

Abstract
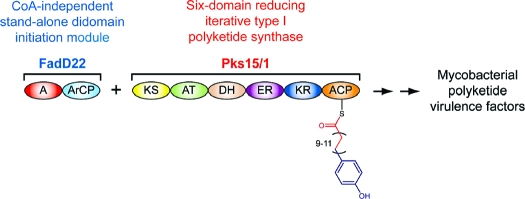
Several Mycobacterium tuberculosis strains, Mycobacterium leprae, and other mycobacterial pathogens produce a group of small-molecule virulence factors called phenolic glycolipids (PGLs). PGLs play key roles in pathogenicity and host−pathogen interaction. Thus, elucidation of the PGL biosynthetic pathway will not only expand our understanding of natural product biosynthesis, but may also illuminate routes to novel therapeutics to afford alternative lines of defense against mycobacterial infections. In this study, we report an investigation of the enzymatic requirements for the production of long-chain p-hydroxyphenylalkanoate intermediates of PGL biosynthesis. We demonstrate a functional cooperation between a coenzyme A-independent stand-alone didomain initiation module (FadD22) and a 6-domain reducing iterative type I polyketide synthase (Pks15/1) for production of p-hydroxyphenylalkanoate intermediates in in vitro and in vivo FadD22-Pks15/1 reconstituted systems. Our results suggest that Pks15/1 is an iterative type I polyketide synthase with a relaxed control of catalytic cycle iterations, a mechanistic property that explains the origin of a characteristic alkyl chain length variability seen in mycobacterial PGLs. The FadD22-Pks15/1 reconstituted systems lay an initial foundation for future efforts to unveil the mechanism of iterative catalysis control by which the structures of the final products of Pks15/1 are defined, and to scrutinize the functional partnerships of the FadD22-Pks15/1 system with downstream enzymes of the PGL biosynthetic pathway.
Introduction
Mycobacterial infections cause serious diseases. In particular, tuberculosis, produced by Mycobacterium tuberculosis, is responsible for nearly two million deaths per year, and leprosy, produced by Mycobacterium leprae, accounts for over three million people with neuropathy-derived disabilities worldwide. Several M. tuberculosis strains (e.g., strains of the W-Beijing family), M. leprae, and other mycobacterial pathogens produce a group of small-molecule virulence factors called phenolic glycolipids (PGLs) (Figure 1).(1) PGLs play key roles in pathogenicity and host−pathogen interaction by virtue of their immunomodulatory properties or, in the case of leprosy, their ability to cause peripheral nerve degeneration.(2) Moreover, PGL production has been suggested as a trait predisposing M. tuberculosis strains of the W-Beijing family to their characteristic epidemic spread and increased likelihood of developing drug resistance.(3) Thus, elucidation of the PGL biosynthetic pathway will not only expand our understanding of natural product biosynthesis, but may also illuminate routes to novel therapeutics to afford alternative lines of defense against mycobacterial infections.(4)
Figure 1.

Representative PGLs. The carbon-chain variability shown is that of the PGLs produced by the opportunistic human pathogen Mycobacterium marinum. The glycosyl substituent in PGLs is strain/species-specific. The involvement of the FadD22-Pks15/1 enzyme system in the biosynthesis of the phenolphthiocerol moiety of PGLs is illustrated according to the model proposed herein based on the results of in vitro and in vivo studies carried out with the proteins from M. marinum.
We have recently demonstrated that the protein FadD22 is required for PGL biosynthesis and reported a FadD22 inhibitor that blocks PGL production.(4b) FadD22 is comprised of a p-hydroxybenzoic acid (pHBA) adenylation (A) domain and an aroyl carrier protein (ArCP) domain.(4b) This didomain protein forms a p-hydroxybenzoyl-S-FadD22 thioester intermediate from pHBA and ATP in a coenzyme A (CoA)-independent manner.(4b) We hypothesize that this intermediate primes the biosynthesis of the long-chain β-diol-containing moiety of PGLs [phenolphthiocerol (PPOL)] by presenting the pHBA starter unit for elongation by Pks15/1 to form p-hydroxyphenylalkanoate (PHPA) intermediates during PPOL biosynthesis (Scheme 1). Genetic studies have demonstrated that the pks15/1 gene is required for PGL biosynthesis,2c,5 and our sequence analysis indicates that Pks15/1 is well-conserved among PGL producers (2104−2118 amino acid residues, 79−100% sequence identity; Figure S1, Supporting Information). Pks15/1 is a 6-domain protein with homology to reducing iterative type I polyketide synthases (PKSs),(1) a poorly understood family of enzymes that synthesize highly reduced products such as lovastatin, T-toxin, and fumonisin.(6) The proposed PHPA intermediates synthesized by the FadD22-Pks15/1 system are likely to be further extended by the PpsA-E noniterative type I PKS system to complete PPOL biosynthesis.(1) Subsequently, PPOL is esterified with characteristic long-chain multimethyl-branched fatty acids (Figure 1) by the acyltransferase PapA5.(7)
Scheme 1. CoA-Independent Loading of the pHBA Starter Unit and Chain Elongation for Biosynthesis of PHPA Intermediates by the FadD22-Pks15/1 System during PGL Assembly.

