

Abstract
Calpain-mediated degradation of the cytoskeletal protein alpha-II-spectrin has been implicated in the pathobiology of experimental and human traumatic brain injury (TBI). Spectrin proteolysis following diffuse/widespread TBI uncomplicated by either subtle or overt contusion and/or mass lesions, i.e. mild to moderate TBI, has not previously been evaluated. To determine the spatiotemporal pattern and cellular localization of calpain-mediated spectrin proteolysis (CMSP) following diffuse/widespread TBI and the extent to which parenchymal changes in CMSP are reflected in the cerebrospinal fluid (CSF), adult rats were subjected to a moderate midline fluid percussion injury and allowed to survive for 3 hours to 7 days post-injury. Light and electron microscopic immunocytochemical and Western blot analyses were performed to identify the calpain-specific 145-kDa breakdown product of alpha-II-spectrin (SBDP145). Following diffuse TBI, enhanced levels of SBDP145 immunoreactivity were observed in the neocortex, subcortical white matter, thalamus and hippocampus, peaking between 24 and 48 hours post-injury. Immunoreactivity was localized almost exclusively to damaged axons and axonal terminal debris. Heightened levels of SBDP145 were also observed in the CSF at 24 hours post-injury. These results confirm the widespread occurrence of CMSP following diffuse TBI without contusion and support the potential utility of SBDPs as biomarkers of a diffusely injured brain.
Keywords: Biomarker, Calpain, Cytoskeleton, Diffuse axonal injury, Spectrin, Traumatic brain injury
INTRODUCTION
Traumatic brain injury (TBI) is a major national and global health care problem that extracts a staggering personal and societal toll (1). Over the past 25 years both basic science and clinical discovery have provided unique insights into this complex disease and have identified a series of pathobiological cascades evoked by the traumatic insult that are capable of causing further progressive damage to the already mechanically perturbed brain (2-5). Of the multiple pathways identified to date, the disruption of calcium homeostasis within the injured brain followed by calcium-induced activation of various proteases has been consistently implicated in neuronal damage and death processes (6-13). Particular significance has been attached to the proteolytic cleavage of neuronal-cytoskeletal elements by the non-lysosomal, calcium-activated protease, calpain (6-13). Calpain-mediated proteolytic degradation of the essential cytoskeletal protein alpha-II-spectrin has been extensively described in experimental brain injury models and in human severe TBI cases (9-11, 13-16). Because of the consistent observation of calpain-mediated processes following TBI, it has been suggested that the identification of calpain-mediated proteolytic degradation products may serve as a valuable biomarker for the assessment of injury severity when harvested from either the blood or cerebrospinal fluid (CSF) of TBI patients (17, 18); several recent studies analyzing CSF from TBI patients have supported this premise (19-21). Despite the extensive experimental and preliminary clinical data supporting the role of calpain-mediated proteolytic degradation and its utility as a biomarker, however, the majority of studies to date have been done either in animal models or in humans in which the animals/patients had significant contusional damage (9, 14, 22, 23). Although contusion is common and clinically significant, contusional changes typically involve large, destructive and ultimately necrotic cortical lesions, consistent with the activation of calpain-mediated proteolytic degradation independent of the presence of blood (9, 14, 22, 23). Therefore, the question remains as to whether comparable calpain-mediated proteolytic degradation would occur following diffuse/widespread TBI in the absence of contusional change and/or mass lesion formation. Calpain-mediated proteolytic degradation in uncomplicated TBI is important because diffuse/widespread brain injury occurs across the spectrum of TBI; moreover it likely is the dominant form of injury in mild to moderate TBI that occurs in significantly larger numbers of individuals than severe TBI (1). Studies of these processes would be of further benefit, in that, they allow parallel assessment of the CSF for the presence of calpain-mediated protein degradation products to determine whether CSF changes correlate with ongoing processes in the injured brain parenchyma.
