

Abstract
The cellular environment is abuzz with noise originating from the inherent random motion of reacting molecules in the living cell. In this noisy environment, clonal cell populations show cell-to-cell variability that can manifest significant phenotypic differences. Noise-induced stochastic fluctuations in cellular constituents can be measured and their statistics quantified. We show that these random fluctuations carry within them valuable information about the underlying genetic network. Far from being a nuisance, the ever-present cellular noise acts as a rich source of excitation that, when processed through a gene network, carries its distinctive fingerprint that encodes a wealth of information about that network. We show that in some cases the analysis of these random fluctuations enables the full identification of network parameters, including those that may otherwise be difficult to measure. This establishes a potentially powerful approach for the identification of gene networks and offers a new window into the workings of these networks.
Keywords: gene regulatory networks, stochastic biological processes, system identification
Introduction
Computational modeling in biology seeks to reduce complex systems to their essential components and functions, thereby arriving at a deeper understanding of biological phenomena. However, measuring or estimating key model parameters can be difficult when measurement noise corrupts experimental data. Thus, when cellular variability or ‘noise' (Elowitz et al, 2002) leads to measurement fluctuations, it may appear deleterious. However, this is not the case. Just as white noise inputs help to identify dynamical system parameters (Ljung, 1999), so too can characterization of noise dynamics elucidate natural mechanisms. For example, steady state noise characteristics can distinguish between different logical structures, such as AND or OR gates (Warmflash and Dinner, 2008). At the same time, temporal measurements of transient dynamics can aid in the construction of reaction pathways (Arkin et al, 1997). In combination, noise and temporal analyses yield powerful tools for parameter identification. For example, the averages of correlations in cell expression at many time points reveal feed-forward loops in the galactose metabolism genes of Escherichia coli (Dunlop et al, 2008). Similarly, manipulating certain gene network transcription rates while observing the response of statistical cumulants can help to identify reaction rates for some gene regulatory networks (Raffard et al, 2008). In this study, we examine the possibility of identifying system parameters and mechanisms directly from single-cell distributions, such as those obtainable with flow cytometry, without time-varying control and at only a handful of different time points. We prove that the analysis of variability provides more information than the mean behavior alone. Furthermore, we illustrate potential of our approach using numerical and experimental analyses of common gene regulatory networks.
Results and discussion
Gene expression model
We adopt the gene expression model used in the study carried out by Thattai and van Oudenaarden (2001), which is characterized by random integer numbers of mRNA and protein molecules: R and P, respectively. Transcription, translation, and degradation events change the system state by altering these numbers. mRNA changes are modeled as random events that occur according to exponentially distributed waiting times that depend on the transcription and degradation rates kr and γr. Thus, given the state of r mRNA molecules, the probability that a single mRNA molecule is degraded within the time increment dt is given by r·(γr·dt). Similarly, translation and degradation of proteins are dictated by rates kp and γp. The resulting stochastic model is represented by a continuous time, discrete state Markov process. The probability of finding the system in a given state (R(t)=r, P(t)=p) is fully characterized by the system's master equation from which the evolution of moments E[R(t), E[P(t), E[R2(t)],… can be described (see Supplementary Section 1).
Our first finding is that all parameters of this model are identifiable from cell population distributions of mRNA/protein measured at least at two time points. In contrast, two time point measurements of mRNA/protein population averages are never sufficient for identifiability. To show this, the use of first and second-order moments, or equivalently means, variances, and covariances of proteins and mRNAs is sufficient, instead of the use of full distributions. At a given time point, t, each such measurement yields a vector: v(t)=(E[R(t)], E[P(t)], E[R(t)2], E[P(t)2], E[R(t)P(t)]). Given v(t0) and v(t1) at two distinct time points t0<t1, there generically exists a set of parameters kr, kp, γr, γp that uniquely gives these measurements—all other parameter sets yield different measurements (see Figures 1E and 2A). We illustrate this here only for transcription (Supplementary Section 3 provides an implicit expression for the parameters of the full model). Suppose that {μ0, μ1} and σ02, σ12 represent the measured mRNA mean and variance at two time points t0<t1<∞. Then the parameters, {kr, γr} are fully identifiable, and
Figure 1.

