intensities for the entire Z stack were calculated, background subtracted, and normalized to control values. Expression levels, in exogenous subunit experiments, was controlled for by measuring the average pixel intensity in a small box drawn outside of the Golgi, where subunits displayed an even distribution. A Golgi-to-soma (non-Golgi) ratio was calculated for each cell, and normalized to control ratio values. Experiments were conducted from a minimum of 2 independent culture preparations, with 10 neurons per experimental group.
Analysis of Golgi outpost localization was performed as above, except that averaged Z projections for all channels were used for quantification. A combined Golgi and mutant ARF1 binary mask was created (Golgi alone for control fields), ensuring the analysis of outposts with impaired COPI function. Averaged NR1 and NR2B pixel intensities in the thresholded regions were measured, background subtracted, and normalized to control values. For exogenous NR1-GFP experiments, differences in protein expression levels across cells were controlled for by measuring average pixel intensities in a small region of the immediate proximal dendrite where subunit distribution was uniform, calculating an outpost to dendrite ratio for each cell analyzed, and normalizing to control ratio values. Experiments were conducted from a minimum of 2 independent culture preparations, and 5 fields per condition.
Synaptic NR1 expression was quantified by setting Z-plane limits using the synaptophysin signal, then acquiring both channels. Averaged Z-projections of image stacks were created, thresholded and scored for co-localization and signal intensity using the cell-scoring feature in MetaMorph. Averaged intensities in each field were background-subtracted and normalized, and the ratio of NR1 to synaptophysin intensities was analyzed in Excel. Experiments were conducted from a minimum of 2 independent culture preparations, and 10 fields per condition.
Analysis of the co-localization of DsRed-ER-containing vesicles with endogenous NR2B or GluR1 were carried out by background-subtracting and thresholding image fields so that only puncta greater than 2 fold above background were selected. Co-localizing puncta were evaluated using the Analyze Particles function in Image J.
Analysis of the effects of mutant ARF1 expression on GFP-GluR1 dendritic distribution (Fig. S2) was performed on Z-stacks of dendrites. Averaged pixel intensities along the first 20μm were calculated, background-subtracted, and normalized to control values. Expression levels in individual cells were controlled for as described above. Experiments were conducted from a minimum of 2 independent culture preparations, with 10 neurons per experimental group.
Synaptic density experiments (Figs. S3, S6) utilized co-localization of pre- and post-synaptic markers to denote synaptic sites. Thresholded puncta were analyzed using the Analyze Particle function in Image J, and the number of co-localizing puncta were divided by the length of dendrite analyzed. Experiments were conducted from a minimum of 2 independent culture preparations, with 5–10 neurons per experimental group.
Statistics
Statistical comparisons for all confocal analyses were made using two-tailed, Student’s t-tests. Flow cytometric comparisons were made using ANOVA/Tukey post hoc analysis.
Spectral separation of GFP and YFP
Samples were sequentially excited in spectral scan mode (Leica), at reduced levels, using the 488 and 514 nm laser lines, and emission windows were visually optimized. GFP bleed-through into the YFP channel was assessed by scanning neurons only expressing the GFP-fusion protein, exciting with the 514 nm laser line, and collecting data with the emission window set at 535–565nm. Under these conditions GFP fluorescence was negligible. YFP cross-talk into the GFP channel was assessed and controlled for by scanning neurons only expressing the YFP fusion protein with the 488 line, and setting the emission window at 490–515. At the laser intensities used, YFP cross-talk was negligible when the emission window did not extend beyond 515nm. The effectiveness of this protocol was confirmed by the presence of mutually exclusive puncta in overlay images from co-transfected neurons.
Time-lapse Imaging
For the time-lapse imaging experiments, the culture medium was replaced with a 10 mM Hepes buffer solution (pH 7.3). Neurons were visualized under an inverted microscope (IX81, Olympus) and a 60x objective (NA 1.4, Olympus) using standard filter sets and a Hg lamp. Culture slips were maintained at ~37°C by placing dishes in a heating chamber affixed to the microscope stage. Sequential images were acquired, 100–1000ms exposures, on a Retiga EXi chilled charged coupled device (QImaging, Burnaby, BC, Canada), under the control of MetaMorph software.
