

Abstract
Cdc37 is a molecular chaperone that physically stabilizes the catalytic domains found in protein kinases and is therefore a wide-spectrum regulator of protein phosphorylation. It is also an overexpressed oncogene that mediates carcinogenesis by stabilizing the compromised structures of mutant and/or overexpressed oncogenic kinases. Recent work shows that such dependency of malignant cells on elevated Cdc37 expression is a vulnerability that can be targeted in cancer by agents that deplete or inhibit Cdc37. Cdc37 is thus a candidate for broad-spectrum molecular cancer therapy.
Although Cdc37 is overexpressed in a number of cancers, it is an unusual oncoprotein whose transforming character does not readily snap into place in the conventional oncogenic pathways. Instead, it is a molecular chaperone, a protein that facilitates folding/refolding of other proteins, often described as its “clients”. The role of molecular chaperones in cancer was first characterized with regard to Hsp90 (for review see5). Hsp90 is a facilitator of oncogenesis that stabilizes a wide range of overexpressed or mutated oncogenic proteins and permits their tumorigenic influence to arise3,7. Tumors thus appear to become addicted to chaperones and can be treated by withdrawing the chaperoning power of Hsp904–6. Cdc37 interacts with a subset of client proteins within Hsp90 complexes and plays a specialized role as primary partner in kinome maintenance3,8,9. This specialized property of Cdc37 fuels the acceleration of cell proliferation observed in cancer by promoting the activities of a broad array of protein kinases1–3. These include receptor tyrosine kinases such as EGFR and MET, non-receptor tyrosine kinases v-src and Lck, and intracellular serine/threonine kinases Raf-1, Ksr, Akt, IKK, Cdc2, Cdk2 and Cdk4. A comprehensive list of Cdc37 client kinases, including the ones listed above and others can be found at the website of Dr Didier Picard: http://www.picard.ch/downloads/Cdc37interactors.pdf. The degree of dependency of the cancer cell kinome on Cdc37 expression thus appears to be extensive and in a recent yeast screen, at least 90 % of the protein kinases examined required Cdc37 for function10.
The defining cellular role for Cdc37, which becomes misappropriated in cancer, resides in its promotion of cell proliferation. Indeed this protein was first discovered in a yeast screen for cell division cycle proteins, hence its name11. Thus when mammalian tissues undergo rapid proliferation during development, the normally scarce levels of Cdc37 are rapidly expanded. Elevated levels of Hsp90/Cdc37 complexes mediate the maturation and stabilization of protein kinases required for proliferation or perhaps for repeated rounds of kinase re-activation9,12,13. For instance, increased levels of Cdc37 in vivo correlate with the upregulation of the G1-specific cyclin D1 in proliferating normal tissues3. Cyclin D1 is the partner of Cdk4, a Cdc37-dependent cell cycle kinase. Elevated Cdc37 levels also mediate cancer cell growth, although the degree to which changes in Cdc37 regulation in proliferating normal tissues resemble those in malignant tissues is not clear. The role of Cdk4 as a key Cdc37 client in cancer is compelling, although the much larger cast of oncogenic signaling molecules mentioned above also require Cdc37. Proliferating tumor cells may also have an expanded dependency on Cdc37 due to the increased repertoire of overexpressed/mutated kinases that mediate their growth (in accordance with the addicted to chaperones hypothesis)5. However, the degree to which Cdc37 responds to the quality or quantity of clients in these contrasting proliferative states is yet to be determined.
The amplified levels of Cdc37 in prostate cancer suggest that it may be of particular significance in this type of neoplasm8,14,15 Indeed, in recent immunohistological studies, Cdc37 overexpression was found in 100% of neoplastic prostate tissue, compared with normal prostate8. Although the rationale for this is not certain, one rather compelling suggestion involves the role of Cdc37 in androgen signaling, a key pathway in prostatic cell growth. The androgen receptor (AR) is a rare non-kinase Cdc37 client16. Although Hsp90 plays a general role in maintaining the function of nuclear receptors, Cdc37 specifically associates with AR in contrast to closely-related nuclear receptors including the glucocorticoid receptor (GR)16. Cdc37 associates with the ligand-binding domain of AR permitting the structural changes that accompany ligand encounter, factor activation and cell proliferation.
