

Abstract
Viruses differ markedly in their specificity toward host organisms. Here, we test the level of general sequence adaptation that viruses display toward their hosts. We compiled a representative data set of viruses that infect hosts ranging from bacteria to humans. We consider their respective amino acid and codon usages and compare them among the viruses and their hosts. We show that bacteria-infecting viruses are strongly adapted to their specific hosts, but that they differ from other unrelated bacterial hosts. Viruses that infect humans, but not those that infect other mammals or aves, show a strong resemblance to most mammalian and avian hosts, in terms of both amino acid and codon preferences. In groups of viruses that infect humans or other mammals, the highest observed level of adaptation of viral proteins to host codon usages is for those proteins that appear abundantly in the virion. In contrast, proteins that are known to participate in host-specific recognition do not necessarily adapt to their respective hosts. The implication for the potential of viral infectivity is discussed.
Keywords: capsid, codon usage, host tropism, protein classification, UniProt database, viral proteome
Introduction
Viruses show appreciable variation in the selectivity with which they infect host organisms. Some viruses infect a broad range of species, whereas others infect only a single host. A successful viral infection requires that the virus possess the capability to enter the host cell and take over cellular functions and direct them toward the efficient production of new viruses. Most viruses recognize their respective hosts through membrane receptors that have a role in host physiology. Examples of such receptors are gangliosides, heparan sulfate moieties, and integrins (Garrigues et al, 2008), which act as the cell receptors for simian virus 40 (SV40), human cytomegalovirus (HHV-5), and human herpesvirus 8 (HHV8), respectively. In stark contrast, for some viruses, host range is not limited to the recognition stage (McFadden, 2005). For example, poxviruses bind to and enter a wide range of mammalian cells, but a fruitful replication cycle occurs only in a restricted set of hosts. Replication of poxviruses involves the host cell cycle, signal transduction, transcription factors, phosphatases, and interferon-induced mediators. Therefore, the features that govern the host range for poxvirus seem to involve a rich collection of host genes (McFadden, 2005).
All viruses are characterized by very high natural mutation rates, with the RNA viruses displaying an exceptionally high rate (Drake, 1993). Co-evolution and adaptation of viruses to their hosts were mostly studied by comparing mutations at synonymous and non-synonymous coding sites in specific genes. The fast adaptation of human immunodeficiency virus-1 (HIV-1) to specific HLA-1 epitopes validates the importance of viral evolution at a population level (Kawashima et al, 2009). As of yet, the study of adaptation of viruses toward their hosts has been undertaken for specific viral families, including retroviruses (Bronson and Anderson, 1994), astroviridae (van Hemert et al, 2007), mimivirus (Sau et al, 2006), and bacteriophages (Lucks et al, 2008), but this has not been systematically investigated for all known viral proteomes.
The degeneracy of the genetic code implies that multiple triplets code for the same amino acid. The frequencies with which different codons are used vary significantly between organisms and between proteins within the same organism (Akashi, 2001). Many studies have focused on the bias in codon usage among species. In single cell organisms (prokaryotes, archaea, and some fungi), the codon usage is strongly tuned for highly expressed genes and was thus concluded to be optimized for translational efficiency (Sharp et al, 1988). However, the main trends in multicellular organism codon usage were attributed to the isochore-dependent genome composition (GC) content, gene architecture, and chromosomal locations (see discussion in Costantini et al, 2009). Still, evidence for codon usage bias toward highly expressed genes and its correlation to tRNA abundance argues that translational efficiency does have a role for some plant, fly, and worm proteomes (Duret, 2000 and references within). Evolutionary forces and multiple molecular processes (e.g., unbiased gene conversion, mutation rates, and genetic drift) have also participated in shaping codon usage in higher eukaryotes (Bernardi, 1986; Duret, 2002). The molecular determinants that have globally influenced the translational efficiency in Escherichia coli (Kudla et al, 2009) and the evolution of polymerase genes in the influenza A virus (Brower-Sinning et al, 2009) indicate that, in addition to GC content, RNA folding processes also affect the adaptability and translational capacity of viral sequences.
Viruses do not have tRNAs, and consequently the translation of viral proteins relies entirely on the pool of host tRNAs. An exception is the Paramecium bursaria chlorella virus, which contains a partial set of tRNAs and other host-like properties (Van Etten and Meints, 1999). In a recent study that tested the codon usage adaptation for over 100 bacteriophages infecting 10 different bacterial hosts, it was shown that the bacteriophage genomes are under codon-selective pressure imposed by the translational biases of their respective hosts (Carbone, 2008). The reasoning underlying this codon selection hypothesis argues that it provides an advantage for viral protein synthesis at the level of translational efficiency.
In viruses infecting multicellular animals, such translational biases may lead to increased virion production rates within the infected cell and reduce the accessibility of viruses to the immune response of the host (Bonhoeffer and Nowak, 1994). However, to the best of our knowledge, the analysis of codon biases of eukaryotic (alongside prokaryotic) viruses compared with their hosts has yet to be undertaken on a large scale. However, related phenomena have been described. Specifically, the codon usage bias in the poxviridae family (dsDNA viruses) was determined by measuring the effective number of codons in the viral proteome. Neither the expression level nor the gene size was shown to be a determinant of the measured codon usage biases. Nonetheless, for most poxviruses, the codon usage was close to the value predicted based on the GC content (Barrett et al, 2006). Similar results were shown for coronavirus (Gu et al, 2004) and other vertebrate-infecting DNA viruses (Shackelton et al, 2006). In papillomavirus, the codon bias was attributed to the AT content rather than to host specificity (Zhao et al, 2003). In the case of retroviruses, it was shown that strong discrimination against CpG sequences directly shapes the codon usage and, as a result, even indirectly restricts the choice of amino acids (Berkhout et al, 2002). Thus, in general, GC and, specifically, the GC content were thus far found to be the major determinants of codon usage in vertebrate DNA viruses (Shackelton et al, 2006).
It has been found that for many viruses, genome-wide mutational pressures override the selection for specific codons (Jenkins and Holmes, 2003). Studies of the evolutionary history of viral adaptation propose a cross talk between codon usage, replication mode, genome size, and host range (Koonin et al, 2006). Furthermore, the observation that there exist both eukaryotic viruses that have adapted their codon usage toward their hosts and those that show little evidence for such adaptation recently prompted the hypothesis that this simply reflects the limited time of the latter for optimization toward their hosts (Barrai et al, 2008). A contrary view would suggest that the extremely high mutation rates in viruses (especially in RNA viruses) outpace the evolutionary processes of selection that drive such optimization of the virus to the host.
In this paper, we set out to determine whether, despite the enormous diversity among viruses, a high-level, generalized trend of adaptation of viruses toward their hosts can be observed. To this end, we provide a strict virus-to-host mapping using a non-redundant set of representative viruses and hosts, ranging from human to bacteria. We develop a statistical framework for the unbiased assessment of the mutual pairwise distances between all viruses and all recognized hosts. To test the hypothesis of general molecular adaptation of a virus toward its hosts, we focus on codon usage and amino acid preferences within groups of viruses that are unified at varying taxonomical granularities. We observe that all bacteriophages are strongly tuned to match their unique hosts and this correspondence is also evident in their GC contents. However, somewhat surprisingly, viruses that infect humans resemble all mammalian hosts equally, and this similarity even extends to aves and several insects. This observation does not hold for viruses of other mammals, despite a strong similarity among the codon usages of most mammals. Finally, we show that viral selection of codon usage toward that of the host has not occurred uniformly for all proteins of the virus, but it is mainly dominated by the set of proteins expressed in high abundance. The implications of these observations for viral evolution and on the potential for zoonotic epidemics are discussed.