Flow cytometry
Surface HA expression was assayed by labeling live cells 24 hours post-transfection with an anti-HA antibody, washing 2X in PBS, and labeling with a fluorescently-conjugated secondary antibody. Fluorescence quantification was carried out using an LSRII Flow Cytometer (488 and 647 nm laser; BD Biosciences, San Jose, CA) and analysis of flow data was performed using FlowJo 7.2.4 software.
Electron and Light Microscopy
Two Sprague-Dawley rats, postnatal day 7, were deeply anesthetized with Nembutal (50 mg/kg). Brains were fixed by transcardial perfusion with a mixture of aldehydes (4% paraformaldehyde and 1% glutaraldehyde in PBS). Vibratome-sections from these brains were incubated in 1% hydrogen-peroxide and then 1% albumin-bovine serum albumin in PBS for 30 minutes each, in order to minimize background staining. Sections were incubated in polyclonal rabbit anti-SAP97 (1:500), and incubations were for 2 days at room temperature. Antigen-antibody complexes were visualized by applying 0.8 nm gold-conjugated secondary antibodies, diluted 1:100. The gold particles were silver intensified (Silver IntensEM kit, Amersham). Digital images were captured at a magnification of 40x for light microscopy.
The SAP97-immunolabeled vibratome sections were processed through osmium-free tissue processing48, dehydrated, and embedded in resin (Embed 812; EMS). The CA1 field of hippocampus was sectioned at a thickness of 80 nm and immunolabeled for the NR2B subunit of NMDARs using post-embed gold (PEG) procedure49. Briefly, the grids holding the sections were incubated in 1:20 dilution of rabbit anti-NR2B antibody (Upstate) overnight, followed by 1 hr incubation in 1:40 dilution of 10 nm gold-conjugated secondary antibody (Aurion, PA). The grids were counterstained with lead citrate to enhance contrast.
Pyramidal cell layer of CA1 was examined under electron microscope (JEOL, Japan) along the tissue-Epon interface. Micrographs containing somata and proximal dendrites were captured digitally (Hamamatsu CCD Camera, Japan; AMT, MA) at a magnification of 40,000x. In order to determine whether SAP97 co-localized with the NR2B-subunit at a frequency above chance level, we drew a 60 nm-radius circle (the maximum distance between two primary antibody-secondary antibody complexes associated with a single antigenic site) around every SIG label (SAP97, circles in Fig. 5) that represented the region that could be ascribed to the co-existence of NR2Bs with SAP97.
Immunoprecipitation and Western blot
Cells were pelleted by brief centrifugation, resuspended, washed once with PBS and solubilized in lysis buffer (150 mM NaCl, 5 mM EDTA pH 7.4, 50 mM Tris pH 7.4, 0.02% NaN3) containing 1% Triton X-100 (TX-100)+NEM (2 mM), PMSF (2 mM), leupeptin (10 μg/ml), TLCK (10 μg/ml), chymotrypsin (10 μg/ml) and pepstatin (10 μg/ml). Following a 1h solubilization (4°C), samples were centrifuged at 14 000 g for 30 min at 4°C. Subsequent analyses were performed using the TX-100-soluble fraction. Immunoprecipitations were performed by overnight antibody incubation at 4°C. Protein antibody complexes were isolated with Protein G-Sepharose for 3 hr at 4°C.
For immunoblotting, proteins separated by SDS-PAGE were transferred to nitrocellulose membranes. After transfer, the nitrocellulose was blocked with 3% milk in wash buffer (10 mM Tris, pH 7.4, 0.05% Tween 20, and 150 mM NaCl). Membranes were washed briefly in wash buffer and then incubated for 1h with primary antibodies. The blots were washed and incubated with secondary antibody (goat anti-mouse or rabbit HRP) at the appropriate dilution for 1 hr. After washing, membranes were treated with an enhanced chemiluminescent reagent (ECL; Amersham) according to the manufacturer’s protocol and exposed to film.
Lentivirus and shRNA
The short hairpin RNA (shRNA) target sequences for SAP97, 5′-GCAAGATACCCAGAGAGCA-3′ (rat, N-terminal), and CASK, 5′-ATCCATGAGCAGGGGCTGA -3′ (rat) were subcloned into the lentiviral vector, FUGWH1(+). This is a modified version of the FUGW vector, containing an H1 promoter which drives expression of the hairpin sequences50.