Structural mechanisms of Cdc37-kinase interaction
Strategies for targeting Cdc37 in cancer treatment will be aided by considering the biochemical and ultrastructural nature of the Cdc37-client interaction. The two best known properties of Cdc37 are its abilities to: (1) bind to clients and (2) associate with Hsp90, functions which involve two major structural domains2,9,10 (Fig. 1). The kinase binding domain (human, 1–126 aa) is the most highly conserved region among Cdc37 homologs and contains several key residues whose mutation or deletion each alter kinase binding2. The residue with most regulatory significance is serine (S) 13 whose phosphorylation permits stable interaction of Cdc37 with client kinases17. In turn, the kinase which phosphorylates S13, casein kinase II is in itself a Cdc37 client, and mediates a positive feedback loop between the client kinase and Cdc37 chaperoning18. A second region within Cdc37 (127–282 aa) interacts with the N-terminal ATP-binding domain of Hsp90. This interaction is predicted not only to mediate recruitment of client kinases to Hsp90 but also to regulate the Hsp90 ATPase activity required for chaperone activity2. By contrast the C-terminal domain (282–378 aa) of Cdc37 remains relatively uncharacterized. In both S. cereviseae and S. pombe, only the amino terminal “client interface” domain is required to support growth, suggesting that Hsp90 is dispensable for the growth-promoting functions of Cdc3719,20. However, in mammalian cells the homologous construct functions as a dominant negative inhibitor of wild type Cdc37 and a growth antagonist in prostate carcinoma cells15. This of course does not rule out Hsp90 independent actions of Cdc37 but rather suggests a shift in emphasis towards Hsp90-dependent interactions in mammalian cells. Such differences between these classes of eukaryote may reflect the rather meager degree of sequence conservation between the mammalian and yeast molecules, which is less than 20% and make inferences derived from yeast studies somewhat uncertain for prediction of Cdc37 behavior in cancer2. Of particular note, the affinity of mammalian Cdc37 for Hsp90 exceeds that of the S. cereviseae homolog by greater than tenfold2. Mammalian Cdc37 in tumor cells may thus have an increased tendency to function within a ternary complex containing the client kinase and Hsp90 (Fig. 1), although independent functions for Cdc37 cannot be ruled out.
Figure 1. Interaction domains in Cdc37.
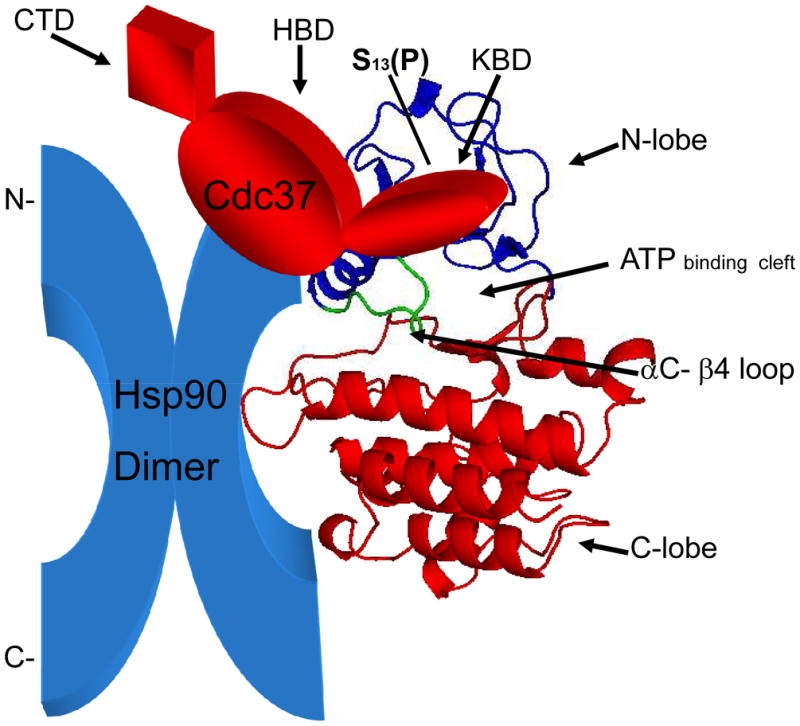
The Cdc37 gene contains three major domains including: an N-terminal client kinase interaction domain (KDB) containing the key phosphoresidue S13, a central domain required for Hsp90 binding (HDB) and a C-terminal domain of unknown function (CTD). The catalytic domains of protein kinases depicted in the ribbon diagram contain N (blue) and C (red) lobes. The KDB of Cdc37 (red) binds to the N lobe while Hsp90 (blue) binds to both N and C lobes while interacting with the HDB of Cdc37.