Pks15/1 is shown loaded with the malonyl extender unit. The indicated range of iterative cycles and consequent carbon-chain variability in the p-hydroxyphenylalkanoyl-S-Pks15/1 thioester intermediate is that expected during biosynthesis of PGLs from M. marinum. Domain abbreviations: A, adenylation; ACP, acyl carrier protein; ArCP, aroyl carrier protein; AT, acyltransferase; DH dehydratase; ER, enoylreductase; KR, ketoreductase; KS, ketosynthase. The thiols of the phosphopantetheinyl group in the carrier domains and the catalytic cysteine in the KS domain are shown.
Herein, we report in vitro and in vivo investigations that demonstrate the functional partnership between FadD22 and Pks15/1 for production of PHPA intermediates from the PGL biosynthetic pathway.
Results and Discussion
Loading of Starter and Extender Units onto the FadD22-Pks15/1 System
We first evaluated whether Pks15/1 could be loaded with pHBA in a FadD22-dependent manner in vitro. To this end, incorporation of covalently bound [14C]pHBA-derived label onto Pks15/1 and FadD22 was monitored by standard sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by autoradiography (Figure 2).(8) Phosphopantetheinylated (holo)-FadD22 was readily autoacylated in an ATP-dependent and CoA-independent manner (cf., lanes 1, 3−6, and 8). Conversely, neither FadD22(S576A) (lane 7), a phosphopantetheinylation site-deficient mutant with Ser-576 substituted by Ala,(4b) nor apo-FadD22 (not shown) were capable of autoacylation, indicating that a functional ArCP domain is required for autoacylation. These results are in agreement with FadD22 autoacylation studies reported previously.(4b) Holo-Pks15/1 was robustly radiolabeled in the reaction containing holo-FadD22 (lane 3). Conversely, holo-Pks15/1 radiolabeling was not detected in reactions lacking holo-FadD22 (lane 2), containing FadD22(S576A) in place of holo-FadD22 (lane 7), or lacking ATP (lane 8).
Figure 2.

FadD22-dependent loading of pHBA onto Pks15/1. (a) Image of a coomassie blue-stained tris-glycine polyacrylamide gel run under denaturing conditions (7.5%, SDS-PAGE). Lanes 1−8 were loaded with acylation reaction mixtures with the indicated composition of key components. Lane M, molecular weight marker. (b) Autoradiograph image of the gel showing incorporation of [14C]pHBA-derived covalent label onto the proteins.
Starter unit loading onto three Pks15/1 site-directed mutants was investigated as well. These mutants had Ala substitutions at the predicted catalytic Cys (Cys-211) of the β-ketoacyl synthase (KS) domain and/or the phosphopantetheinylation site (Ser-2039) of the acyl carrier protein (ACP) domain. These key amino acid residues were identified by sequence analysis (Figure S1, Supporting Information). Pks15/1(S2039A), the ACP domain mutant defective for carrier domain phosphopantetheinylation (Figure S2, Supporting Information), was radiolabeled like wild-type (cf., lanes 3 and 5). In contrast, holo-Pks15/1(C211A), the KS domain mutant, and Pks15/1(C211A/S2039A), the double mutant, were not competent for label incorporation (lanes 4 and 6, respectively). These observations indicate that a functional KS domain is required for starter unit loading, whereas a functional ACP domain is not. Overall, the results from the starter unit loading analysis are consistent with a model in which the p-hydroxybenzoyl unit loaded onto the holo-ArCP domain of FadD22 is transferred to the KS domain active-site Cys-211 residue in Pks15/1. Formation of a p-hydroxybenzoyl-Cys-KS domain thioester intermediate would parallel the priming mechanism of other type I PKSs with ordinary acetyl-CoA- or propionyl-CoA-derived starter units.(9) Pks15/1 was also competent for autoacylation with the predicted extender unit, the malonyl group derived from malonyl-CoA. Covalently bound [14C]malonyl-CoA-derived label was readily incorporated onto Pks15/1 (not shown). This labeling is expected to result from AT domain-catalyzed acylation of the ACP domain as seen in other PKSs.(9)