In this study we explored these issues using a well controlled and well characterized animal model of diffuse/widespread TBI uncomplicated by contusional change (24). The neocortex, thalamus and dorsal hippocampus were assessed via both light and electron microscopic immunocytochemical and Western blot methods to confirm the occurrence of calpain-mediated degradation and to identify its cellular localization following diffuse TBI. Parallel CSF analyses were performed to evaluate the fidelity of those changes found in the CSF to those observed in the brain parenchyma. Through these multiple, integrated approaches, we confirmed the traumatic activation of calpain-mediated degradation within the neocortex, subcortical white matter, thalamus and hippocampus in the absence of contusional change. In these domains this calpain-mediated degradation was associated with axonal damage and its downstream anterograde consequences. Further, the identification of calpain-mediated degradation products in the CSF of diffusely injured animals suggested a correlation between the presence of these diffusely injured immunoreactive axonal profiles manifesting calpain-mediated degradation and the degradation products found in the CSF.
MATERIALS AND METHODS
Surgical Preparation and Injury Induction
Adult male Sprague-Dawley rats (375-400g) were subjected to midline fluid percussion injury (FPI) consistent with methods described previously (25, 26). Briefly, rats were anesthetized with 4% isoflurane in 70% N2O and 30% O2, intubated, and maintained on a ventilator with 2% isoflurane. During surgery, body temperature was measured with a rectal probe and maintained at 37° C with a thermostat controlled heating pad (Harvard Apparatus, Holliston, MA). Rats were positioned in a head holder assembly (Kopf Instruments, Tujunga, CA) and a midline sagittal incision was made to expose the skull from bregma to lambda. The skull was cleaned and dried, and 2 holes 1 mm in diameter were drilled into the right frontal and occipital bones 1 mm rostral and caudal to bregma and lambda, respectively, for the subsequent insertion of fixation screws. A 4.8-mm circular craniotomy was performed (centered on the sagittal suture midway between bregma and lambda) without disrupting the underlying dura or superior sagittal sinus. A Leur-Loc syringe hub was then cut away from a 20-gauge needle (Becton-Dickinson, Franklin Lakes, NJ) and affixed to the craniotomy site using cyanoacrylate. After confirming the integrity of the seal between the hub and the skull, two fixation screws (round machine screws; 3/16 inch long) were inserted into the 1-mm holes. Dental acrylic (methyl-methacrylate; Hygienic Corp., Akron, OH) was then applied over the screws and around the hub to provide stability during the induction of injury. After the dental acrylic hardened, the scalp was sutured closed over the hub, topical lidocaine ointment was applied, and the animal was extubated and monitored until ambulatory (approximately 60-90 minutes). For injury induction, animals were re-anesthetized with 4% isoflurane. The incision was reopened and the male end of a spacing tube was inserted into the injury hub. The female end of the spacer-hub assembly, filled with normal saline, was attached to the male end of the fluid percussion device (Custom Design and Fabrication, Virginia Commonwealth University, Richmond, VA). An injury of moderate severity (2.01-2.04 atmospheres) was administered by releasing the pendulum onto the fluid-filled piston before reflexive responses returned (26). After injury, the injury hub assembly was removed en bloc; gel foam (Pharmacia, Kalamazoo, MI) was applied extradurally to all rats to control any local bleeding; the incision was sutured and topical lidocaine and bacitracin ointments were applied to the wound. Animals were monitored for spontaneous respiration and the return of the following reflexes: toe pinch, tail pinch, corneal blink, pinnal, and righting. After recovery of the righting reflex, animals were placed in a warmed holding cage to ensure the maintenance of normothermia and monitored during recovery before being returned to the vivarium. Identical surgical procedures were followed for sham-injured animals, barring the induction of the injury.
Experiments were conducted in accordance with National Institutes of Health and Virginia Commonwealth University institutional guidelines concerning the care and use of laboratory animals (Institutional Animal Care and Use Committee).