Protein kinases in all cells become associated with Cdc37 and Hsp90 in a stepwise process based largely on the paradigm first established for GR folding and maturation21. Initial kinase folding involves association with the chaperones Hsp70 and Hsp40 followed by a series of ATP-fueled steps that lead to assembly of a folding complex containing Hsp90 and its cohorts including Cdc37 bound to the kinase4,5. The more stable clients then exit the Cdc37-Hsp90 complex, although particularly sensitive kinases bind persistently to the Hsp90 complexes. Such unstable clients proliferate in tumor cell populations due to competition and selection for cells expressing variant and super-abundant mutant kinase variants that mediate tumor progression (2).
Considering the interaction from the viewpoint of the kinase, both Cdc37 and Hsp90 bind directly to the catalytic domain2 (Fig. 1). The conserved catalytic domain is comprised of two lobes separated by a deep ATP-binding cleft. Cdc37 associates with the N-terminal lobe while Hsp90 interacts with both lobes2,22,23. The N- lobe is more loosely packed in terms of polypeptide chain folding and can thus comprise a hazard in terms of tendency to unfold, lose function and nucleate degradation cascades22,23. Moreover, each kinase-chaperone interaction shows characteristic properties despite the catalytic domain being the third most common in the human genome24. Indeed, due to subtle differences in N-lobe structure, some kinases may need persistent interactions with Hsp90 and Cdc37 for function and stability2. Other kinases may be stabilized by additional intramolecular domains and/or by regulatory partner proteins that bring stability along with substrate specificity23. For instance Cdk4 binds initially to Hsp90 and Cdc37 until encountering cyclin D1 and forming a Cdk4-Cyclin D1 complex sufficiently stable to be independent of further chaperone interaction25. Other kinases such as the oncogenic c-erb-B2 bind persistently to chaperone complexes even in the active form (reviewed2). It is predicted that the proliferation of mutation-activated kinases in tumors will lead to a greater dependency on Cdc37 chaperoning. Indeed, individual kinase targets may undergo continuous evolution in terms of Cdc37 dependency due to the emergence of novel mutant forms. Future approaches to individualized care may involve screening for the emergence of Cdc37 dependent kinase mutants. Drugs that inhibit the ATPase activity of Hsp90 further destabilize these quasi-stable clients5. This concept has been reaffirmed in studies with anti-cancer drugs that bind and stabilize the ATP binding groove of client kinases and are able to prevent their proteasomal degradation via Hsp90 inhibition. Indeed, the exchange of ATP in activated kinases induces their destabilization and interaction with Cdc37 and Hsp90. Thus mutations that allow a kinase to develop “fast” activity also result in the kinase becoming structurally “loose,” with Cdc37 and Hsp90 helping maintain this “fast and loose” existence. Hsp90 inhibitors are currently undergoing clinical trial4. Understanding the dynamics of kinase-Cdc37-Hsp90 interaction may be of particular significance for prediction of kinases in cancer cells most adversely affected by Hsp90 and/or Cdc37 inhibition and facilitation of anti-Cdc37 drug design. (For a definitive review of Cdc37 and protein kinase interactions see:2).