Results
Viral proteomes are biased and poorly annotated
Viruses comprise the largest group of parasitic organisms for which cross talk between the proteomes and their cognate hosts can be studied.
The huge diversity among viruses encompasses their mode of replication, shape, stability, proteome size, and infectivity. These factors impose an inherent difficulty in the classification of viruses into taxonomical groupings. Currently, ∼10% of all sequences in the UniProtKB database (Boutet et al, 2007) (release 14.6) are viral proteins (718 000 proteins). Actually, full-length proteins account for only a third of these, and, following the elimination of sequence redundancy (at the level of 90% identity), the number of proteins is reduced to only ∼10% of the original number (72 992 proteins) (Figure 1). In addition, the low fraction of these proteins that are manually reviewed (based on the SwissProt database) results in only 1% of the initial collection (7416 proteins). Furthermore, the relevance of specific virus families to human health has led to a strong bias in the quality and reliability of genome annotation. The majority of viral sequences in the public databases are derived from only a few viral families, whereas most families remain poorly represented. This point is illustrated for the HIV, which makes up 36% of all viral protein entries (Figure 1). Half of all viral proteins are either from the HIV or hepatitis (Hepadnaviridae) viruses, two families with an indisputable impact on human health. An additional source of bias in analyzing the viral world stems from data that originate from incomplete genomes. The UniProtKB annotation of ‘complete proteome' covers only 0.5% of all viral sequences.
Figure 1.

Viral proteins from UniProtKB. (A) Total number of UniProtKB viral proteins [Virus], following filtration by removal of proteins with the database term ‘polyprotein' [Virus-(PP)], proteins that are marked as fragments [Virus-(PP+Fr)], and after removal of redundancy at the level of 90% sequence identity [(Virus-(PP+Fr))*0.9]. The fraction of viral proteins of the human immunodeficiency virus (HIV) is in yellow, and the number of proteins is as indicated (in thousands). (B) Partition of all proteins of 121 human-infecting viruses (from 50 virus genera) by viral classification into the 7 Baltimore classes and by the number of proteins in each class. Note the significant change in the fraction of proteins in each class when the manually reviewed data resource (SwissProt) or all data (UniProtKB) are considered. Source data and additional clinical information can be found in Supplementary Table S1.
The collection of proteins from ViralZone, a manually reviewed virus–host web portal that provides information on all known virus genera, overcomes some of these biases. ViralZone lists ∼300 genera of viruses belonging to 80 major families. Associated with each genus is information on the host range and tissue tropism. All viruses are classified by their taxonomical order as well as by the accepted index that divides them into seven classes (Baltimore index I–VII), based on their genetic material and mode of replication. One hundred twenty-one human-infecting viruses that belong to 50 genera are currently known (Supplementary Table S1). The uneven partition for human-infecting viruses among the seven classes is shown (Figure 1B). Class I (dsDNA) and class V (ssRNA(−)) account for 70% of the proteins, but all other classes are also represented among human viruses. By considering all proteins that are known from UniProtKB (a unification of SwissProt and TrEMBL), only 25% of the relevant proteomes are included in classes I and V, whereas the dominating class in terms of the quantity of protein sequences is class VI (ssRNA (RT), including HIV). Proteins belonging to class IV account for ∼50% of the proteins of human-infecting viruses (total ∼568 000). We used the manually compiled set from SwisProt for analyzing the human viruses throughout this study. Thus, in summary, we chose to focus only on complete proteomes of the representative species to ensure an unbiased and unabridged data set for subsequent analysis, as an uneven representation of viral protein sequences will affect most statistical properties (e.g., codon usage, GC content, and amino acid composition).
Ambiguity in mapping of viruses to their respective hosts
Ambiguity in virus-to-host mappings in publicly available databases often reflects missing information regarding a specific host. For example, a virus may be assigned to several hosts described at various levels of the species taxonomical tree (e.g., rodents, primates, and insects). However, only rarely do members of the same virus genus infect hosts differing above the level of class (e.g., mammals), phylum (e.g., chordata), or regnum (e.g., animals). An example of such an uncommon case is the Iridoviridae family (dsDNA viruses), which infects frog, snakes, insects, and fish. To overcome the ambiguities resulting from virus–host assignments, we adopt a mapping that focuses on the host taxonomical level of interest, which then groups together viruses that infect a unique group of hosts at that particular level.
As an illustrative example (Figure 2), we depict the viruses that infect mammals (excluding humans and other primates). Critically, these mappings account both for the virus under study and its hosts, with respect to the underlying host taxonomical tree. There are 10 host organisms that are infected by 17 viruses. These 17 viruses are represented by 7 types of viruses (Figure 2, V1–V7) that are identical in terms of their defined host range. We show that for the case in which the host-species level is considered (level A), only a restricted virus-to-host mapping can be applied. However, higher taxonomical views (levels B, C, or D) are consistent with a mapping of additional viruses. All further analyses herein will follow such a mapping (see Materials and methods). Note that resolving the ambiguity of assignment of viruses to their hosts is a fundamental precondition for studying virus–host evolution on a large scale.
Figure 2.

Mapping of viruses to hosts. (Top) a tree is drawn according to the hierarchical taxonomy of the hosts (from class to genus, based on NCBI taxonomy). The hosts that are unified at the suborder level are framed with an identical color. The four levels (A–D) represent the host grouping at the genus, suborder, order, and class levels, respectively. Below each host, the viruses that infect it are listed. (Bottom) for each taxonomy level, the virus-to-host mapping resulting from the tree is shown. Ambiguity in mapping of viruses to their hosts results from viruses that are annotated to infect a group of hosts that are not uniquely defined at the taxonomical level of interest (e.g., V5 not uniquely defined at level B). In this real-life example, V1–V7 are Mokola virus, Woodchuck hepatitis B virus, Hamster polyomavirus, Murine coronavirus, Sendai virus, Artic squirrel hepatitis virus, and Ground squirrel hepatitis virus, respectively.
Amino acid distribution and codon usage signature
We set out to test the preference of amino acids in viral proteomes vis-a-vis their hosts. To this end, we compiled an exhaustive representative set (see Materials and methods) and applied the virus-to-host mapping at a high taxonomical level (Figure 2, level C). To start with, we focused on two taxonomical groups: mammals (subdivided into human and nonhuman hosts) and bacteria. This analysis is based on 481 779 and 312 201 amino acids from the respective virus groups. The proteomes of virus representatives that infect humans and those that infect bacteria (bacteriophages) are compared (Figure 3A). It is evident that some amino acids strongly deviate between these two groups. For example, arginine (R) is more prevalent in the viruses of humans (P<10−6, t-test with Bonferroni correction), whereas lysine (K) appears more in bacterial proteomes (P<10−6). A similar trend is seen for isoleucine (I, P<10−6) and leucine (L, P<10−6). The source and biological significance of such differences are under study and beyond the scope of this study.
Figure 3.