SBDP145 Fragment-Specific Antibody Generation
For analysis of immunoblots and immunocytochemistry, a novel second alpha-II-spectrin SBDP145 cleavage site (G*SAHEVQ) fragment-specific antibody was raised in rabbit (polyclonal) and affinity-purified using a method reported in previous studies (27, 28) (Fig. 1).
Figure 1.

Fragment-specific SBDP145 Antibody tool characterization. Naïve rat brain lysate (10 μg) was either undigested (1) or digested with rat calpain-2 (1/50 ratio) (2) or with recombinant human caspase-3 (1/50 ratio) (3) for 30 minutes. The samples were subjected to SDS-PAGE, immunoblotting probed with either mouse anti-total alpha-II-spectrin (left) or rabbit anti-SBDP145 (right) and substrate development. In addition to intact alpha-II-spectrin, calpain-generated SBDP150 and SBDP145 fragments and caspase-generated SBDP150i and SBDP120 fragments are demonstrated.
Immunocytochemistry
Following injury, animals were allowed to recover for times varying from 3 hours to 7 days (n = 3-4/time point). At the predetermined times, the rats were injected i.p. with an overdose of sodium pentobarbital and then transcardially perfused with 4% paraformaldehyde and 0.1% glutaraldehyde in Millonig's buffer for immunohistochemistry. After perfusion, the brains were removed and coronally blocked at the optic chiasm and midbrain to include the parietal and temporal cortices, hippocampus, and thalamus. The blocked brains were then post-fixed in the perfusion fixative for 24 hours. For tissue sectioning, the brain blocks were flat mounted with cyanoacrylate and embedded in agar. Blocks were then coronally sectioned in 0.1 M phosphate buffer with a Leica VT1000S (Leica Microsystems, Bannockburn, IL) at a thickness of 40 μm. Sections were serially collected in alternating wells, such that each well contained adjacent sections that, after immunocytochemical processing, could be compared to permit spatiotemporal characterization of calpain mediated spectrin proteolysis. The tissue was stored in Millonig's buffer in 12-well culture plates (Falcon, Newark, DE). To identify and characterize calpain-mediated spectrin proteolysis following TBI, parallel light microscopic (LM) and electron microscopic (EM) immunocytochemical studies were performed using an antibody to the 145-kDa spectrin breakdown product. To this end, endogenous peroxidase activity was blocked with 0.5% H2O2 in PBS for 30 minutes. Sections were then processed using a temperature-controlled microwave antigen retrieval approach described previously (29). After microwave antigen retrieval, sections were pre-incubated for 45 minutes in 10% normal goat serum (NGS; Vector, Burlingame, CA) with 0.2% Triton X-100 in PBS. The tissue was then incubated overnight in a 1:1000 dilution of rabbit anti-SBDP145-fragment specific antibody (see above) in 1% NGS in PBS. Following several washes, sections were incubated for 1 hour in biotinylated goat anti-rabbit secondary antibody (IgG; Vector) diluted 1:1000 in 1% NGS in PBS and then for 1 hour in a 1:200 dilution of an avidin-horseradish peroxidase complex (ABC Standard Elite Kit, Vector). The reaction product was visualized with 0.05% diaminobenzidine, 0.01% hydrogen peroxide, and 0.3% imidazole in 0.1 M phosphate buffer for 10 minutes. Sections were mounted on gelatin-coated slides, dehydrated, and coverslipped. All qualitative light microscopic analyses and capture were performed using a Nikon Eclipse 800 (Tokyo, Japan) fitted with an Olympus DP71 digital camera (Tokyo, Japan).
Electron Microscopy
Selected SBDP145-labeled sections from animals allowed to survive for 24 hours post-injury (n = 4) were subjected to further processing for EM analysis in order to ascertain the precise cellular localization of calpain-mediated spectrin proteolysis. The tissue was osmicated, dehydrated, and flat embedded between plastic slides in medcast resin (Ted Pella, Redding, CA). The embedded slides were then scanned to identify SBDP145-positive regions. Once identified, these sites were removed, mounted on plastic studs, and thick sectioned to the depth of the immunoreactive sites of interest. Serial 70-nm sections were then cut and picked up on Formvar-coated, slotted grids. The grids were then stained in 5% uranyl acetate in 50% methanol for 2 minutes and in 0.5% lead citrate for 1 min. Ultrastructural analysis was performed using a JEOL 1200 electron microscope (Tokyo, Japan).