(A) Simple gene expression model representing gene transcription and translation. (B, C) Simulations of mRNA (green) and protein (blue) populations. The solid red lines denote the mean values and the dashed lines are one s.d. value above and below that mean. (D, E) mRNA (green) and protein (blue) distributions at (D) t=5000 s and (E) t=1000 s for two different parameter (solid or dotted lines) sets but with the same initial conditions.
Figure 2.

Comparison of strategies for the identification of the gene expression model. (A) Minimum number of measurements needed for full parameter identification. (B) The log-normally distributed parameters of 100 simulated models, which combined with different unknown initial distributions at time t=0 define 100 different moment trajectories. (C) Percent identification success rates (within 5% for all parameters) for different identification strategies, assuming that measurements had unknown errors of ±10% and were taken every 100 s.
where τ:=t1−t0.
Thus, the statistics, {μ0, σ02, μ1, σ12} contain sufficient information to identify the model parameters. However, measurement of just the population averages, for example, E[R], is insufficient for identifiability, and there exists an infinite set of parameters {kr, γr}, that is consistent with the same two mean measurements μ0 and μ1.
Although parameters are identifiable from transient moment measurements, we find that it is impossible to identify all parameters from stationary moments. Measuring means, variances, and other statistics after all the transients have died represents a lost opportunity to peek into the cell's inner workings and to recover the network parameters. For example, two different parameter sets may produce very different protein distributions after a short interval time (Figure 1E), but indistinguishable distributions after a longer interval (Figure 1D). Supplementary Section 2 provides a proof that stationary moments of any arbitrary order are insufficient to uniquely identify the model parameters kr, kp, γr, γp. Such stationary distributions will only enable the determination of relative parameter values, but any positive scaling of these values would produce the exact same measurements for v∞. We note that stationary correlations, for example, E[R(t)R(t+τ)] for small time intervals, τ, could also provide the necessary dynamic information (Cinquemani et al, 2009), but taking such measurements is more difficult and requires the tracking of individual cells between measurement times.
After having determined that full identification is achievable using two measurements of all first and second order moments, we now explore the effect of partial moment measurements. We consider two new scenarios: (a) only {E[R], E[P]} measurements are available; and (b) only {E[P], E[P2]} measurements are available. For each scenario, Figure 2A shows the number of measurements needed for parameter identifiability and demonstrates the advantage of using full second order statistics. Furthermore, the performance with partial information depends on which partial information is being used. When protein and mRNA mean measurements alone are used, full parameter identifiability is possible using three measurements. However, with only protein mean and variance measurements, at least, five time measurements are needed. When only protein mean measurements are available, full identifiability is impossible, regardless of the number of measurements (see Supplementary Section 4).
Time measurements of moment dynamics impose nonlinear algebraic constraints on model parameters. The above results can be understood by exploring the number of such constraints that is needed to uniquely solve for the unknown parameters. The gene expression model has four unknown parameters (p=4) and five unknown initial conditions (moments at t=0). Thus, one would expect that, at least, nine independent measurements are needed to identify these unknowns. The five elements of v at t0 and t1 provide ten pieces of information and are generally sufficient (see Figure 2A). Conversely, in a model for just the mean values {E[R(t)], E[P(t)]}, there are four parameters (p=4) and two initial conditions, and one expects that, at least, six independent pieces of information would be needed for the identification. Indeed, at least three time measurements are required and two measurements are never enough (see Figure 2A). However, for a model that describes only protein mean and variance measurements, at least five time measurements are needed for full parameter identifiability. In this case, the dynamics of {E[P], E[P2]} are coupled to those of {E[R], E[R2], E[RP]}, and additional measurements are needed to identify the initial values for these. Finally, we note that in these cases, the number of measurements needed for parameter identification are far fewer than the 2p+1 measurements that were shown (Sontag, 2002) to be sufficient for the identification of the p unknown parameters of a general nonlinear dynamical system.
The results above establish the principle that transient measurements of full second order moments carry information that allows one to identify all model parameters, at least, assuming noise-free measurements. If the measurements are corrupted by noise, it is often possible to compensate with a larger number of measurements. To illustrate this, we have conducted 100 simulated identification studies in which the unknown parameters were taken from a broad lognormal distribution (Figure 2B). For these, we supposed that vj:=v(tj) could be measured at m equally separated time points {t0, ….,tm−1}, and that each measurement had unknown errors of ±10% To explore the effect of incomplete measurements, we performed the identification method for the three data scenarios considered earlier: (1) all moments; (2) only the means; and (3) only the protein means and variances. For each scenario, we investigated the impact on parameter identification of using an increasing number of noisy measurements obtained from a different number of independent experiments (with different randomly chosen unknown initial conditions).