Synthesis of PHPA Products in the in Vitro and in Vivo FadD22-Pks15/1 Reconstituted Systems
Having demonstrated FadD22-dependent loading of the starter unit onto Pks15/1 and the ability of Pks15/1 for autoacylation with the extender unit, we next evaluated the ability of Pks15/1 to synthesize PHPA products in the in vitro and in vivo FadD22-Pks15/1 reconstituted systems. In these studies, PHPA formation was assessed by liquid chromatography−mass spectrometry (LC−MS) analysis of extracted products released from the enzyme after alkaline hydrolysis of in vitro enzymatic reactions or lysates of engineered E. coli strains expressing the FadD22-Pks15/1 system. Gratifyingly, incubation in vitro of holo-FadD22 and holo-Pks15/1 in the presence of pHBA, malonyl-CoA, ATP, and NADPH led to formation of a characteristic product series. The products in this series had exact masses consistent with predicted PHPAs differing by multiples of the 28-amu unit (−CH2CH2−) expected from the presence of different numbers of two-carbon extender units (Figure 3a; Table 1; Figure S3 in Supporting Information). The retention time and the observed ion [M − H+]−m/z of a synthetic 21-(4-hydroxyphenyl)henicosanoic acid PHPA standard (C27H46O3, calculated exact mass: 418.3447; experimental neutral mass 418.3453, where the experimental neutral mass is the observed ion [M − H+]−m/z plus H+, 1.00728 Da) matched the retention time and the ion [M − H+]−m/z of the corresponding PHPA product (C27H46O3) (peak 6 in Figure 3; Table 1; Figure S3 in Supporting Information). This PHPA was one of the most abundant in the PHPA product series. The compounds in the PHPA product series also had the same MS/MS fragmentation pattern of the synthetic PHPA standard. This pattern was consistent with loss of H2O (m/z [M − H+ − 18, M − H+ − H2O]−) or CO2 (m/z [M − H+ − 44, M − H+ − CO2]−) by the [M − H+]− parent ion (Figure S3, Supporting Information).
Figure 3.

Analysis of PHPA products synthesized by FadD22-Pks15/1 reconstituted systems. Extracted ion chromatograms for the [M − H+]− ion exact masses (listed above) of the expected PHPA products from in vitro (a) and in vivo (b) reconstituted systems are shown. The PHPA structures are illustrated. Data representative of three experiments are shown.
Table 1. Mass Spectrometry Data for p-Hydroxyphenylalkanoate Products.
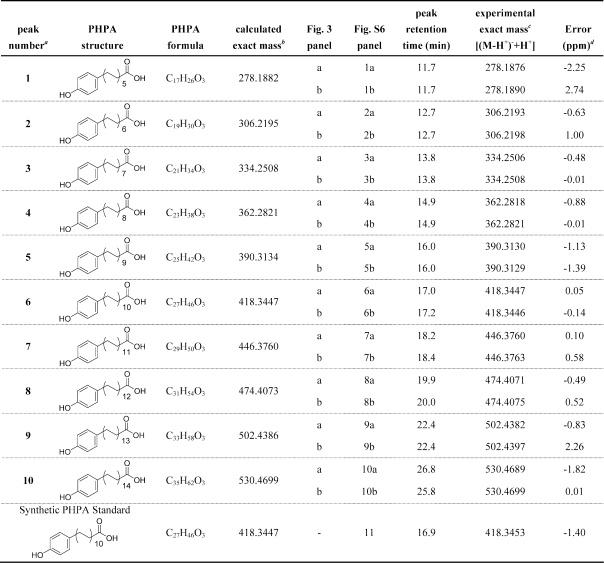 |
Peak numbers correspond to those shown in Figure 3.
The calculated exact masses for the molecular formulas shown were determined using Agilent’s MassHunter Qualitative Analysis Software package version B.02.00 (Agilent Technologies, Inc.).
The experimental exact masses were determined by processing the data using Agilent’s MassHunter Qualitative Analysis Software’s “Find Compounds by Formula” feature. The software searches the data for ions based on a given molecular formula and, in this case, extracts the ions for the [M − H+]− ion for the molecular formulas of interest. The experimental exact mass is calculated by the addition of a proton (H+; 1.00728 Da) to the measured m/z value. The experimental exact mass is used to calculate the ppm difference from the calculated exact mass for a given molecular formula.