Supplementary Material
Acknowledgments
We would like to thank Sergio Leal-Ortiz for assistance with the generation of lentivirus and shRNA, Leigh A. Needleman and Jeremy Marks. Also, thanks to Ben Margolis for the CASK constructs, Jennifer Lippincott-Schwartz for the Galtase-GFP construct, and Vytas Bindokas for help and advice with imaging protocols. This work was supported by NIH grant # DA016758 and the Nancy Pritzker Family to CCG, DFG postdoctoral fellowship to CGS, and The Marsden Fund (Royal Society of New Zealand) to JMM. NIH grant #s NS043782, DA13602 and DA019695 to WNG and the Albert & Ellen Grass Faculty Award to WNG and JM.
Footnotes
Author contributions
O.J. conducted the majority of experiments and data analysis, except those detailed below, and co-wrote the manuscript. C.L.W. contributed to the shRNA experiments and helped in the editing of the manuscript. C.G.S designed, constructed, and aided in the testing of the shRNAs. S.F. performed the EM studies. M.S. conducted temperature block, BFA, and some of the live-imaging experiments. E.L performed flow cytometry experiments. J.M. provided advice and aided in the interpretation of data. C.A. supervised the EM studies. T.S. assisted with the temperature block experiments. J.M.M. supervised and designed studies, and helped with the editing of the manuscript. C.C.G and W.N. supervised the project and co-wrote the manuscript.
References
- 1.Dumas TC. Developmental regulation of cognitive abilities: modified composition of a molecular switch turns on associative learning. Prog Neurobiol. 2005;76:189–211. doi: 10.1016/j.pneurobio.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 2.Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–26. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 3.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 4.Rao A, Kim E, Sheng M, Craig AM. Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. J Neurosci. 1998;18:1217–29. doi: 10.1523/JNEUROSCI.18-04-01217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedman HV, Bresler T, Garner CC, Ziv NE. Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron. 2000;27:57–69. doi: 10.1016/s0896-6273(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 6.Washbourne P, Bennett JE, McAllister AK. Rapid recruitment of NMDA receptor transport packets to nascent synapses. Nat Neurosci. 2002;5:751–9. doi: 10.1038/nn883. [DOI] [PubMed] [Google Scholar]
- 7.Montgomery JM, Zamorano PL, Garner CC. MAGUKs in synapse assembly and function: an emerging view. Cell Mol Life Sci. 2004;61:911–29. doi: 10.1007/s00018-003-3364-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kennedy MJ, Ehlers MD. Organelles and trafficking machinery for postsynaptic plasticity. Annu Rev Neurosci. 2006;29:325–62. doi: 10.1146/annurev.neuro.29.051605.112808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greger IH, Esteban JA. AMPA receptor biogenesis and trafficking. Curr Opin Neurobiol. 2007;17:289–97. doi: 10.1016/j.conb.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 10.Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–5. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- 11.Yudowski GA, et al. Real-time imaging of discrete exocytic events mediating surface delivery of AMPA receptors. J Neurosci. 2007;27:11112–21. doi: 10.1523/JNEUROSCI.2465-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cognet L, Groc L, Lounis B, Choquet D. Multiple routes for glutamate receptor trafficking: surface diffusion and membrane traffic cooperate to bring receptors to synapses. Sci STKE. 2006:pe13. doi: 10.1126/stke.3272006pe13. [DOI] [PubMed] [Google Scholar]
- 13.Sheng M, Hoogenraad CC. The Postsynaptic Architecture of Excitatory Synapses: A More Quantitative View. Annu Rev Biochem. 2006 doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- 14.Setou M, Nakagawa T, Seog DH, Hirokawa N. Kinesin superfamily motor protein KIF17 and mLin-10 in NMDA receptor-containing vesicle transport. Science. 2000;288:1796–802. doi: 10.1126/science.288.5472.1796. [DOI] [PubMed] [Google Scholar]