Targeting Cdc37 in cancer
As mentioned earlier, Cdc37 is an overexpressed oncogene. Indeed, under forced Cdc37 expression cells become transformed, proliferate and tumors arise. When Cdc37 is overexpressed along with the proto-oncogene c-Myc, the effects of both agents become amplified and more and larger tumors arise in transgenic mice15,8,14. Indeed, Cdc37 levels are elevated in many clinical cancers8. Although Cdc37 may be regarded as a specialized partner of Hsp90, dealing with structurally compromised kinase clients, it may also offer a preferable target for cancer therapy. Hsp90 is expressed with similar abundance in normal and malignant cells and includes among its clientele proteins required for the viability of normal cells26. In its favor as a therapeutic target, some studies indicate that tumor Hsp90 is selectively vulnerable due to the proliferation of its oncogenic clients and the formation of mature Hsp90/client/co-chaperone complexes that bind Hsp90 drugs with increased affinity27. However this phenomenon is not universally observed28. Cdc37, by contrast is increased in proliferating tissues, and is heavily expressed in certain cancers including prostate carcinoma, anaplastic large cell lymphoma, acute myelocytic leukemia, hepatocellular carcinoma and multiple myeloma29–32. As most normal tissues do not proliferate or appear to require Cdc37, a potential therapeutic window is available, which is not as obvious in the case of Hsp90.
The mechanisms underlying increased Cdc37 expression in malignant cells are not known. The Cdc37 promoter contains multiple binding sites for transcription factor Mzf1, implicated in myeloid tumorigenesis, although evidence for a dynamic role in Cdc37 expression is not available33. The addicted to chaperones hypothesis might predict that chaperone levels should increase as clients proliferate. Indeed, there is a clear rationale for this mechanism in the case of Hsp90 whose transcription is activated by the factor HSF1 which undergoes induction by increased accumulation of unfolded proteins34. However, the Cdc37 promoter is devoid of canonical HSF1 binding elements and does not respond to HSF1 activation. A further clue that warrants exploration is that Cdc37 is overexpressed in prostate cancer and such cancers are induced by the forced expression of Cdc378,14. As AR is one of the rare non-kinase clients for Cdc37, and is essential for proliferation and differentiation of prostatic epithelium, a role for AR would fit the profile8.
Recent studies address the feasibility of targeting Cdc37 in prostate carcinoma15,35. Indeed, depletion of Cdc37 in prostate cancer cell lines by RNA interference techniques leads to permanent growth inhibition both in androgen-dependent and androgen-independent cell lines35. Growth inhibition was correlated with decreased signaling through the anabolic ERK and Akt kinase cascades as well as reduced AR-dependent transcriptional signaling35 (Fig 2). In addition, Cdc37 targeting inhibits HSF1 activity and Hsp70 expression, components of a common resistance pathway that mediates insensitivity to therapeutics such as Hsp90 inhibitors and proteasomal targeting agents35,36 (Fig 2). Inactivation of Cdc37 may thus decrease cell survival through a range of pathways, permitting effective inhibition of anabolic signaling and breaking resistance to cytotoxic therapy. It is significant that the consequences of Cdc37 depletion and Hsp90 inhibition are divergent at the molecular level, with Hsp90 inactivation triggering degradation of the abandoned clients while Cdc37 depletion does not35 (Fig 3). Cdc37 knockdown instead causes profound inhibition of the activities of multiple kinase cascades and loss of the transcriptional activity of AR35. These data suggest intrinsic differences in the relationships between Hsp90, Cdc37 and their clients that become unmasked during therapy. Cdc37 is evidently not essential for the intracellular survival of its clients while functional Hsp90 is required to prevent degradation37 (Fig 3). However, in order for protein kinases to transfer phosphate to their substrates or for AR to interact productively with its ligand, Cdc37 is indispensable. Further underlining the contrasting mechanisms of Hsp90- and Cdc37-dependent prostate cancer growth, functional inhibition of both proteins in tumor cells is supra-additive35. The question of independent, non-Hsp90 requiring interactions for Cdc37 observed in recent studies also arises20.
Figure 2. Effects of Cdc37 depletion on anabolic signaling and cell stress pathways.

Cdc37 depletion inhibits signal flux through at least three pathways in prostate carcinoma. These include the (1) ERK, (2) Akt and (3) AR pathways. Depletion of Cdc37 also inhibits (4) the HSF1-mediated heat shock stress response that can be activated by many anti-cancer agents. Well-characterized molecular clients for Cdc37 are indicated by blue coloration.