The ppm error was determined by Agilent’s MassHunter Qualitative Analysis Software. PHPA, p-hydroxyphenylalkanoate.
Conversely, the PHPA product series was not detected in control reactions lacking a single enzyme or the substrates malonyl-CoA or NADPH (not shown). Traces of the most prominent product peaks in the series were seen in controls lacking pHBA or ATP (not shown). This may result from purified FadD22 preparations containing traces of the p-hydroxybenzoyl-S-FadD22 covalent intermediate or the stable p-hydroxybenzoyl-AMP·FadD22 noncovalent intermediate.(4b) These intermediates could form in E. coli using endogenous pHBA, an intermediate in ubiquinone synthesis.(10) Reactions in which holo-Pks15/1 was replaced with holo-Pks15/1(C211A), Pks15/1(S2039A), or Pks15/1(C211A/S2039A) were not competent for PHPA formation either (not shown). These results are consistent with the fact that Pks15/1(C211A) and Pks15/1(C211A/S2039A) cannot be loaded with the starter unit (Figure 2) and Pks15/1(S2039A) is defective for ACP domain phosphopantetheinylation (Figure S2, Supporting Information). The results obtained with the mutant proteins are in agreement with the hypothesized functions of Cys-211 and Ser-2039.
Encouragingly, FadD22-Pks15/1-dependent biosynthesis of the PHPA product series was achieved also in an E. coli strain engineered to express FadD22 and Pks15/1 from plasmids pCDF-FadD22 and pETDuet-Pks15/1 (Figure 3b; Table 1; Figure S3 in Supporting Information). This strain also contained the plasmid pSU20-Sfp. pSU20-Sfp expresses Bacillus subtilis phosphopantetheinyl transferase Sfp,(11) which was used herein to obtain FadD22 and Pks15/1 in their holo forms. Conversely, the PHPA product series was not detected in control strains in which pETDuet-Pks15/1 was replaced with the pETDuet-1 vector, pCDF-FadD22 was replaced with the pCDF-1 vector, pSU20-Sfp was omitted, or the culture medium was not supplemented with the starter unit, pHBA (not shown).
The results of the PHPA production studies support the idea that the FadD22-Pks15/1 reconstituted systems produce PHPAs with alkyl chain length variability consistent with a 5-to-14 range of iterative extension cycles with the malonyl-CoA-derived extender unit. This suggests that Pks15/1 is a reducing iterative type I PKS with a relaxed control of catalytic cycle iterations. Importantly, such a mechanistic property would explain the origin of the characteristic alkyl chain length variability seen in the PPOL moiety of PGLs.(1) Notably, one of the most prominent PHPA peaks observed in the product series (peak 7, Figure 3) corresponds to 23-(4-hydroxyphenyl)tricosanoic acid (C29H50O3; calculated exact mass, 446.3760; experimental neutral mass in the in vitro and in vivo reconstituted systems, 446.3760 and 446.3763, respectively; Table 1). This PHPA product is predicted to arise from elongation of the starter unit with 11 extender units, and its generation will require a total of 68 catalytic operations by the FadD22-Pks15/1 system [68 = 1 (pHBA adenylation) + 1 (pHB-S-ArCP formation) + 1 (pHB-S-KS formation) + 11 × 4 (condensation, reduction, dehydration, enoyl reduction) + 11 (AT-catalyzed malonyl unit loading onto the ACP at each of 11 cycles) + 10 (transfer of intermediate from ACP to KS, except in the last cycle)]. Coincidently, 23-(4-hydroxyphenyl)tricosanoic acid would be the hypothesized PHPA intermediate extended by the PpsA-E PKS system to form the PPOL with the largest carbon chain reported in M. marinum (Figure 1).
The 5-to-14 range of extension cycles observed in the reconstituted systems is wider than the 9-to-11 range deduced from reported M. marinum PPOL structures.(1) In connection with this observation, it is worth noting that, in nonreducing iterative type I PKS systems, a product template (PT) domain and a thioesterase (TE) domain are proposed to be involved in the control of product chain length, cyclization, and release.6,12 In contrast, reducing iterative type I PKSs, such as Pks15/1, lack the PT and TE domains, implying a distinct mechanism for the control of the structure of the final products. In fact, Du and co-workers recently suggested that the final size of highly reduced polyketide products of iterative type I PKSs is not determined by the PKSs alone.(13) It is possible that, in the case of Pks15/1, the complete information for final product size is “programmed” via the interaction of Pks15/1 with other enzymes of the PGL pathway. In this regard, our FadD22-Pks15/1 reconstituted systems lay a first foundation for future efforts to unveil the mechanism of iterative catalysis control by which the structures of the final products of Pks15/1 are defined, and to scrutinize the functional partnerships of Pks15/1 with downstream enzymes of the PGL pathway to achieve PPOL biosynthesis.