As more data were gathered, the effects of measurement error were overcome and the probability of successful identification increased for every strategy (see Figure 2C). Using many measurements, the parameters and the unknown initial conditions of mRNAs and proteins could be resolved even from inaccurate protein data alone—provided that it included information on the protein variance. All of the above numerical experiments were conducted assuming that the initial conditions were unknown; for known or specified initial conditions, we found that the identification was even more successful (see Supplementary Figure 5). We have thus shown that for the simple gene expression model, cellular noise enhances the opportunity for system parameter identification, whereas measurement noise impedes it. The deleterious effects of measurement noise can be overcome by increasing the number of measurements.
Experimental identification of lac induction
Among the most studied gene regulatory elements is the lac operon of Escherichia coli. This mechanism has been used to construct toggle switches (Gardner et al, 2000; Kobayashi et al, 2004), genetic oscillators (Elowitz and Leibler, 2000; Atkinson et al, 2003) and logical circuits (Weiss, 2001). Despite its ubiquitous use, precise in vivo single-cell quantification of the system remains insufficient. Indeed, most such quantification attempts have come from in vitro experiments or population level studies. For example, the lac repressor dissociation constant has been estimated to be Kd=10−11–10−9 M (Oehler et al, 1990). In an E. coli cell with a volume of 10−15 l, such dissociation constants mean the occupancy of the lac promoter is 94–99.94% when there are ten such molecules. At best, such measurements have only a probabilistic meaning at the level of single cells; at worst, they have no relevance at all as other mechanisms, such as nonspecific binding (Kao-Huang et al, 1977), take on much greater significance.
We used flow cytometry experiments and computational analyses to identify a parameter set to describe the in vivo single-cell dynamics of green fluorescent protein (GFP) controlled by the lac operon under isopropyl-β-D-thiogalactoside (IPTG) induction (see Figure 3A and Materials and methods section). We explored the response of the system at several IPTG levels and at multiple time points. Although many mechanistic models may capture the available data, we focused on the simplest consistent model, which consists of diffusion of IPTG into the cell, [IPTG]IN=[IPTG]OUT·(1−exp(−rt)), and four basic reactions, R1, R2, R3, and R4 corresponding to production and degradation of LacI and GFP.
Figure 3.

Experimental identification of a simple construct (A) in which IPTG induces the production of GFP. (B) Experimentally measured histograms of gfp expression on two different days (solid blue and green lines—in arbitrary units) and the best determined parameter fit (red dashed lines). Here, each column corresponds to a different measurement time (0, 3, 4, or 5 h) after induction, and each row corresponds to a different level of extracellular IPTG induction (5, 10, or 20 μM). In the parameter fits, different weights were applied to each experimental condition, shown as the values {q} in the histograms. (C) Predicted (red) and then measured (blue and green) fluorescence at 40 and 100 μM.
 |
The production of LacI is constant, w1=kL, corresponding to constitutive expression. However, production of GFP is a nonlinear function of the LacI level:
where kG is the unrepressed GFP production rate, α describes LacI occupancy strength, and the Hill coefficient, η, accounts for cooperative binding of LacI. The GFP degradation rate, w4=δG·[GFP], is fixed, but LacI can be degraded or inactivated by IPTG such that the total LacI removal depends on the IPTG concentration and is assumed to have the form w2=δL·[LacI], where δL = δL(0) + δL(1) [IPTG]IN. The model also explicitly characterizes uncertainties in the flow cytometry measurements (see Materials and methods). In total, there are ten unknown positive real parameters for the regulatory system, Λ = {kL,kG, δL(0), δL(1), δG, α η, r, μGFP, σGFP2} ∈ R+10.
The measured fluorescence histograms at different times and different IPTG levels (Figure 3) cannot adequately be captured using low order moments. Furthermore, as wG is a nonlinear function of LacI, there is no known analytical expression for the statistical moments of GFP. Instead, we used a new method, called finite state projection (FSP), to identify the unknown parameters on the basis of their probability densities (see Materials and methods section). In the identification routine, a parameter search was conducted to find parameter sets such that the total predicted fluorescence distribution was as close as possible to the measured distribution in a least squares sense for all time points and IPTG levels.