Amino acid distribution and codon usage in viruses infecting taxonomy-unified hosts. (A) Amino acid distribution for human-infecting viruses (orange) and bacteria-infecting viruses (gray). The analysis is based on the complete proteomes of the mapped viruses. (B) The relative codon usage of the six triplets that code for Arginine (R) and Leucine (L) in human-infecting viruses (yellow) and viruses that infect non-human mammals (blue). Such data, when combined for all codons (excluding triplets for Tryptophan and Methionine), produce a vector of 59 codon frequencies that is subsequently used for quantifying the distance between any pair of virus and host groups.
Similarly, we measured the codon usage for each of the 59 codons that code for 1 of the 18 degenerately encoded amino acids (tryptophan and methionine are encoded by only a single codon). As an illustration, we show the codon preferences for arginine (R, 6 codons) and leucine (L, 6 codons), as measured for human-infecting and mammalian (excluding human) virus groups (Figure 3B). The different usage of each of the amino acids' codon triplets is evident (χ2 test, P<10−6).
Variability among viral proteomes is greater than for their hosts
To test the range of variability in amino acids and in codon usage within the space of the viruses studied, all representative viruses were divided on the basis of their infectivity toward a taxonomical partition of six high-level host groups: humans, mammals (excluding humans), vertebrates (excluding mammals—mainly fish and aves), insects, plants, and bacteria. This partition permitted maximal coverage of the virus representatives.
For a particular group of viruses and a given set of hosts, frequency vectors (20 element vectors for amino acids, and 59 element vectors for codons) were calculated. To compare these vectors, we measure the pairwise distance between codon usage (or amino acid distribution) using a distance metric, where lower values indicate greater similarity. We applied multiple measures to determine the distance between any pair of vectors for virus and host. We will present the results obtained using the L2 norm measure. Additional measures that were applied include the L1 norm and DKL (see Materials and methods), but their use has only a negligible impact on the results, supporting the robustness of the analysis performed hereafter.
Under the scheme outlined above, we consider the ranked distance of each pair of viruses and host groups relative to the L2 variability among the entire set of tested pairs (36 pairs, covering all 6 major taxonomical groups). Figure 4 shows the subset of results for humans (H), non-human mammals (M), and non-mammal vertebrates (V), where we compare pairs of host groups (Ho × Ho), pairs of virus groups (Vir × Vir), and pairs of virus group–host group (Vir × Ho). The results for the amino acid distributions (Figure 4A, left) suggest that the taxonomical host groups are less variable (dominated by blue) than are the respective viral groups (predominantly red). The variability among viruses that infect plants, insects, and bacteria is substantially higher, and so is the variability among the respective host genomes (the full results are found in Supplementary information S1). The resemblance in amino acid preference between viruses and their grouped taxonomical hosts is rather weak except for humans and somewhat for non-mammal vertebrates (Figure 4B, left; Supplementary information S1).
Figure 4.

Distance matrices for the similarity of amino acid distribution and codon usage between viruses and hosts mapped at high-level taxonomies. The analysis is based on the complete proteomes of the mapped viruses following partition into six taxonomical groups. For the complete matrices that include plants, insects, and bacteria, see Supplementary information S1. (A) Viruses that infect humans (H), mammalians excluding human (M), and vertebrates excluding humans (V). aa and codon indicate the L2 distance of amino acids and codon usage, respectively. The pairwise distances among the hosts and among the viruses are marked as Ho × Ho and Vir × Vir, respectively. Color code (1–36) is according to the ranking of the 36 values of all pairs used in the respective analysis, from blue (minimal distance, most similar) to red (maximal distance). (B) The Vir × Ho analysis shows the L2 distances between viruses and hosts. Note that this matrix is not symmetric and that the x- and y-axes show the hosts (Ho) and the viruses (Vir), respectively. Source data is available for this figure at www.nature.com/msb.
The results for a host–host and virus–virus comparison of codon usages (Figure 4A, right) show a trend similar to that for amino acid distributions, namely, substantial similarity among the host groups and enhanced diversity among the corresponding virus groups. Nevertheless, viruses infecting non-human mammals and viruses infecting non-mammal vertebrates show an intermediate level of resemblance to each other (green squares), whereas human viruses differ from these two groups.
Next, we tested the similarity between virus, host amino acid, and codon preferences, as a measure for coherent adaptation of viruses with respect to their host taxonomical groups (Figure 4B, right). Interestingly, viruses that infect humans are not only adapted to the human host, but are also similar in codon preference to host groups comprising mammals excluding humans (M), and vertebrates excluding mammals (V, mostly viruses of fish and birds). On the other hand, the codon usage of viruses infecting vertebrates is highly dissimilar from that of all host groups shown (the opposite of that for human viruses, Supplementary information S1).
Comparison of codon usages between hosts and between viruses
The similarity between the codon and amino acid preferences of human-infecting viruses and a wide variety of host organisms (Figure 4) may reflect the non-unique definition for virus strains that are associated with broad taxonomical host groups. We thus compiled a set of representative viruses derived from an organism-level view of the hosts (Figure 2, level A), where, in this setting, only viruses that uniquely infect a defined host species are included. The 30 hosts infected by virus representatives unique to their respective hosts are listed in Table I. Most viruses are represented with >1000 codons for each host and 10 of the viruses are supported by >20 000 codons (see Supplementary Table S2). A comparison of the codon usage among the viruses themselves is shown (Figure 5A), indicating enormous variability between viral genomes. Note that the colors in the various matrices range from blue (high similarity) to red (maximal distance); also, as data normalization is performed to obtain ranks for the 900 values (30 × 30 pairs) in each matrix, the matrices can be easily compared. Unlike the intra-virus comparisons, when the 30 hosts were compared among themselves (Figure 5B), the internal variability in the groups of mammals, plants, and insects was relatively low (especially among the mammal hosts). Nonetheless, among the 10 bacterial hosts tested, the variability is very high (dominated by red color).
Table 1.
Virus representatives and their unique hosts
| Tax | ID | Virus–hosts | # AAa | # Codonsb | |
|---|---|---|---|---|---|
| AA, amino acids; Tax, taxonomical groups; Mammals, M; aves, A; insects, I; plants, P, bacteria, B. | |||||
| 1 | M | HUM | Human | 471 751 | 793 917 |
| 2 | SQUM | Squirrel monkey | 33 202 | 58 837 | |
| 3 | MAC | Macaque | 1704 | 2335 | |
| 4 | RAT | Rat | 5023 | 17 393 | |
| 5 | MOUS | Mouse | 2754 | 3556 | |
| 6 | PIG | Pig | 93 628 | 125 470 | |
| 7 | BOVN | Bovine | 14 567 | 23 234 | |
| 8 | SHP | Sheep | 8976 | 9004 | |
| 9 | HORS | Horse | 1136 | 2385 | |
| 10 | CAT | Cat | 774 | 776 | |
| 11 | DOG | Dog | 2357 | 5113 | |
| 12 | A | CHK | Chicken | 46 673 | 189 090 |
| 13 | I | MOSQ | Tiger mosquito | 1532 | 1543 |
| 14 | COMO | Codling moth | 36 936 | 37 370 | |
| 15 | CHIL | Rice stem borer | 77 552 | 78 020 | |
| 16 | AMMO | Noctuid moth | 37 391 | 43 519 | |
| 17 | P | ARAB | Arabidopsis | 1624 | 1629 |
| 18 | LET | Lettuce | 3857 | 8186 | |
| 19 | RICE | Rice | 1436 | 1235 | |
| 20 | TOM | Tomato | 3840 | 4993 | |
| 21 | B | BACI | Bacillus | 6064 | 6374 |
| 22 | CHLM | Chlamydia | 1648 | 2268 | |
| 23 | ENCO | Enterococcus | 42 234 | 14 526 | |
| 24 | LACO | Lactococcus | 6900 | 7181 | |
| 25 | MYBC | Mycobacteria | 15 251 | 15 669 | |
| 26 | MYPL | Mycoplasma | 3272 | 3287 | |
| 27 | PSDO | Pseudomonas | 866 | 871 | |
| 28 | SALM | Salmonella | 10 207 | 31 106 | |
| 29 | STRP | Streptomyces | 12 454 | 15 392 | |
| 30 | ECOL | Escherichia coli | 154 167 | 273 272 | |
| Total | 1 099 776 | 1 777 551 | |||
aNumber of amino acids based on complete proteome of the viruses.
bNumber of codons based on UniProtKB mapping to the corresponding coding sequences.