Conclusion
The studies reported herein demonstrate the functional partnership between FadD22, a pHBA-specific CoA-independent stand-alone didomain initiation module, and Pks15/1, a 6-domain reducing iterative type I PKS, for production of PHPA intermediates from the PGL biosynthetic pathway. To the best of our knowledge, this is the first demonstration of aroyl-S-ArCP-dependent priming of an iterative PKS. Our results are consistent with a model in which (1) pHBA is transferred from the ArCP domain in p-hydroxybenzoyl-FadD22 to the KS domain active-site Cys residue of Pks15/1 and (2) the formed p-hydroxybenzoyl-KS transient intermediate serves as a substrate for Pks15/1-dependent acyl chain elongation using the malonyl-CoA-derived extender unit for biosynthesis of PHPA intermediates during PGL production (Scheme 1). The presented data suggest that Pks15/1 has a relaxed control of catalytic cycle iterations, a mechanistic property that explains the origin of a characteristic alkyl chain length variability seen in mycobacterial PGLs. Overall, this study provides novel insight into the biosynthesis of PGLs, an important group of mycobacterial virulence factors.
Experimental Section
Reagents
Unless otherwise stated, all commercially available materials were purchased from Aldrich, Acros Organics, Fisher Scientific, TCI, or Alfa Aesar and were used without any further purification. When necessary, solvents and reagents were dried prior to use, using standard protocols. Radiolabeled [carboxy-14C]p-hydroxybenzoic acid and [2-14C]malonyl-CoA were purchased from American Radiolabeled Chemicals, Inc. 12-N-Biotinyl-(aminododecanoyl)-Coenzyme A (biotinyl-CoA) was purchased from Avanti Polar Lipids, Inc. Monoclonal antibiotin-alkaline phosphatase conjugated antibody was obtained from Sigma-Aldrich. All other molecular biology reagents were from Sigma, Invitrogen, New England Biolabs, Novagen, QIAGEN, or Stratagene. Oligonucleotide primers were purchased from Integrated DNA Technologies, Inc. The synthetic PHPA standard [21-(4-hydroxyphenyl)henicosanoic acid; C27H46O3] was synthesized as described in the Supporting Information and outlined in Figure S4.
Incorporation of Covalently Bound [14C]pHBA-Derived Label onto Pks15/1 and FadD22 in Vitro
For these and all other experiments with FadD22 and Pks15/1 described in this study, we used the proteins from the opportunistic human pathogen Mycobacterium marinum.(14) The proteins utilized in the experiments were recombinantly produced and purified as described in the Supporting Information (Figure S5). The starter unit loading experiments were conducted using reported approaches for monitoring [14C]/[3H]acyl-enzyme thioester intermediate formation.4b,15 The standard reaction mixture (20 μL) contained 12 μM [14C]p-hydroxybenzoic acid (pHBA), 100 mM sodium phosphate buffer (pH 7.2), 10% glycerol, 1 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP), 0.5 mM MgCl2, 1 mM ATP, 4 μM FadD22 (wild-type or mutant), and 8 μM Pks15/1 (wild-type or mutant). In control reactions, selected components were omitted as indicated in Figure 2. After incubation (30 °C, 2 h), the reactions were quenched by the addition of SDS-PAGE loading buffer(16) (10 μL, 2× concentrated, lacking reducing reagent) and analyzed by standard SDS-PAGE (7.5%).(16) For detection of 14C-labeled proteins, the polyacrylamide gels were treated with Amplify Fluorographic Reagent (GE Healthcare) as recommended in the manufacturer’s manual. The treated polyacrylamide gels were exposed to storage phosphor screens (GE Healthcare), which were subsequently scanned using a Typhoon Trio Imager (Amersham Biosciences).
Incorporation of Covalently Bound [14C]Malonyl-CoA-Derived Label onto Pks15/1
The extender unit loading experiments were conducted using reported approaches for monitoring [14C]/[3H]acyl-enzyme thioester intermediate formation.4b,15 The proteins utilized were recombinantly produced and purified as described in the Supporting Information. The standard reaction mixture (20 μL) contained 50 μM [14C]malonyl-CoA, 100 mM sodium phosphate buffer (pH 7.2), 10% glycerol, 1 mM TCEP, 0.5 mM MgCl2, 1 mM ATP, and 8 μM Pks15/1 (wild-type or mutant). After incubation (30 °C, 3 min), the reactions were quenched by the addition of SDS-PAGE loading buffer(16) (7 μL, 3× concentrated, lacking reducing reagent) and analyzed by standard SDS-PAGE (7.5%).(16) For detection of 14C-labeled proteins, the polyacrylamide gels were treated and exposed to storage phosphor screens as described in the preceding paragraph. The screens were then scanned using a Typhoon Trio Imager.