Figure 3B shows that the identified model results match the experimentally measured distributions exceptionally well. However, with the full set of ten unknowns in Λ, this identification is not unique, and we found multiple parameter sets that provided equally good fits. However, by utilizing additional information about the system, we could reduce the uncertainty of identification. In particular, assuming that GFP is lost solely to dilution, we could specify the rate δG=3.8 × 10−4 N−1 s−1, corresponding to a half-life of 30 min. The remaining nine parameters could then be identified as:
 |
where N refers to molecule number.
As the assumed model represents a simplified description of multiple events (folding dynamics, elongation, etc.), these parameters are best viewed as model-specific empirical measurements. Even so, it is possible to make some comparisons between the identified parameters and previous analyses. First, the production and degradation rates of LacI yield a mean number of kL/ γL(0) ≈ 5 molecules per cell at steady state in the absence of IPTG, on the same magnitude of wild-type levels of about ten per cell. Second, the level of LacI required for half occupancy of the lac operon is [LacI]1/2=(1/α)1/η=0.012, which compares well to values 0.006–0.6 molecules (10−11–10−9 M, Oehler et al, 1990). Third, a Hill coefficient of 2.1 is reasonable considering that LacI binds to the operon as a tetramer. Finally, the degradation rate LacI, δL(0) is close to the dilution rate of 3.8 × 10−4 N−1 s−1, reflecting the high stability of that protein. In addition to comparing the parameters to values in the literature, we have used the parameter set identified from {5, 10, 20} μM IPTG induction to predict the fluorescence under {40, 100} μM IPTG. Figure 3C shows that these predictions match the subsequent experimental measurements very well despite the vastly different shapes observed at the high induction levels.
Using single-cell experimental techniques, it has become possible to efficiently measure fluctuations in cell constituents. When properly extracted and processed with rapidly improving computational tools, these measurements contain sufficiently rich information as to enable the unique identification of parameters. We have shown that transient dynamics are important to this effort, and in principle, identification can be accomplished when accurate distributions are measured at only two distinct time points. More time points are needed if the distributions are poorly measured, but the idea remains the same. We have show the potential of our approach by experimentally identifying a predictive model of lac regulation from flow cytometry data. Hence, the proposed integration of single-cell measurements and stochastic analyses establishes a promising approach that offers new windows into the workings of cellular networks.
Materials and methods
Medium and reagents
Cells were grown in Luria–Bertani (LB) medium supplemented with 1% tryptone, 0.5% yeast extract, and 0.4% NaCl and containing IPTG at the concentrations noted. To select for plasmid maintenance, antibiotics were used at the following concentrations: 100 μg/ml ampicillin (amp); 40 μg/ml kanamycin (kan); and 12.5 μg/ml tetracycline (tet).
Bacterial strains and plasmids
The E. coli strain used was DL5905—E. coli K-12 (isolate MC4100) containing [F′ proAB lacIqZM15 Tn10 (Tetr)] from strain XL-1 Blue (Stratagene) and plasmid pDAL812. To construct plasmid pDAL812, GFP(LVA) (Anderdson et al, 1998) was PCR amplified from plasmid pRK9 (a gift from John Cronan) using the forward primer (5′-CAACAAAGATCTATTAAAGAGGAGAAATTAAGCATGAGTAAAGGAGAAGAACTTTTCA-3′) that includes a BglII site and removes an SphI site from the original pRK9 sequence, and the reverse primer (5′-CAACAAGCATGCATTAAGCTACTAAAGCGTAGTTTTCGTCGTTTGC-3′) that adds an SphI site. This fragment was digested using BglII and SphI and cloned into BglII and SphI sites of pLAC33 (Warren et al, 2000), removing a portion of the TetR cassette.
Fluorescence induction experiments
Twenty-four separate cell cultures were allowed to grow in LB broth containing the appropriate antibiotics to an approximate OD600 of 0.2, and were then induced with {0, 5, 10, 20, 40, 100} μM concentrations of IPTG at 5, 4, 3, and 0 h before flow cytometry measurements. Flow cytometry was carried out using a BD Biosciences FACSAria instrument with a 100-μm sorting nozzle at low pressure. GFP(LVA) was excited using a 488-nm blue laser and detected using 530/30-nm filter. For each sample, 1 000 000 events were collected. To ensure repeatability, experiments were conducted twice, each on a separate day.