Figure 3. Contrasting effects of targeting Hsp90 and Cdc37.

Kinases (▭) interact with both Hsp90 (□) and Cdc37 (○). However, while pharmacological inhibition of Hsp90 (red rectangle) leads to client degradation, depletion of Cdc37 (red circle) causes a block to client maturation and activity that does not involve such extensive client destruction.
Recent work also shows that depletion of another Hsp90 co-chaperone AHA1 leads to a similar reduction in activity of Hsp90 client kinases without marked increases in degradation of such proteins. These findings suggest that degradation of clients may require direct interaction between drugged, inactive Hsp90 and its bound clients and cannot be mimicked by the inactivation of co-chaperones38. Targeting Hsp90 co-chaperones may therefore lead to radically different consequences compared to the direct inhibition of Hsp90. Future strategies for Cdc37 drug design and selection would likely aim at the inhibition of either: (1) the Cdc37-client interaction or (2) Cdc37-Hsp90 association (Fig 1). Indeed recent studies using the triterpine drug celastrol indicate cancer cell growth reduction through mechanism (2) the inhibition of Hsp90-Cdc37 interaction39. These natural plant products cause pleiotropic changes in cancer cells and can lead to tumor regression39. However, the cellular responses to celastrol resemble those provoked by Hsp90 inhibitors, and include client protein destruction and HSF1 activation and may not provide a radical departure from current treatments. Further studies might concentrate on agents with more novel mechanisms of action such as Cdc37-client interaction.
In conclusion, inhibition of Cdc37 activity is a promising new approach to the treatment of cancer due to its multi-targeting nature, the elevated expression of Cdc37 in dividing cells and the ability of Cdc37 depletion to arrest growth in both AR+ and AR− prostate cancer. Cdc37 targeting also has the potential ability to deter the evolution of new traits that depend on kinase mutation and such a block on adaptation might limit the ability of tumors to respond to therapy and a deteriorating microenvironment. Such effects are proposed to occur in tumors treated with Hsp90-targetting drugs5. As to the ultimate selectivity of Cdc37 inhibition for treating malignant cells, this chaperone is not expressed in quiescent cells but appears necessary for the growth of normal epithelial cells. Therefore treating prostate cancer through the inhibition of Cdc37 might involve sacrificing this non-essential population15. However, this drawback would not differ markedly from current therapies used to treat prostate cancer, which also focus on eradicating normal prostate tissue as well as cancer. One potential concern with this therapy could however be the unknown potential for effects on other normally proliferating epithelium including that in the GI tract and skin.
Future Directions
More remains to be learned regarding the oncogenic role of Cdc37 and its utility as a target for cancer therapy. Thus we remain uncertain of the full extent of types and stages of cancer mediated by Cdc37 overexpression and such a gap in knowledge will hopefully be rectified in subsequent studies. Future studies will also ask- by which molecular mechanism(s) does Cdc37 become overexpressed during tumor progression? Promising approaches to Cdc37 targeting of malignant tissues that will likely be explored include pharmacological disruption of Cdc37-client interactions and modulation of phosphorylation of the key residue Serine 13 within the client interaction domain of Cdc37.
Acknowledgments
We acknowledge the support of the Department of Radiation Oncology at BIDMC. These studies were also supported by NIH grants 5RO1CA047407 and 3RO1CA094397 (SKC) and a Howard Hughes Medical Institute Medical Student Research Training Fellowship (PJG).