Figure 5.

Distance matrix for the similarity in codon usage between pairs of viruses and pairs of hosts. Color code is based on the ranking of all 900 L2 values, as calculated from all pairs of 30 viruses and 30 unique hosts. The matrix is organized by groups according to Table I. (A) Symmetric L2 distance matrix for all 30 viruses (B) Symmetric L2 distance matrix for all 30 hosts. The analysis is based on the complete proteomes of the mapped viruses. The sub-matrices indicate the partition into groups of mammals (1–11), aves (12), insects (13–16), plants (17–20), and bacteria (21–30). Note the large diversity among viruses infecting mammals, insects, and bacteria (A) and the strong resemblance among the mammalian hosts (B). Source data is available for this figure at www.nature.com/msb.
Adaptation of viruses toward their hosts is shown by GC content and codon usage
It is known that the GC content is a strong determinant in shaping codon usage, specifically in the higher multicellular eukaryotes. As a control experiment, a comparison of the GC content between viruses and their cognate hosts shows that viruses have an overall weak, but significant (R2=0.575, P<10−5), correlation with their host GC content (Figure 6A). In fact, for bacteria, the partition by host GC content provides a very strong linear association (Figure 6A, blue points, R2=0.927, P<10−5). However, no significant associations are found between the GC contents of viruses and their hosts for other taxonomic groups. For example, for the 11 mammals analyzed in this study, the correlation was extremely poor (R2=0.065). This can be explained by the fact that although the GC content in mammal-infecting viruses ranges between 35 and 56%, the GC content of the proteomes of the mammal hosts studied (Supplementary Table S3) is rather narrow (50–53%). Thus, we conclude that the correlation between the GC contents of the viruses and their hosts (Figure 6A) is dominated by the bacteriophages matching their unique bacteria.
Figure 6.

Similarity in GC content and codon usage between pairs of viruses and hosts. The GC content from the proteomes of all viruses and their hosts was compiled. (A) Analysis of the GC content correlation between the hosts (x-axis) and viruses (F-test for linear regression), color coded by their taxonomical grouping to mammals, aves, insects, plants, and bacteria (according to Table I). (B) Codon usage distance matrix for all pairs of hosts and viruses is shown. Color code is according to the ranking of all 900 values as calculated from all pairs of 30 viruses and 30 unique hosts. The matrix is organized by groups according to Table I. L2 distance matrix for all 30 viruses (y-axis) and 30 unique hosts (x-axis). The analysis is based on the complete proteomes of the mapped viruses. The sub-matrices indicate the partition to groups of mammals (1–11), aves (12), insects (13–16), plants (17–20), and bacteria (21–30). Note the strong resemblance in human and rat viruses relative to all other mammals and the resemblance among all viruses infecting plants. For data of the complete matrix, see Supplementary information S2.
As we did not find virus-to-host adaptation of GC content with respect to the entire taxonomical spectrum, we proceeded to test the codon usage distances for all pairs of virus and host (Figure 6B); the similarity of the viruses toward their specific hosts (the diagonal of the matrix in Figure 6B) is also summarized in Table II. The adaptation among the bacterial set is very prominent, especially in light of the extreme differences among the different bacterial hosts themselves (Figure 5B; Supplementary information S1). In fact, each bacterial virus shows a very different pattern relative to all other bacterial viruses. In addition, significant levels of resemblance are evident among the different plant viruses and their hosts.
Table 2.
Relative L2 percentile of viral codon adaptation to their hosts
| Tax | ID | Virus–hosts | % L2 similaritya | |
|---|---|---|---|---|
| 1 | M | HUM | Human | 4 |
| 2 | SQUM | Squirrel monkey | 77 | |
| 3 | MAC | Macaque | 53 | |
| 4 | RAT | Rat | 8 | |
| 5 | MOUS | Mouse | 67 | |
| 6 | PIG | Pig | 72 | |
| 7 | BOVN | Bovine | 53 | |
| 8 | SHP | Sheep | 86 | |
| 9 | HORS | Horse | 54 | |
| 10 | CAT | Cat | 45 | |
| 11 | DOG | Dog | 57 | |
| 12 | A | CHK | Chicken | 51 |
| 13 | I | MOSQ | Tiger mosquito | 63 |
| 14 | COMO | Codling moth | 44 | |
| 15 | CHIL | Rice stem borer | 69 | |
| 16 | AMMO | Noctuid moth | 15 | |
| 17 | P | ARAB | Arabidopsis | 9 |
| 18 | LET | Lettuce | 12 | |
| 19 | RICE | Rice | 18 | |
| 20 | TOM | Tomato | 1 | |
| 21 | B | BACI | Bacillus | 1 |
| 22 | CHLM | Chlamydia | 57 | |
| 23 | ENCO | Enterococcus | 11 | |
| 24 | LACO | Lactococcus | 13 | |
| 25 | MYBC | Mycobacteria | 2 | |
| 26 | MYPL | Mycoplasma | 7 | |
| 27 | PSDO | Pseudomonas | 19 | |
| 28 | SALM | Salmonella | 12 | |
| 29 | STRP | Streptomyces | 36 | |
| 30 | ECOL | Escherichia coli | 36 |
aRelative L2 percentile values calculated between virus and host, where lower values indicate higher observed similarity (see Figure 6B). The most adapted virus–host pairs are indicated in bold.
However, the strongest signal observed is the resemblance of human viruses to all mammalian hosts; at the same time, these viruses remain rather different from any of the other mammalian viruses (Figure 5A). Furthermore, the strong similarity of the codon usage of human viruses to all 11 mammalian hosts reaches substantially farther into the taxonomic realm, approaching the insect and bird host species as well (Supplementary Table S3). Interestingly, the viruses that actually infect birds do not show strong adaptation to their hosts (based on viruses that infect chickens). We have shown that human viruses show an unexpected similarity to a broad range of host taxonomical groups, including mammals, avians, most insects, and some plants. Among all tested mammals, only human and rat viruses share strong resemblance in their codon usage profiles. However, owing to the relatively weak support for rat-infecting viruses (i.e., few proteins, narrower virus representatives), we will focus only on the adaptation of human viruses.