GFP induction model
The stochastic model for the IPTG–GFP induction is composed of four nonlinear production/degradation reactions given in the main text. The rates of these reactions depend on the integer populations of the proteins LacI and GFP, as well as the set of nonnegative parameters, kL, k G, δ(0,1), δG, α,r,η} ∈ R8. For the stochastic system modeled here, the joint (LacI, GFP) probability distributions of both proteins evolve according to the infinite dimensional chemical master equation (CME; van Kampen, 2007). This can in turn be expressed as an infinite set of linear ordinary differential equations—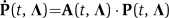 . Unlike in the simple transcription/translation model, the toggle reactions are nonlinear, and the CME has no known exact solution. We use a finite state projection approach (Munsky and Khammash, 2006) that makes it possible to approximate the solution to any degree of accuracy. For any error tolerance ɛ>0, we systematically find a finite-dimensional projected system—
. Unlike in the simple transcription/translation model, the toggle reactions are nonlinear, and the CME has no known exact solution. We use a finite state projection approach (Munsky and Khammash, 2006) that makes it possible to approximate the solution to any degree of accuracy. For any error tolerance ɛ>0, we systematically find a finite-dimensional projected system—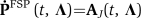 —the solution for which is within the desired tolerance. More precisely,
—the solution for which is within the desired tolerance. More precisely,
where the index vector J denotes the set of states included in the projection, PJ is the corresponding probability of those states, and AJ is the corresponding principle sub-matrix of A (Munsky and Khammash, 2006). The one-norm measure is used to ensure that absolute sum of the probability density error is guaranteed to lie within the tolerance. The solution of each projected master equation is found using the stiff ode solver ode23s in MathWorks Matlab.
Modeling flow cytometry data
In addition to modeling the regulatory dynamics of the system, one must also account for the inherent uncertainty within measured levels of fluorescence activity. The process used to account for this uncertainty has three components. First, in an effort to remove outliers in cell volume and density, and thereby reduce the effects of unmodeled dynamics, each cell population was gated separately using forward and side scatter data. Specifically, the forward and side scatter measurements were used to form a two-dimensional joint histogram with 50 × 50 logarithmically distributed bins (see Supplementary Figure 6). The maximum point in this histogram was recorded and then the gating region was chosen to include every bin that had, at least, one third as many counts as the maximal bin. Second, flow cytometry measurements in the absence of IPTG have been used to calibrate the background fluorescence of cell populations at various instances in time, and it has been assumed that the background fluorescence distribution, fBG(x), is independent of the levels of IPTG, LacI, and GFP. Third, each GFP molecule is assumed to emit a normally distributed random amount of fluorescence with unknown mean, μGFP, and variance, σGFP2, both of which are to be identified. Thus, if pn=pn(t, Λ, [IPTG]) denotes the probability of having exactly n={0, 1, 2, …} molecules of GFP, then the probability density of having exactly x arbitrary units of fluorescence because of GFP is computed as:
 |
Finally, the total observable fluorescence is the sum of the GFP florescence plus the background noise, and the distribution of total fluorescence is found using the convolution:
 |
Identification procedure
With the FSP solution and the computation of the expected fluorescence, the identification procedure is carried out by finding the parameter vector Λ* that minimizes the one norm difference between the experimentally measured distribution fMeas(i) (t,[rm IPTG]) and the numerical solution of that distribution:
 |
where the summation is taken over all of the different experimental conditions of different induction times and IPTG levels, and the weight qi specifies the relative importance to each of these measurements. These weights have been chosen such that each IPTG level has the same total importance and so that greater importance is placed on measurements that differ the most from the background fluorescence. The values for these weights are given in Figure 3. The parameter identification is accomplished by starting with an initial parameter guess, Λ0, and then this set is updated iteratively using gradient-based and simulated annealing searches until the computed distribution matches the experimental distribution as closely as possible. The optimization procedure is repeated for multiple, randomly generated initial parameter guesses. An optimal parameter set is regarded as unique if the given solution yields the smallest achieved value for the objective function, and if that parameter has been achieved during many such identification runs each beginning with different parameter guesses.