- 15.Sans N, et al. Synapse-associated protein 97 selectively associates with a subset of AMPA receptors early in their biosynthetic pathway. J Neurosci. 2001;21:7506–16. doi: 10.1523/JNEUROSCI.21-19-07506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wyszynski M, et al. Interaction between GRIP and liprin-alpha/SYD2 is required for AMPA receptor targeting. Neuron. 2002;34:39–52. doi: 10.1016/s0896-6273(02)00640-2. [DOI] [PubMed] [Google Scholar]
- 17.Guillaud L, Setou M, Hirokawa N. KIF17 dynamics and regulation of NR2B trafficking in hippocampal neurons. J Neurosci. 2003;23:131–40. doi: 10.1523/JNEUROSCI.23-01-00131.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lise MF, et al. Involvement of myosin Vb in glutamate receptor trafficking. J Biol Chem. 2006;281:3669–78. doi: 10.1074/jbc.M511725200. [DOI] [PubMed] [Google Scholar]
- 19.Setou M, et al. Glutamate-receptor-interacting protein GRIP1 directly steers kinesin to dendrites. Nature. 2002;417:83–7. doi: 10.1038/nature743. [DOI] [PubMed] [Google Scholar]
- 20.Perestenko PV, Henley JM. Characterization of the intracellular transport of GluR1 and GluR2 alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits in hippocampal neurons. J Biol Chem. 2003;278:43525–32. doi: 10.1074/jbc.M306206200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sans N, et al. NMDA receptor trafficking through an interaction between PDZ proteins and the exocyst complex. Nat Cell Biol. 2003;5:520–30. doi: 10.1038/ncb990. [DOI] [PubMed] [Google Scholar]
- 22.Sans N, et al. mPins modulates PSD-95 and SAP102 trafficking and influences NMDA receptor surface expression. Nat Cell Biol. 2005;7:1179–90. doi: 10.1038/ncb1325. [DOI] [PubMed] [Google Scholar]
- 23.Bassand P, Bernard A, Rafiki A, Gayet D, Khrestchatisky M. Differential interaction of the tSXV motifs of the NR1 and NR2A NMDA receptor subunits with PSD-95 and SAP97. Eur J Neurosci. 1999;11:2031–43. doi: 10.1046/j.1460-9568.1999.00611.x. [DOI] [PubMed] [Google Scholar]
- 24.Gardoni F, et al. CaMKII-dependent phosphorylation regulates SAP97/NR2A interaction. J Biol Chem. 2003;278:44745–52. doi: 10.1074/jbc.M303576200. [DOI] [PubMed] [Google Scholar]
- 25.Mauceri D, Gardoni F, Marcello E, Di Luca M. Dual role of CaMKII-dependent SAP97 phosphorylation in mediating trafficking and insertion of NMDA receptor subunit NR2A. J Neurochem. 2007;100:1032–46. doi: 10.1111/j.1471-4159.2006.04267.x. [DOI] [PubMed] [Google Scholar]
- 26.Pierce JP, Mayer T, McCarthy JB. Evidence for a satellite secretory pathway in neuronal dendritic spines. Curr Biol. 2001;11:351–5. doi: 10.1016/s0960-9822(01)00077-x. [DOI] [PubMed] [Google Scholar]
- 27.Pfeffer S. Membrane domains in the secretory and endocytic pathways. Cell. 2003;112:507–17. doi: 10.1016/s0092-8674(03)00118-1. [DOI] [PubMed] [Google Scholar]
- 28.Dascher C, Balch WE. Dominant inhibitory mutants of ARF1 block endoplasmic reticulum to Golgi transport and trigger disassembly of the Golgi apparatus. J Biol Chem. 1994;269:1437–48. [PubMed] [Google Scholar]
- 29.Horton AC, Ehlers MD. Dual modes of endoplasmic reticulum-to-Golgi transport in dendrites revealed by live-cell imaging. J Neurosci. 2003;23:6188–99. doi: 10.1523/JNEUROSCI.23-15-06188.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schuman EM, Dynes JL, Steward O. Synaptic regulation of translation of dendritic mRNAs. J Neurosci. 2006;26:7143–6. doi: 10.1523/JNEUROSCI.1796-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bannai H, Inoue T, Nakayama T, Hattori M, Mikoshiba K. Kinesin dependent, rapid, bi-directional transport of ER sub-compartment in dendrites of hippocampal neurons. J Cell Sci. 2004;117:163–75. doi: 10.1242/jcs.00854. [DOI] [PubMed] [Google Scholar]
- 32.Mironov SL, Symonchuk N. ER vesicles and mitochondria move and communicate at synapses. J Cell Sci. 2006;119:4926–34. doi: 10.1242/jcs.03254. [DOI] [PubMed] [Google Scholar]