Western Blot
A separate population of animals was used for Western blot analyses. Based on our previous observations of scattered, delayed axotomy in the neocortex, thalamus and hippocampus of diffusely injured animals, our analyses were confined to these brain regions. At various survival time points (3 hours to 7 days) following central FPI or sham-injury, rats (n = 6-8/time point) were anesthetized for 4 minutes with 4% isoflurane in 70% N2O/30% O2. Animals were then quickly decapitated and their brains were removed and placed on an ice-cold marble-dissecting block for rapid subregion isolation. Specifically, each brain was blocked into 1-mm coronal sections and the mediodorsal neocortex, hippocampus, and thalamus were bilaterally isolated, rinsed in ice cold PBS, snap-frozen in liquid nitrogen and frozen at -80°C until further use. For Western blot analysis, the brain samples were pulverized to a fine powder with a small mortar and pestle set over dry ice. The pulverized brain tissue was then lysed for 90 minutes at 4°C with 50 mM Tris (pH 7.4), 5 mM EDTA, 1% (v/v) Triton X-100, 1 mM DTT, and 1x protease inhibitor cocktail (Roche Biochemicals). The brain lysates were then centrifuged at 8000 x g for 5 minutes at 4°C to clear and remove insoluble debris, 10 μl of each sample were collected for protein assay and the remaining sample was snap-frozen and stored at -80°C until further use. Total protein concentration in each supernatant was then determined via the Bradford method (Bio-Rad, Hercules, CA) using duplicate samples of each supernatant assayed in triplicate. Samples of the specific region of interest from each animal were then reduced and prepared for SDS-PAGE by mixing in a 1:1 mixture with Laemmli's sample buffer (Bio-Rad) and 5% β-mercaptoethanol (Bio-Rad) followed by heating to 70°C for 10 minutes. Twenty μg of each sample were loaded in separate lanes onto 4% to 20% Tris-HCl gels (Bio-Rad) and electrophoresed at a constant voltage of 10 0mV for 90 minutes. The proteins were then transferred to a nitrocellulose membrane (0.45 μm pore size; Bio-Rad) and gels were subsequently stained for total protein with Coomassie Blue (Sigma) to ensure complete and equal transfer. After three 10 minutes rinses in Tris-buffered saline (TBS), blots were incubated for 1 hour in 5% nonfat dry milk in TBS with 1% Tween 20 (TBST). Blots were then incubated overnight in a primary antibody solution containing 5% milk in TBST and one of the following primary antibodies: mouse anti-fodrin (1:10,000, Biomol International, Plymouth Meeting, PA) or rabbit anti-SBDP145 fragment specific antibody (1:10,000). Following three-10 minute rinses in TBST, blots were incubated in a 1:10,000 dilution of secondary antibody for 90 minutes (horseradish peroxidase-conjugated rabbit anti-mouse secondary antibody, Santa Cruz Biotechnology, Santa Cruz, CA, or horseradish peroxidase-conjugated goat anti-rabbit secondary antibody, Invitrogen, Carlsbad, CA). Blots were then rinsed again 3 times for 10 minutes in TBS and immunoreactive bands were identified using an enhanced chemiluminescence system (Amersham Biosciences, Piscataway, NJ). To control for loading, membranes were stripped with Restore Western Blot Stripping Buffer (Pierce, Rockford, IL) and re-probed for cyclophilin B at a dilution of 1:10,000 (Abcam, Cambridge, MA). Densitometry was performed (NIH Image) to quantitate the optical density of each band and to detect any injury-induced alterations in spectrin proteolytic breakdown products relative to sham-injured animals. Data were analyzed by ANOVA with a Tukey-Kramer post hoc with significance deemed at p < 0.05.