Supplementary Material
Supplementary text, Supplementary figures S1-6
Acknowledgments
The authors thank John Little and Hana El-Samad for thoughtful discussions and David Low for advice, assistance, and materials required in the experimental aspects of this study. This study is funded by the LANL LDRD program and NSF through Grants ECCS-0835847 and ECCS-0802008, and by ICB Grant DAAD19-03-D-0004 from the US Army Research Office.
Footnotes
The authors declare that they have no conflict of interest.
References
- Andersen J, Sternberg C, Poulsen L, Bjorn S, Givskov M, Molin S (1998) New unstable variants of green fluorescent protein for studies of transient gene expression in bacteria. Appl Environ Microbiol 64: 2240–2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin A, Shen P, Ross J (1997) A test case of correlation metric construction of a reaction pathway from measurements. Science 227: 1275–1279 [Google Scholar]
- Atkinson M, Savageau M, Myers J, Ninfa A (2003) Development of genetic circuitry exhibiting toggle switch or oscillatory behaviour in E. coli. Cell 113: 597–607 [DOI] [PubMed] [Google Scholar]
- Cinquemani E, Milias-Argeitis A, Summers S, Lygeros J (2009) Local identification of piecewise deterministic models of genetic networks, lecture notes in computer science. Springer 5469: 105–119 [Google Scholar]
- Dunlop M, Cox R III, Levine J, Murray R, Elowitz M (2008) Regulatory activity revealed by dynamic correlations in gene expression noise. Nat Genet 40: 1493–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz M, Leibler S (2000) A synthetic oscillatory network of transcriptional regulators. Nature 403: 335–338 [DOI] [PubMed] [Google Scholar]
- Elowitz M, Levine A, Siggia E, Swain P (2002) Stochastic gene expression in a single cell. Science 297: 1183–1186 [DOI] [PubMed] [Google Scholar]
- Gardner T, Cantor C, Collins J (2000) Construction of a genetic toggle switch in escherichia coli. Nature 403: 339–342 [DOI] [PubMed] [Google Scholar]
- Kao-Huang Y, Revzin A, Butler AP, O'Conner P, Noble DW, von Hippel PH (1977) Nonspecific DNA binding of genome-regulating proteins as a biological control mechanism: Measurement of DNA-bound E. coli lac repressor in vivo. Proc Natl Acad Sci USA 74: 4228–4232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Kaern M, Araki M, Chung K, Gardner T, Cantor C, Collins J (2004) Programmable cells: interfacing natural and engineered gene networks. Proc Natl Acad Sci 101: 8414–8419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljung L (1999) System Identification, Theory for the User. Upper Saddle River, NJ, USA: Prentice-Hall [Google Scholar]
- Munsky B, Khammash M (2006) The finite state projection algorithm for the solution of the chemical master equation. J Chem Phys 124: 044104. [DOI] [PubMed] [Google Scholar]
- Oehler S, Eismann E, Krämer H, Müller-Hill B (1990) The three operators of the lac operon cooperate in repression. EMBO J 9: 973–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffard R, Lipan O, Wong W, Tomlin C (2008) Optimal discovery of a stochastic genetic network. Proc 2008 Amer Contr Conf, Vol. 1, pp 2773–2779, 11–13 June 2008, Seattle, WA, USA [Google Scholar]
- Sontag E (2002) For differential equations with r parameters, 2r+1 experiments are enough for identification. J Nonlinear Sci 12: 553–583 [Google Scholar]
- Thattai M, van Oudenaarden A (2001) Intrinsic noise in gene regulatory networks. Proc Natl Acad Sci USA 98: 8614–8619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kampen N (2007) Stochastic Processes in Physics and Chemistry, 3rd edn. Amsterdam: Elsevier [Google Scholar]
- Warmflash A, Dinner A (2008) Signatures of combinatorial regulation in intrinsic biological noise. Proc Nat Acad Sci USA 105: 17262–17267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren J, Walker J, Roth J, Altman E (2000) Construction and characterization of a highly regulable expression vector, pLAC11, and its multipurpose derivatives, pLAC22 and pLAC33. Plasmid 44: 138–151 [DOI] [PubMed] [Google Scholar]
- Weiss R (2001) Cellular Computation and Communications using Engineered Genetic Regular Networks. PhD thesis, MIT, 2001 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary text, Supplementary figures S1-6