We tested whether the above phenomenon is perhaps dominated by the virus classification scheme. Human-infecting viruses are found in each of the seven classes (see Materials and methods). However, only for four of the seven classes do there exist three or more proteins derived from viruses that exclusively infect humans. Overall, all four of these human virus classes provide an almost identical codon usage profile when compared with mammals, insects, and plants (not shown), thus precluding such reasoning.
Codon usage resemblance is stronger for structural viral proteins
Most virus proteomes are rather simple and include <10 proteins. A minimal set of proteins comprises the virion structure by building the atomic units of the capsomers. Similarly, most viruses have a replication enzyme such as reverse transcriptase and RNA or DNA polymerase, according to their mode of replication, transcription, and regulation. In some instances (e.g., small DNA viruses and hepadnaviruses), the involvement of host polymerases is essential for the initial phase of viral replication. The rest of the proteome encodes diverse functions that are mostly uncharacterized and are often specialized to the life cycle of the particular virus. We tested the hypothesis that the evolutionary forces underlying codon usage adaptation of the virus may not be determined at the overall genomic level but may instead reflect some functional properties of its proteins.
The variability in viral structure, size, complexity, and shape is enormous. Despite such diversity, we assigned all viral proteins to four mutually exclusive functional sets (Figure 7; see Materials and methods). Figure 7C shows that structural proteins (‘H') that do not function as host recognition elements are characterized by the highest levels of codon usage similarity with their respective host (i.e., lower L2 distance measure). This diverse group includes proteins that participate in packing and covering the DNA, as well as the structural proteins that build the core of the virions. On the other hand, proteins that are expressed on the surface (‘R'), which are molecules that participate in recognition of the host receptors, show the largest deviation from the host relative to the other defined groups. The polymerases and additional nucleic acid-related enzymes (‘EC') show an intermediate level of resemblance to host codon usage.
Figure 7.

Codon usage adaptation for functional groups of viral proteins. Viral proteins annotated as ‘complete proteome' were classified according to the taxonomic view of their hosts—for humans and mammals (excluding human). The analysis for human-infecting viruses includes 2186 proteins and for mammals (excluding humans) 513 proteins. (A) Graphical schemes of enveloped viruses (adapted from the ViralZone illustrations) are shown. Proteins that are exposed on the virus surface and are part of the host receptor recognition include proteins annotated as glycoprotein, coat, spike, and fiber (marked ‘R', orange). Capsids, core, and structural proteins are characterized by high expression (marked ‘H', light blue). Capsids may appear in multiple layers (intermediate and inner capsids). Other proteins expressed in large quantities include core, matrix, tegument, DNA-packing proteins, and nucleoproteins. (B) Partition of the proteomes to functional groups of surface-protein recognition (orange), structural protein with high copy number in the virion (light blue), enzymes as defined by the E.C. enzyme annotation (purple), and uncharacterized viral proteins (gray). (C) The overall similarity, as measured by L2 distance (see Materials and methods), is shown according to each of the functional partitions of proteins. Lower value indicates higher resemblance. The distance measure for the entire set of viral proteomes is marked by the dashed line. Source data is available for this figure at www.nature.com/msb.
Discussion
As early as 20 years ago, a correlation was detected between the prevalence of dinucleotides in viruses and their hosts (Barrai et al, 1990). Although these data were based on a very limited set of sequences, the main conclusion remains accurate in view of the current scale of sequenced data, which suggests an active adaptation process of viruses toward their hosts. We found that the huge amounts of data regarding viral genomes and the genomes of their respective hosts have enabled the compilation of a balanced data set for further analysis (Figure 1).
The viral space displays enormous diversity but high redundancy
In this study, we set out to analyze the overall potential adaptability of known virus families by using the complete set of virus representatives that reflect the current knowledge of all major viruses. Clearly, our analyses are strongly dependent on the correct mapping of viruses to their hosts. We analyzed 122 viruses at the higher taxonomical levels and 64 viruses at the host-species level, where each such virus represents a different viral genus (Table I; Supplementary Table S2). By the strict mapping of a set of 30 virus genera that exclusively infect 30 different hosts, we limited the set of viruses and often remained with only one or very few representatives for a unique host. It is possible that the set of viruses that have a restricted range of hosts is skewed. They may reflect (i) poorly studied cases, which leads to partial information regarding the virus and its hosts; and (ii) cases where the dependency on virus–host pair is stronger because of a specific molecular barrier that restricts the host range. We cannot separate these two instances, but for many cases the restricted host assignment is supported by a large body of literature. For example, the observed overwhelming similarity in amino acid distributions and in the codon usage among viruses infecting the tomato, lettuce, rice, and arabidopsis plants (Figure 4) seems more likely to be a result of incomplete annotation in the viral database, where each of these viruses overlap and, in reality, infect the other plants but are simply not yet annotated as such. This is in accord with the current view of plant-infecting viruses (Roossinck, 1997). Although the statistical power for some analyses may be affected by this ‘reduction to representatives,' we argue that the trends observed in this study hold and will be further substantiated when additional viruses with accurate annotations become available.
We analyzed a representative set of viruses irrespective of their mode of replication according to a partition to the 7 standard classes (Melnick, 1972) that are indicated by I–VII. Note that host specificity does not determine the class. For example, although most viruses that infect humans (Supplementary Table S1) belong to class I, human-infecting viruses are represented in all other classes, including the well-known health-threatening viruses, such as the Coronavirus family (SARS, class IV), Lentivirus (AIDS virus, class VI), and Ebola virus (class V). The genomic structures, nucleotide composition, replicative mode, replication time, and rates of mutation in the different classes are estimated to show great differences, where, for example, RNA viruses mutate much faster than other groups. Despite these differences, the analysis of human-infecting viruses based on this classification showed that the observed adaptation of human viruses characterizes viruses from a broad range of life cycles and replication modes.
Testing codon and amino acids usage (rather than more direct measures of substitution and mutation rate) has the advantage that it provides a view on the variability of the viral proteome relative to its potential hosts (Figures 4, 5 and 6). Nonetheless, our observations cannot provide insights into the dynamics or rates of viral evolution. Studies that estimate the diversity among viruses and their hosts often focus on those having high enough mutation rates or short generation times, resulting in increased genetic diversity (van Hemert et al, 2007). Our analysis is thus complementary to such studies.
Adaptation of viruses toward their hosts
In this paper, we observed that all mammalian genomes have similar codon usage. Furthermore, we found that human viruses share this common codon usage with their human host; on the other hand, other mammalian viruses do not. Theoretically, this could derive from a situation where, for some reason, only human viruses are required to adapt their codon usage to successfully infect their host, whereas this adaptation does not seem critical for the viruses of other mammals. More likely explanations may be related to the recent expansion of humans and the co-evolution of their viruses, or to the hypothesis that large portions of the human genome are actually of viral origin (Kazazian, 2004).
A high similarity was reported earlier between the codon usage of bacteriophages and their hosts (Lucks et al, 2008). In that study, the authors analyzed a large set of bacteriophages and isolated the effect of the GC (i.e., GC content) and the adaptation of specific viral codons toward the primary bacterial host. Interestingly, for about 40% of the viruses, host-preferred codons were selected, which suggests that adaptation toward the host has a strong role in viral evolution. In addition, they found that structural proteins show maximal similarity toward the host-preferred codon, in accordance with our finding regarding the high degree of adaptation for highly abundant proteins (Figure 7C).