Footnotes
References
- 1.Caplan AJ, Ma’ayan A, Willis IM. Multiple Kinases and System Robustness: A Link Between Cdc37 and Genome Integrity. Cell Cycle. 2007;6 doi: 10.4161/cc.6.24.5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caplan AJ, Mandal AK, Theodoraki MA. Molecular chaperones and protein kinase quality control. Trends Cell Biol. 2007;17:87–92. doi: 10.1016/j.tcb.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 3.Pearl LH. Hsp90 and Cdc37 -- a chaperone cancer conspiracy. Curr Opin Genet Dev. 2005;15:55–61. doi: 10.1016/j.gde.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 4.Neckers L, Ivy SP. Heat shock protein 90. Curr Opin Oncol. 2003;15:419–24. doi: 10.1097/00001622-200311000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–72. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 6.Workman P. Altered states: selectively drugging the Hsp90 cancer chaperone. Trends Mol Med. 2004;10:47–51. doi: 10.1016/j.molmed.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 7.Calderwood SK, Khaleque MA, Sawyer DB, Ciocca DR. Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem Sci. 2006;31:164–72. doi: 10.1016/j.tibs.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 8.Stepanova L, et al. Induction of human Cdc37 in prostate cancer correlates with the ability of targeted Cdc37 expression to promote prostatic hyperplasia. Oncogene. 2000;19:2186–93. doi: 10.1038/sj.onc.1203561. [DOI] [PubMed] [Google Scholar]
- 9.Vaughan CK, et al. Structure of an Hsp90-Cdc37-Cdk4 complex. Mol Cell. 2006;23:697–707. doi: 10.1016/j.molcel.2006.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mandal AK, et al. Cdc37 has distinct roles in protein kinase quality control that protect nascent chains from degradation and promote posttranslational maturation. J Cell Biol. 2007;176:319–28. doi: 10.1083/jcb.200604106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reed SI. The selection of S. cerevisiae mutants defective in the start event of cell division. Genetics. 1980;95:561–77. doi: 10.1093/genetics/95.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pascale RM, et al. Role of HSP90, CDC37, and CRM1 as modulators of P16(INK4A) activity in rat liver carcinogenesis and human liver cancer. Hepatology. 2005;42:1310–9. doi: 10.1002/hep.20962. [DOI] [PubMed] [Google Scholar]
- 13.Prince T, Sun L, Matts RL. Cdk2: a genuine protein kinase client of Hsp90 and Cdc37. Biochemistry. 2005;44:15287–95. doi: 10.1021/bi051423m. [DOI] [PubMed] [Google Scholar]
- 14.Stepanova L, Finegold M, DeMayo F, Schmidt EV, Harper JW. The oncoprotein kinase chaperone CDC37 functions as an oncogene in mice and collaborates with both c-myc and cyclin D1 in transformation of multiple tissues. Mol Cell Biol. 2000;20:4462–73. doi: 10.1128/mcb.20.12.4462-4473.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwarze SR, Fu VX, Jarrard DF. Cdc37 enhances proliferation and is necessary for normal human prostate epithelial cell survival. Cancer Res. 2003;63:4614–9. [PubMed] [Google Scholar]
- 16.Robzyk K, et al. Uncoupling of hormone-dependence from chaperone-dependence in the L701H mutation of the androgen receptor. Mol Cell Endocrinol. 2007;268:67–74. doi: 10.1016/j.mce.2007.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shao J, Prince T, Hartson SD, Matts RL. Phosphorylation of serine 13 is required for the proper function of the Hsp90 co-chaperone, Cdc37. J Biol Chem. 2003;278:38117–20. doi: 10.1074/jbc.C300330200. [DOI] [PubMed] [Google Scholar]
- 18.Miyata Y, Nishida E. CK2 binds, phosphorylates, and regulates its pivotal substrate Cdc37, an Hsp90-cochaperone. Mol Cell Biochem. 2005;274:171–9. doi: 10.1007/s11010-005-2949-8. [DOI] [PubMed] [Google Scholar]
- 19.Turnbull EL, Martin IV, Fantes PA. Cdc37 maintains cellular viability in Schizosaccharomyces pombe independently of interactions with heat-shock protein 90. Febs J. 2005;272:4129–40. doi: 10.1111/j.1742-4658.2005.04825.x. [DOI] [PubMed] [Google Scholar]