- 33.Jo K, Derin R, Li M, Bredt DS. Characterization of MALS/Velis-1, -2, and -3: a family of mammalian LIN-7 homologs enriched at brain synapses in association with the postsynaptic density-95/NMDA receptor postsynaptic complex. J Neurosci. 1999;19:4189–99. doi: 10.1523/JNEUROSCI.19-11-04189.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Butz S, Okamoto M, Sudhof TC. A tripartite protein complex with the potential to couple synaptic vesicle exocytosis to cell adhesion in brain. Cell. 1998;94:773–82. doi: 10.1016/s0092-8674(00)81736-5. [DOI] [PubMed] [Google Scholar]
- 35.Lee S, Fan S, Makarova O, Straight S, Margolis B. A novel and conserved protein-protein interaction domain of mammalian Lin-2/CASK binds and recruits SAP97 to the lateral surface of epithelia. Mol Cell Biol. 2002;22:1778–91. doi: 10.1128/MCB.22.6.1778-1791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leonoudakis D, Conti LR, Radeke CM, McGuire LM, Vandenberg CA. A multiprotein trafficking complex composed of SAP97, CASK, Veli, and Mint1 is associated with inward rectifier Kir2 potassium channels. J Biol Chem. 2004;279:19051–63. doi: 10.1074/jbc.M400284200. [DOI] [PubMed] [Google Scholar]
- 37.Tiffany AM, et al. PSD-95 and SAP97 exhibit distinct mechanisms for regulating K(+) channel surface expression and clustering. J Cell Biol. 2000;148:147–58. doi: 10.1083/jcb.148.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leonard AS, Davare MA, Horne MC, Garner CC, Hell JW. SAP97 is associated with the alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit. J Biol Chem. 1998;273:19518–24. doi: 10.1074/jbc.273.31.19518. [DOI] [PubMed] [Google Scholar]
- 39.Nakagawa T, et al. Quaternary structure, protein dynamics, and synaptic function of SAP97 controlled by L27 domain interactions. Neuron. 2004;44:453–67. doi: 10.1016/j.neuron.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 40.Dotti CG, Poo MM. Neuronal polarization: building fences for molecular segregation. Nat Cell Biol. 2003;5:591–4. doi: 10.1038/ncb0703-591b. [DOI] [PubMed] [Google Scholar]
- 41.Dresbach T, et al. Assembly of active zone precursor vesicles: obligatory trafficking of presynaptic cytomatrix proteins Bassoon and Piccolo via a trans-Golgi compartment. J Biol Chem. 2006;281:6038–47. doi: 10.1074/jbc.M508784200. [DOI] [PubMed] [Google Scholar]
- 42.Tang BL. Protein trafficking mechanisms associated with neurite outgrowth and polarized sorting in neurons. J Neurochem. 2001;79:923–30. doi: 10.1046/j.1471-4159.2001.00674.x. [DOI] [PubMed] [Google Scholar]
- 43.Claudio T. Stable expression of heterologous multisubunit protein complexes established by calcium phosphate- or lipid-mediated cotransfection. Methods Enzymol. 1992;207:391–408. doi: 10.1016/0076-6879(92)07028-m. [DOI] [PubMed] [Google Scholar]
- 44.Banker G, Goslin K. Developments in neuronal cell culture. Nature. 1988;336:185–6. doi: 10.1038/336185a0. [DOI] [PubMed] [Google Scholar]
- 45.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–9. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 46.Wu H, Reuver SM, Kuhlendahl S, Chung WJ, Garner CC. Subcellular targeting and cytoskeletal attachment of SAP97 to the epithelial lateral membrane. J Cell Sci. 1998;111 ( Pt 16):2365–76. doi: 10.1242/jcs.111.16.2365. [DOI] [PubMed] [Google Scholar]
- 47.Bresler T, et al. Postsynaptic density assembly is fundamentally different from presynaptic active zone assembly. J Neurosci. 2004;24:1507–20. doi: 10.1523/JNEUROSCI.3819-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Phend KD, Rustioni A, Weinberg RJ. An osmium-free method of epon embedment that preserves both ultrastructure and antigenicity for post-embedding immunocytochemistry. J Histochem Cytochem. 1995;43:283–92. doi: 10.1177/43.3.7532656. [DOI] [PubMed] [Google Scholar]
- 49.Fujisawa S, Aoki C. In vivo blockade of N-methyl-D-aspartate receptors induces rapid trafficking of NR2B subunits away from synapses and out of spines and terminals in adult cortex. Neuroscience. 2003;121:51–63. doi: 10.1016/s0306-4522(03)00341-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leal-Ortiz S, et al. Piccolo modulation of Synapsin1a dynamics regulates synaptic vesicle exocytosis. J Cell Biol. 2008;181:831–46. doi: 10.1083/jcb.200711167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.