Protein Phosphopantetheinylation Analysis
Phosphopantetheinylation was assessed as incorporation of the biotinylated phosphopantetheinyl group derived from the CoA analog biotinyl-CoA (Avanti Polar Lipids) onto the proteins. The promiscuous Bacillus subtilis phosphopantetheinyl transferase Sfp(11) was utilized to phosphopantetheinylate Pks15/1 proteins in vitro. Sfp was recombinantly produced and purified as previously reported.(11) The Pks15/1 proteins were produced recombinantly and purified as described in the Supporting Information. The standard reaction mixture (20 μL) contained 4 μM Pks15/1 (wild-type or mutant), 100 mM sodium phosphate buffer (pH 7.2), 10% glycerol, 2 mM TCEP, 0.5 mM MgCl2, 50 nM Sfp, and 50 μM biotinyl-CoA. After incubation (30 °C, 15 min), the reactions were quenched by addition of SDS-PAGE loading buffer(16) (7 μL, 3× concentrated, lacking reducing reagent). The samples were analyzed by standard SDS-PAGE(16) (5%) and by Western blot to detect Sfp-dependent biotinylation of Pks15/1 proteins. Western blot was carried out using standard methodologies.(16) Briefly, proteins were transferred from the polyacrylamide gel to a hydrophobic polyvinylidene difluoride (PVDF) membrane (Hybond-P, GE Healthcare) using a Mini Trans-Blot electrophoretic transfer cell as recommended by the manufacturer (Bio-Rad Laboratories). After the electrophoretic transfer (3 h, 200 mA), the membrane was washed with TBS-T buffer (50 mM Tris, pH 7.6; 150 mM NaCl; 0.1% Tween-20) and blocked in 5% skim milk powder in TBS-T. Covalently incorporated biotinylated phosphopantetheinyl group onto the proteins was detected using a commercial mouse monoclonal antibiotin-alkaline phosphatase conjugated antibody as recommended by the manufacturer (Sigma-Aldrich). The antibody was used at a dilution of 1:10 000 relative to the original stock. The colorimetric detection of biotinylated proteins was achieved using alkaline phosphatase BCIP/NBT (5-bromo-4-chloro-3′-indolyphosphate p-toluidine salt/nitro-blue tetrazolium chloride) liquid substrate system as recommended by the manufacturer (Sigma-Aldrich).
FadD22-Pks15/1 Reconstituted System in Vitro
The proteins utilized in the reconstitution experiments were produced recombinantly and purified as described in the Supporting Information. The standard reaction mixture (1 mL) contained 8 μM pHBA, 100 mM sodium phosphate buffer (pH 7.2), 1 mM TCEP, 0.5 mM MgCl2, 1 mM ATP, 0.5 mM malonyl-CoA, 1 mM NADPH, 0.8 μM FadD22 (wild-type or mutant), and 8 μM Pks15/1 (wild-type or mutant). In control reactions, selected components were omitted as indicated in the Results and Discussion. After incubation (30 °C, 5 h), the reactions were quenched by the addition of 200 μL of 1 M NaOH and incubated at 65 °C for 20 min to release covalently bound products from the enzymes. After acidification with 200 μL of 2 M HCl, the products were extracted with ethyl acetate (1.4 mL × 5), and the recovered organic layer was evaporated to dryness. The residual material was dissolved in 50 μL of dichloromethane/methanol (3:1, vol/vol) and analyzed by LC−MS as described below.