Here, we found similar codon usages among viruses, hosts, and for virus–host pairs. Similarity in codon usage in different viruses can somewhat be explained by the occurrence of lateral gene transfer (LGT) and other modes of genetic material exchange. Accordingly, recent recombination events between the host and the virus may leave behind similar codon frequencies. Yet we do not believe this phenomenon to be a major determinant in codon usage adaptation as (i) it is unlikely that the codon usage of some functional groups but not of the entire proteome will show differences in the patterns observed (Figure 7C); (ii) there is no evidence that among the mammals we tested here some are more likely to be affected by LGT than others, yet human viruses show a significantly different pattern than other mammals; (iii) different classes of viruses (class I–VII) have similar adaptation trends, despite substantial differences in the potential for the exchange of genetic material with the host in RNA and DNA viruses. Thus, although it is unlikely that LGT dominates the observed resemblance of codon usage between eukaryotic viruses and their hosts, this does not hold for bacteria and archaea, which are exposed to high frequencies of LGT events.
An interesting case of co-evolution with expected restrictions on infectivity is that of viruses that infect hosts that use alternative genetic code assignments. Indeed, studies on mitoviruses that infect fungal mitochondria led to insights on host limitation that are imposed by the use of a specialized genetic code (Shackelton and Holmes, 2008).
Possible selection for translational efficiency in mammalian viruses
In our study, the similarity between the codon usage of human viruses and that of mammals, birds, and some insects is not duplicated for other mammalian viruses (Figure 6). Furthermore, the signal observed for codon usage exceeds that detected for amino acid distributions, potentially indicating selection for translational efficiency.
The number of protein products in the viral capsid can reach thousands; for example, the mature HIV-1 contains 1572 capsid proteins. The African swine fever virus (family Asfarviridae) consists of ∼1900–2200 capsomers. On the other hand, recognition proteins on the viral surface are not necessarily expressed in such large amounts. A partition of structural proteins and enzymes is based on ‘virion properties' from the ICTV database (http://www.ncbi.nlm.nih.gov/ICTVdb). Currently, on the basis of 3D structure, sparse data on the stoichiometry of virion composition are available. For example, the Adenoviridae virus genome encodes 10 structural proteins and ∼30 non-structural proteins. The capsid is composed of 720 copies of the major hexon protein (protein II, 988 aa), 64 and 60 copies that build the penton (proteins III and IIIa, respectively), 180 copies of the minor core (protein V), but only 12 copies of the recognition fiber (protein IV, 582 aa).
We found that for mammalian viruses, the proteins that appear in virion in high numbers (Figure 7, marked ‘H') are the ones with codon usage most similar to that of their hosts. In the case of human viruses, we can see that highly expressed genes in different viruses that infect the same host preferentially use codons similar to that of humans and of each other (Figure 7C). On the other hand, the surface proteins that participate in recognition are often expressed in lower quantities displaying a rather low adaptation level toward their hosts (marked ‘R'). A complementary explanation may rely on the positive selection paradigm that was proposed in virus–host recognition (Sawyer et al, 2005). The enzymes (marked ‘EC'), which are generally expressed in minute amounts, show only an intermediate codon usage similarity. Thus, overall, these results further strengthen the case for translational selection. Note that earlier studies did not find evidence for translational selection operating on mammalian genes (see discussion in (dos Reis and Wernisch, 2009; Semon et al, 2006 #544) and references within). It may be possible that such selection does exist, but these phenomena are weak because of the low effective mammalian population sizes. On the other hand, viruses affecting mammals have larger effective population sizes and a shorter generation time (dos Reis and Wernisch, 2009). Thus, similar analysis to that performed here may be able to identify translational selection in genomes in which it was impossible to do so earlier.
In the case of bacterial viruses (Lucks et al, 2008), we were unable to consistently and reliably partition the proteins that are involved in recognition from those that are abundant, because of the enormous variability in shape and recognition mode among bacteriophages. Our results agree with a role of translational selection and extend it toward mammalian viruses, where it may have a role in their evolutionary fitness. However, this adaptation may be of lesser importance, as a critical obstacle for viruses that infect mammals is the need to invade their host cells, while bypassing an active immune system (whereas no such extensive system exists in bacterial hosts). For example, the HIV virus has adopted recognition strategies that overcome the immune barrier (Holmes et al, 1992).
Host range, tissue specificity, and codon usage similarity
It is known that a change in only a few amino acids of viral proteins can lead to a shift in the host infectivity range. Such a shift occurs through a genetic adaptation process that overcomes the hurdles of viral entry and replication in a new cellular environment. ΦX174 bacteriophage, which normally grows on E. coli, was switched to infect Salmonella, where this shift was attributed to only a very few mutations (2–3) in the major capsid gene (Crill et al, 2000). This phenomenon is not unique to bacterial viruses, as this has occurred in canine parvovirus, which appeared in the late 1970s as a variant of a feline parvovirus. The host shift was attributed to only two to three substitutions (Truyen et al, 1995). A shift in host recognition was also shown in the case of HIV-1, where a single mutation in the envelope gene was sufficient to alter cell specificity (Rambaut et al, 2004). In all these strategies, virus–host shift is based on modifications in the virus receptor recognition step. However, it has been shown that host range is not entirely dependent on the initial recognition stage (McFadden, 2005).
Our results on the high adaptation in codon usage, especially for human viruses, suggest that viral envelope/capsid proteins have the potential to be a factor in infectivity and efficiency. Furthermore, our observation that some viruses are adapted toward multiple hosts, in terms of their codon usage, can even possibly permit the expansion of host infectivity.
In multicellular organisms, viruses do not infect the organism but rather are restricted to a specific organ, tissue, or cell type (Gallagher and Buchmeier, 2001). Throughout this study, we presented data that use the average codon usage of the organism as a reference measure to study adaptation. With the fast growth of high-quality mass spectrometry proteomics data from different tissues and cell types, the notion of resemblance between viruses and their hosts under the assumption of translational (and not transcriptional) efficiency at the tissue and cell-type levels will be of great interest.
Adaptation and human health
Studying the evolution of viral codon usage and amino acid preferences in view of their hosts is fundamental in developing strategies for managing viral infections in the scope of human health, agriculture, and the environment. Insight into such phenomena was used in the laboratory, for example, when unfavorable codon pairs of capsid poxvirus proteins were injected into infected mice, resulting in virus attenuation (Coleman et al, 2008). Similarly, neuroattenuated phenotype was associated with codon preference deoptimization in polioviruses (Mueller et al, 2006). In a common vaccination practice, a live, attenuated virus is produced by adaptation to a new host, thereby eliminating its virulence to humans. As we have found that human-infecting viruses have conserved and unique codon usages, we propose that a fine-tuning of codon deoptimization may allow the alteration of tissue tropism and virulence attenuation.
In addition, shifts in hosts have huge implications on human health and on the world economy, for example, zoonotic epidemics. Known examples of naturally occurring host–virus shifts are the introduction of HIV-1 to humans in the early 1950s and the shift in the SARS (CoV) virus that crossed over to infect humans only very recently. The worldwide threat of influenza-based epidemics, such as the transmission of avian flu (Influenza A virus, H5N1) to humans and the latest outbreak of swine influenza (H1N1, April 2009) in Mexico, is heightened by the rapid evolution of the Influenza virus witnessed during the last decade; recently, H3N2 and H3N8 were introduced from humans to pigs and from horses to dogs, respectively (Campitelli et al, 1997). It is likely that the domestication and close interaction between humans, rats, and farm animals for thousands of years has led to the evolution of viruses that infect humans and are adapted toward a broad range of hosts. The similarities in codon usage and amino acid composition that we have observed in this work can somewhat relate to the potential for zoonosis. Although, as discussed above, these molecular properties are neither necessary nor sufficient conditions for host shifts, our analysis can nevertheless contribute to a framework that would permit analysis of the potential of certain viruses to adapt to new host species.