- 20.MacLean M, Picard D. Cdc37 goes beyond Hsp90 and kinases. Cell Stress Chaperones. 2003;8:114–9. doi: 10.1379/1466-1268(2003)008<0114:cgbhak>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cox MB, et al. Fkbp52 Phosphorylation: a Potential Mechanism for Regulating Steroid Hormone Receptor Activity. Mol Endocrinol. 2007 doi: 10.1210/me.2006-0547. [DOI] [PubMed] [Google Scholar]
- 22.Prince T, Matts RL. Definition of protein kinase sequence motifs that trigger high affinity binding of Hsp90 and Cdc37. J Biol Chem. 2004;279:39975–81. doi: 10.1074/jbc.M406882200. [DOI] [PubMed] [Google Scholar]
- 23.Prince T, Matts RL. Exposure of protein kinase motifs that trigger binding of Hsp90 and Cdc37. Biochem Biophys Res Commun. 2005;338:1447–54. doi: 10.1016/j.bbrc.2005.10.100. [DOI] [PubMed] [Google Scholar]
- 24.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–34. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 25.Stepanova L, Leng X, Parker SB, Harper JW. Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes Dev. 1996;10:1491–502. doi: 10.1101/gad.10.12.1491. [DOI] [PubMed] [Google Scholar]
- 26.Waza M, et al. Modulation of Hsp90 function in neurodegenerative disorders: a molecular-targeted therapy against disease-causing protein. J Mol Med. 2006;84:635–46. doi: 10.1007/s00109-006-0066-0. [DOI] [PubMed] [Google Scholar]
- 27.Kamal A, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–10. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 28.Theodoraki MA, Kunjappu M, Sternberg DW, Caplan AJ. Akt shows variable sensitivity to an Hsp90 inhibitor depending on cell context. Exp Cell Res. 2007 doi: 10.1016/j.yexcr.2007.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thompson MA, et al. Differential gene expression in anaplastic lymphoma kinase-positive and anaplastic lymphoma kinase-negative anaplastic large cell lymphomas. Hum Pathol. 2005;36:494–504. doi: 10.1016/j.humpath.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 30.Katayama Y, et al. Cyclin D1 overexpression is not a specific grouping marker, but may collaborate with CDC37 in myeloma cells. Int J Oncol. 2004;25:579–95. [PubMed] [Google Scholar]
- 31.Feo F, et al. Hepatocellular carcinoma as a complex polygenic disease. Interpretive analysis of recent developments on genetic predisposition. Biochim Biophys Acta. 2006;1765:126–47. doi: 10.1016/j.bbcan.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 32.Casas S, et al. Changes in apoptosis-related pathways in acute myelocytic leukemia. Cancer Genet Cytogenet. 2003;146:89–101. doi: 10.1016/s0165-4608(03)00102-x. [DOI] [PubMed] [Google Scholar]
- 33.Gaboli M, et al. Mzf1 controls cell proliferation and tumorigenesis. Genes Dev. 2001;15:1625–30. doi: 10.1101/gad.902301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94:471–80. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
- 35.Gray PJ, Jr, Stevenson MA, Calderwood SK. Targeting Cdc37 inhibits multiple signaling pathways and induces growth arrest in prostate cancer cells. Cancer Res. 2007;67:11942–50. doi: 10.1158/0008-5472.CAN-07-3162. [DOI] [PubMed] [Google Scholar]
- 36.Zaarur N, Gabai VL, Porco JA, Jr, Calderwood S, Sherman MY. Targeting heat shock response to sensitize cancer cells to proteasome and Hsp90 inhibitors. Cancer Res. 2006;66:1783–91. doi: 10.1158/0008-5472.CAN-05-3692. [DOI] [PubMed] [Google Scholar]
- 37.Roiniotis J, Masendycz P, Ho S, Scholz GM. Domain-mediated dimerization of the Hsp90 cochaperones Harc and Cdc37. Biochemistry. 2005;44:6662–9. doi: 10.1021/bi047406z. [DOI] [PubMed] [Google Scholar]
- 38.Holmes JL, Sharp SY, Hobbs S, Workman P. Silencing of HSP90 cochaperone AHA1 expression decreases client protein activation and increases cellular sensitivity to the HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2008;68:1188–97. doi: 10.1158/0008-5472.CAN-07-3268. [DOI] [PubMed] [Google Scholar]
- 39.Zhang T, et al. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol Cancer Ther. 2008;7:162–70. doi: 10.1158/1535-7163.MCT-07-0484. [DOI] [PubMed] [Google Scholar]