FadD22-Pks15/1 Reconstituted System in Vivo
The engineered E. coli strains used in the in vivo reconstitution experiments were generated by introducing selected expression plasmids into the expression host E. coli BL21(DE3) (Stratagene). The plasmids were constructed as described in the Supporting Information and outlined in Figure S6. They were introduced into E. coli BL21(DE3) using standard electroporation.(16)E. coli strains were routinely cultured in Luria−Bertani (LB) media(16) containing 50 μg/mL ampicillin (pETDuet-Pks15/1 or pETDuet-1 containing strains), 20 μg/mL chloramphenicol (pSU20-Sfp containing strains), 30 μg/mL kanamycin (pRSF-ACC containing strains), and 50 μg/mL streptomycin (pCDF-FadD22 or pCDF-1 containing strains). Cultures (30 mL) were incubated with orbital shaking (220 rpm) at 37 °C. When cultures reached an OD600nm of 0.6, recombinant protein production was induced by addition of isopropyl-β-d-1-thiogalactopyranoside (IPTG, 0.1 mM). pHBA (5 μM) was added to the cultures concomitantly with IPTG, except in selected control cultures. After 24 h of additional incubation at a reduced temperature optimized for protein expression (18 °C, 220 rpm), cells were harvested by centrifugation (6000g, 20 min) and washed with 100 mM sodium phosphate buffer pH 7.2 (5 mL × 3). After being washed, the cells were resuspended in 500 μL of 100 mM sodium phosphate buffer (pH 7.2) containing 1 mM TCEP, and cell lysates were prepared using a Mini-Bead Beater cell disrupter (BioSpec Products, Inc.) according to the manufacturer’s instructions. The cell lysates were treated by addition of 100 μL of 1 M NaOH and incubated at 65 °C for 20 min to release covalently bound products from the enzymes. After acidification with 100 μL of 2 M HCl, the products were extracted with ethyl acetate (700 μL × 5), and the recovered organic layer was evaporated to dryness. The residual material was dissolved in 50 μL of dichloromethane/methanol (3:1, vol/vol) and analyzed by LC−MS as described below.
LC−MS Instrumentation and Analysis
Mass spectral data were collected at the City University of New York (CUNY) Mass Spectrometry Facility at Hunter College (New York, NY) on an Agilent Technologies G6520A high-resolution Q-TOF mass spectrometer attached to an Agilent Technologies 1200 Capillary HPLC system. Samples were ionized by electrospray ionization in negative mode. Chromatography was performed on a Zorbax 2.1 × 30 mm SB-C18 3.5 μm column (part no. 873700-902) using water containing 0.1% formic acid and 50 μM ammonium formate (solvent A) and 95% acetonitrile containing 0.1% formic acid and 50 μM ammonium formate (solvent B) at a flow rate of 350 μL/min. The gradient program was as follows: 2% B (0−3 min), 2−100% B (3−15 min), 100% B (15−30 min). Total analysis time was 30 min. The HPLC flow was diverted to waste for the first 2.5 min. The temperature of the column was held at 45 °C for the entire analysis. Instrument parameters were as follows: fragmentor = 140 V; drying gas temperature = 300 °C; drying gas flow = 12 L/min; nebulizer pressure = 40 psi; and capillary voltage = 3500 V. Data were collected with the instrument set to low mass range (100−1700 m/z) under high-resolution conditions at 4 GHz, and data were stored as both centroid and profile mode. The mass spectra were collected over a range of 115−1600 m/z at 1 spectra/s, and MS/MS spectra were collected over a range of 60−800 m/z at 1 spectra/s with an isolation width of ∼4 m/z. The collision energy was ramped using a slope of 6.5 V/100 Da and an offset of 5 V using nitrogen as the collision gas. The reference masses used were purine with (M − H+)− at 119.03632 m/z and HP-922 with (M + formate)− ion at 966.00072 m/z. They were infused into the spray chamber using Agilent’s calibrant delivery system. The instrument was controlled with Agilent MassHunter Workstation Acquisition Software, and data were analyzed using Agilent MassHunter Workstation Qualitative Analysis Software (Agilent Technologies, Santa Clara, CA). Funding for this instrument system was provided by NIH Shared Instrumentation Grant 1S10RR022649-01 and the CUNY Instrumentation Fund.
Acknowledgments
This work was supported by NIH Grant AI069209 (L.E.N.Q.) and a Milstein Program in Chemical Biology Grant (L.E.N.Q.). We are grateful to R. Moy and G. Sadhanandan (L.E.N.Q. laboratory) for assistance with construction of recombinant plasmids.
Two names were added to the list of authors of this publication on October 30, 2009.