Materials and methods
Data collection
Proteins for all organisms were collected from UniProt (Apweiler et al, 2004). Virus proteins were collected from ViralZone (http://www.expasy.ch/viralzone, coordinated by UniProt/SwissProt), which holds 314 reference strain viruses that belong to 80 families and 291 genera. ViralZone provides reviewed data that cover molecular information (shape, genome and replication mode, and capsomer composition), epidemiological data, cell tropism, and host range. Each genus is specified by a manually selected representative (in some cases, >1). All viruses are classified into seven classes: (I) double-stranded DNA viruses, (II) single-stranded DNA viruses, (III) double-stranded RNA viruses, single-stranded RNA viruses with positive and negative sense (IV, V, respectively), (VI) positive sense single-stranded RNA viruses that replicate through a DNA intermediate and double-stranded DNA viruses that replicate though a single-stranded RNA intermediate (VII). Fragmented proteins and polyproteins were filtered out. Coding sequences were collected from EMBL through an SRS querying system that links UniProt proteins to their respective EMBL coding sequences. As one protein is often associated with multiple sequences, we extracted all data as mapped by EMBL to UniProt ID. This collection of virus proteins in UniProt covers ∼13 000 proteins that are reviewed (SwissProt) and additionally ∼730 000 from a non-reviewed TrEMBL resource.
We selected 30 organisms and 30 matched viruses (Supplementary Table S1) that are unique (i.e., assigned to a specific organism, Figure 2). Taxonomical views that have very little support (<2 proteins, <500 amino acids, or <700 codons) were eliminated. Note that the representative virus (reference strain) corresponds to tens of other viruses that are poorly annotated and thus are not selected as representatives. The mapping of a representative to other viruses is based on the ViralZone mapping.
Data analysis
For each group of (virus or host) genes, codon usage frequencies were independently calculated for each of the amino acids. For each of the 18 degenerately encoded amino acids, the empirical frequencies of its corresponding codons were counted and normalized to sum to 1. The other two amino acids tryptophan (W) and methionine (M) each have a single codon and were not included in the analysis. Thus, each of the 59 redundant codons that account for these 18 amino acids were assigned a number between 0 and 1. The GC content of each virus–host pair was also calculated independently and was assigned a number between 0 and 1.
Divergence between the codon usage of two viruses, two hosts, or virus and host was estimated according to the distances between their usage vectors. Specifically, for each group, a usage vector of 59 coordinates, denoted as F=(f1,…,f59), was calculated as described above. The distance between two such vectors was measured in two different ways: once as the L1 distance 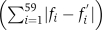 and the second time as the Euclidean (L2) distance
and the second time as the Euclidean (L2) distance 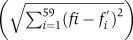 . For differences in the amino acid frequencies between two species, the same method was used, with the corresponding 20-coordinated vectors.
. For differences in the amino acid frequencies between two species, the same method was used, with the corresponding 20-coordinated vectors.
The codon usage differences were also measured in a manner that integrates the amino acid frequencies, where the 59 codons were assigned their empirical frequencies in the data, regardless of their corresponding amino acid frequency. This quantification results in a probability vector P=(p1,…,p59), where 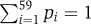 .
.
For this representation, the differences between two codon usages (P=(p1,…,p59), Q=(q1,…,q59)) were measured using their KL divergences (DKL, Kullback–Leibler divergence), where 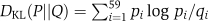 and
and 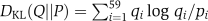 .
.
Virus–host mapping
For each partition of the host taxonomy that we considered, we included a virus in the calculations only if there was not more than one taxonomic class that it is capable of infecting. Formally, for each virus v, define h(v) to be the set of host species that it can infect. And, let C1, …, Ck be a disjoint partition of the host organisms under study. Now, for a particular virus v, consider the least common ancestor (LCA) of the host species of v in the host taxonomic tree: LCA(h(v)). If there exists a single cluster Ci (1⩽i⩽k) such that LCA(h(v)) is a descendant of Ci (possibly Ci itself), then we uniquely map virus v to be among the viruses that infect the taxonomic sub-tree rooted at Ci.
Division of viral proteins into functional categories
We divided all mammalian virus proteins into one of four classes: (i) recognition receptors on the surface, for example, coat, spike, glycoprotein, or envelope (Figure 7, orange frames); (ii) enzymes (as annotated by the EC classification according to UniProt—mostly polymerases, purple frames); (iii) capsomers and structural units, including tegument, nucleoproteins, and capsids in enveloped viruses (Figure 7, blue frame); and (iv) proteins that are either unknown or cannot be uniquely assigned to the other three functional sets (see Supplementary Table S4). This assignment was performed manually, addressing the proteins with multiple functions or non-exclusive functional assignments (mainly in filamentous phage and other bacteriophages).
Supplementary Material
Supplementary tables S2-3, Supplementary data S1-2
Human infecting viruses.
Proteins from human infecting viruses based on ‘complete proteome' are listed according to their unique UniProt accession.
Acknowledgments
We thank Nati Linial for valuable suggestions and a critical view throughout the research. We thank the ProtoNet research group for comments and discussions. YP and MF are fellows of the SCCB, the Sudarsky Center for Computational Biology. This research is partially supported by EU Prospects FR7 and a grant from the Israel Science Foundation (ISF).
Footnotes
The authors declare that they have no conflict of interest.