Supporting Information Available
Experimental procedures for plasmid constructions, protein purification, and PHPA standard synthesis; sequence alignment; and Figures S1−S6. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Onwueme K. C.; Vos C. J.; Zurita J.; Ferreras J. A.; Quadri L. E. Prog. Lipid Res. 2005, 19, 259–302. [DOI] [PubMed] [Google Scholar]
- a Ng V.; Zanazzi G.; Timpl R.; Talts J. F.; Salzer J. L.; Brennan P. J.; Rambukkana A. Cell 2000, 103, 511–524. [DOI] [PubMed] [Google Scholar]; b Rambukkana A.; Zanazzi G.; Tapinos N.; Salzer J. L. Science 2002, 296, 927–931. [DOI] [PubMed] [Google Scholar]; c Reed M. B.; Domenech P.; Manca C.; Su H.; Barczak A. K.; Kreiswirth B. N.; Kaplan G.; Barry C. E. III. Nature 2004, 431, 84–87. [DOI] [PubMed] [Google Scholar]; d Ruley K. M.; Ansede J. H.; Pritchett C. L.; Talaat A. M.; Reimschuessel R.; Trucksis M. FEMS Microbiol. Lett. 2004, 232, 75–81. [DOI] [PubMed] [Google Scholar]; e Collins D. M.; Skou B.; White S.; Bassett S.; Collins L.; For R.; Hurr K.; Hotter G.; de Lisle G. W. Infect. Immun. 2005, 73, 2379–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Tsenova L.; Ellison E.; Harbacheuski R.; Moreira A. L.; Kurepina N.; Reed M. B.; Mathema B.; Barry C. E. III; Kaplan G. J. Infect. Dis. 2005, 192, 98–106. [DOI] [PubMed] [Google Scholar]; g Robinson N.; Kolter T.; Wolke M.; Rybniker J.; Hartmann P.; Plum G. Traffic 2008, 9, 1936–1947. [DOI] [PubMed] [Google Scholar]
- Reed M. B.; Gagneux S.; Deriemer K.; Small P. M.; Barry C. E. III. J. Bacteriol. 2007, 189, 2583–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Quadri L. E. Infect. Disord. Drug Targets 2007, 7, 230–237. [DOI] [PubMed] [Google Scholar]; b Ferreras J. A.; Stirrett K. L.; Lu X.; Ryu J. S.; Soll C. E.; Tan D. S.; Quadri L. E. Chem. Biol. 2008, 15, 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constant P.; Perez E.; Malaga W.; Laneelle M. A.; Saurel O.; Daffe M.; Guilhot C. J. Biol. Chem. 2002, 277, 38148–38158. [DOI] [PubMed] [Google Scholar]
- Cox R. J. Org. Biomol. Chem. 2007, 5, 2010–2026. [DOI] [PubMed] [Google Scholar]
- a Buglino J.; Onwueme K. C.; Ferreras J. A.; Quadri L. E.; Lima C. D. J. Biol. Chem. 2004, 279, 30634–30642. [DOI] [PubMed] [Google Scholar]; b Onwueme K. C.; Ferreras J. A.; Buglino J.; Lima C. D.; Quadri L. E. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 4608–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See the Supporting Information for additional experimental procedures (plasmid constructions, protein purification, and PHPA standard synthesis), sequence alignment, and Figures S1−S6.
- a Staunton J.; Weissman K. J. Nat. Prod. Rep. 2001, 18, 380–416. [DOI] [PubMed] [Google Scholar]; b Fischbach M. A.; Walsh C. T. Chem. Rev. 2006, 106, 3468–3496. [DOI] [PubMed] [Google Scholar]
- Meganathan R. FEMS Microbiol. Lett. 2001, 203, 131–139. [DOI] [PubMed] [Google Scholar]
- Quadri L. E.; Weinreb P. H.; Lei M.; Nakano M. M.; Zuber P.; Walsh C. T. Biochemistry 1998, 37, 1585–1595. [DOI] [PubMed] [Google Scholar]
- Crawford J. M.; Thomas P. M.; Scheerer J. R.; Vagstad A. L.; Kelleher N. L.; Townsend C. A. Science 2008, 320, 243–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; Vogeler C.; Du L. J. Nat. Prod. 2008, 71, 957–960. [DOI] [PubMed] [Google Scholar]
- a Katoch V. M. J. Med. Res. 2004, 120, 290–304. [PubMed] [Google Scholar]; b Jarzembowski J. A.; Young M. B. Arch. Pathol. Lab. Med. 2008, 132, 1333–1341. [DOI] [PubMed] [Google Scholar]
- a Quadri L. E.; Keating T. A.; Patel H. M.; Walsh C. T. Biochemistry 1999, 38, 14941–14954. [DOI] [PubMed] [Google Scholar]; b Quadri L. E.; Sello J.; Keating T. A.; Weinreb P. H.; Walsh C. T. Chem. Biol. 1998, 5, 631–645. [DOI] [PubMed] [Google Scholar]
- Sambrook J.; Russell D. W.. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, 2001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.