References
- Akashi H (2001) Gene expression and molecular evolution. Curr Opin Genet Dev 11: 660–666 [DOI] [PubMed] [Google Scholar]
- Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O'Donovan C, Redaschi N, Yeh LS (2004) UniProt: the Universal Protein knowledgebase. Nucleic Acids Res 32: D115–D119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrai I, Salvatorelli G, Mamolini E, De Lorenzi S, Carrieri A, Rodriguez-Larralde A, Scapoli C (2008) General preadaptation of viral infectors to their hosts. Intervirology 51: 101–111 [DOI] [PubMed] [Google Scholar]
- Barrai I, Scapoli C, Barale R, Volinia S (1990) Oligonucleotide correlations between infector and host genomes hint at evolutionary relationships. Nucleic Acids Res 18: 3021–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JW, Sun Y, Nazarian SH, Belsito TA, Brunetti CR, McFadden G (2006) Optimization of codon usage of poxvirus genes allows for improved transient expression in mammalian cells. Virus Genes 33: 15–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkhout B, Grigoriev A, Bakker M, Lukashov VV (2002) Codon and amino acid usage in retroviral genomes is consistent with virus-specific nucleotide pressure. AIDS Res Hum Retroviruses 18: 133–141 [DOI] [PubMed] [Google Scholar]
- Bernardi G (1986) Compositional constraints and genome evolution. J Mol Evol 24: 1–11 [DOI] [PubMed] [Google Scholar]
- Bonhoeffer S, Nowak MA (1994) Intra-host versus inter-host selection: viral strategies of immune function impairment. Proc Natl Acad Sci USA 91: 8062–8066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutet E, Lieberherr D, Tognolli M, Schneider M, Bairoch A (2007) UniProtKB/Swiss-Prot. Methods Mol Biol 406: 89–112 [DOI] [PubMed] [Google Scholar]
- Bronson EC, Anderson JN (1994) Nucleotide composition as a driving force in the evolution of retroviruses. J Mol Evol 38: 506–532 [DOI] [PubMed] [Google Scholar]
- Brower-Sinning R, Carter DM, Crevar CJ, Ghedin E, Ross TM, Benos PV (2009) The role of RNA folding free energy in the evolution of the polymerase genes of the influenza A virus. Genome Biol 10: R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campitelli L, Donatelli I, Foni E, Castrucci MR, Fabiani C, Kawaoka Y, Krauss S, Webster RG (1997) Continued evolution of H1N1 and H3N2 influenza viruses in pigs in Italy. Virology 232: 310–318 [DOI] [PubMed] [Google Scholar]
- Carbone A (2008) Codon bias is a major factor explaining phage evolution in translationally biased hosts. J Mol Evol 66: 210–223 [DOI] [PubMed] [Google Scholar]
- Coleman JR, Papamichail D, Skiena S, Futcher B, Wimmer E, Mueller S (2008) Virus attenuation by genome-scale changes in codon pair bias. Science 320: 1784–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini M, Cammarano R, Bernardi G (2009) The evolution of isochore patterns in vertebrate genomes. BMC Genomics 10: 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crill WD, Wichman HA, Bull JJ (2000) Evolutionary reversals during viral adaptation to alternating hosts. Genetics 154: 27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Reis M, Wernisch L (2009) Estimating translational selection in eukaryotic genomes. Mol Biol Evol 26: 451–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW (1993) Rates of spontaneous mutation among RNA viruses. Proc Natl Acad Sci USA 90: 4171–4175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L (2000) tRNA gene number and codon usage in the C. elegans genome are co-adapted for optimal translation of highly expressed genes. Trends Genet 16: 287–289 [DOI] [PubMed] [Google Scholar]
- Duret L (2002) Evolution of synonymous codon usage in metazoans. Curr Opin Genet Dev 12: 640–649 [DOI] [PubMed] [Google Scholar]
- Gallagher TM, Buchmeier MJ (2001) Coronavirus spike proteins in viral entry and pathogenesis. Virology 279: 371–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrigues HJ, Rubinchikova YE, Dipersio CM, Rose TM (2008) Integrin alphaVbeta3 Binds to the RGD motif of glycoprotein B of Kaposi's sarcoma-associated herpesvirus and functions as an RGD-dependent entry receptor. J Virol 82: 1570–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Zhou T, Ma J, Sun X, Lu Z (2004) Analysis of synonymous codon usage in SARS Coronavirus and other viruses in the Nidovirales. Virus Res 101: 155–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes EC, Zhang LQ, Simmonds P, Ludlam CA, Brown AJ (1992) Convergent and divergent sequence evolution in the surface envelope glycoprotein of human immunodeficiency virus type 1 within a single infected patient. Proc Natl Acad Sci USA 89: 4835–4839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins GM, Holmes EC (2003) The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res 92: 1–7 [DOI] [PubMed] [Google Scholar]
- Kawashima Y, Pfafferott K, Frater J, Matthews P, Payne R, Addo M, Gatanaga H, Fujiwara M, Hachiya A, Koizumi H, Kuse N, Oka S, Duda A, Prendergast A, Crawford H, Leslie A, Brumme Z, Brumme C, Allen T, Brander C et al. (2009) Adaptation of HIV-1 to human leukocyte antigen class I. Nature 458: 641–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH Jr (2004) Mobile elements: drivers of genome evolution. Science 303: 1626–1632 [DOI] [PubMed] [Google Scholar]
- Koonin EV, Senkevich TG, Dolja VV (2006) The ancient virus world and evolution of cells. Biol Direct 1: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudla G, Murray AW, Tollervey D, Plotkin JB (2009) Coding-sequence determinants of gene expression in Escherichia coli. Science 324: 255–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucks JB, Nelson DR, Kudla GR, Plotkin JB (2008) Genome landscapes and bacteriophage codon usage. PLoS Comput Biol 4: e1000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden G (2005) Poxvirus tropism. Nat Rev Microbiol 3: 201–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnick JL (1972) Classification and nomenclature of viruses, 1972. Prog Med Virol 14: 321–332 [PubMed] [Google Scholar]
- Mueller S, Papamichail D, Coleman JR, Skiena S, Wimmer E (2006) Reduction of the rate of poliovirus protein synthesis through large-scale codon deoptimization causes attenuation of viral virulence by lowering specific infectivity. J Virol 80: 9687–9696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Posada D, Crandall KA, Holmes EC (2004) The causes and consequences of HIV evolution. Nat Rev Genet 5: 52–61 [DOI] [PubMed] [Google Scholar]
- Roossinck MJ (1997) Mechanisms of plant virus evolution. Annu Rev Phytopathol 35: 191–209 [DOI] [PubMed] [Google Scholar]
- Sau K, Gupta SK, Sau S, Mandal SC, Ghosh TC (2006) Factors influencing synonymous codon and amino acid usage biases in Mimivirus. Biosystems 85: 107–113 [DOI] [PubMed] [Google Scholar]
- Sawyer SL, Wu LI, Emerman M, Malik HS (2005) Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci USA 102: 2832–2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semon M, Lobry JR, Duret L (2006) No evidence for tissue-specific adaptation of synonymous codon usage in humans. Mol Biol Evol 23: 523–529 [DOI] [PubMed] [Google Scholar]
- Shackelton LA, Holmes EC (2008) The role of alternative genetic codes in viral evolution and emergence. J Theor Biol 254: 128–134 [DOI] [PubMed] [Google Scholar]
- Shackelton LA, Parrish CR, Holmes EC (2006) Evolutionary basis of codon usage and nucleotide composition bias in vertebrate DNA viruses. J Mol Evol 62: 551–563 [DOI] [PubMed] [Google Scholar]
- Sharp PM, Cowe E, Higgins DG, Shields DC, Wolfe KH, Wright F (1988) Codon usage patterns in Escherichia coli, Bacillus subtilis, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila melanogaster and Homo sapiens; a review of the considerable within-species diversity. Nucleic Acids Res 16: 8207–8211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truyen U, Gruenberg A, Chang SF, Obermaier B, Veijalainen P, Parrish CR (1995) Evolution of the feline-subgroup parvoviruses and the control of canine host range in vivo. J Virol 69: 4702–4710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Etten JL, Meints RH (1999) Giant viruses infecting algae. Annu Rev Microbiol 53: 447–494 [DOI] [PubMed] [Google Scholar]
- van Hemert FJ, Berkhout B, Lukashov VV (2007) Host-related nucleotide composition and codon usage as driving forces in the recent evolution of the Astroviridae. Virology 361: 447–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao KN, Liu WJ, Frazer IH (2003) Codon usage bias and A+T content variation in human papillomavirus genomes. Virus Res 98: 95–104 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary tables S2-3, Supplementary data S1-2
Human infecting viruses.
Proteins from human infecting viruses based on ‘complete proteome' are listed according to their unique UniProt accession.