CSF Collection and SBDP145 Sandwich ELISA Analysis of CSF Samples
Twenty-four hours following midline FPI or sham-injury, rats (n = 5/group) were anesthetized, intubated, and maintained on a ventilator as described above (see surgical preparation) and positioned in a stereotactic frame with the head allowed to move freely along the longitudinal axis. The head was flexed in a manner causing the external occipital protuberance in the neck to be prominent and a dorsal midline incision was made over the cervical vertebrae and occiput. The atlanto-occipital membrane was exposed by blunt dissection and a 25-G needle (Becton-Dickinson) fixed to the stereotactic frame and attached to polyethylene tubing (PE50, Becton-Dickinson) was carefully lowered into the cisterna magna to collect approximately 0.1 to 0.15 mL of CSF from each rat. Following CSF collection, animals were removed from the stereotactic frame and immediately processed for either immunohistochemical or Western blot analyses as described above. CSF samples were centrifuged at 4000 x g for 7 minutes at 4°C to clear any contaminating erythrocytes and cleared CSF was then stored at -80° C until ready for use.
For SBDP145 sandwich ELISA, 96-well plate coating 100 μL/well capture antibody (500 ng/well purified rabbit polyclonal anti-SBDP145-fragment specific antibody) in 0.1 M sodium bicarbonate, pH 9.2 was added to 96-well Corning plate and incubated overnight at °C. The plates were then emptied and 300 μl/well of blocking buffer (Startingblock T20-TBS) was added and incubated for 30 minutes at ambient temperature with gentle shaking. This was followed by addition of antigen standard (recombinant GST-fusion-αII-spectrin (repeat 9 to 15) cleaved with calpain-2 (1/50 ratio) for 30 minutes) to establish a standard curve. Stock solutions of 0.5 ng/mL to 5000 ng/mL of prepared SBDP145 protein (0.005 ng-50 ng in 10 μL) were diluted 1/10 with sample diluent to a final incubation volume of 100 μL/well. The standard curve range was 0.05 ng/mL to 500 ng/mL in the wells. For rat CSF samples, 10 μL was typically used per well and diluted 1/10 with diluent to a final incubation volume of 100 μL/well. (To adjust for the 1/10 dilution, the calculated sample SBDP145 concentration (ng/mL) in the well has been multiplied by a factor of 10 to express the actual rat CSF SBDP145 levels (ng/mL). The plate was incubated for 2 hours at room temperature, followed by washing with automatic plate washer. If amplification was needed, biotinyl-tyramide solution (Perkin Elmer Elast Amplification Kit) was added for 15 minutes at room temperature, washed and followed by 100 μL/well Streptavidin-HRP (1:500) in PBS with 0.02% Tween-20 and 1% BSA for 30 minutes followed by washing. Lastly, the wells were developed with 100 μl/well TMB substrate solution (Ultra-TMB ELISA, Pierce# 34028) with incubation for 5 to 30 minutes and read at 652 nm with a 96-well spectrophotometer (Molecular Device Spectramax 190).
RESULTS
General Physiological Findings
Both sham controls and animals that sustained TBI displayed no alteration in body temperature. All brain-injured animals displayed transient unconsciousness and had righting reflex recovery times greater than 6 minutes (and less than 10 minutes) compared to less than 1 minute for sham-injured animals. No significant differences in transient unconsciousness were noted among injury groups indicating that all groups received injuries of comparable severity (data not shown).
Light Microscopic Immunocytochemistry
All sections processed for LM immunohistochemical analyses revealed macroscopic and microscopic changes consistent with those described with moderate central FPI (24, 26). Specifically, all brain sites assessed exhibited no evidence of contusional change or cavitation and the brain parenchyma was devoid of hemorrhage other than isolated petechial hemorrhages in the corpus callosum.
Immunostaining for SBDP145 revealed widespread increases in immunoreactivity throughout the neocortex, subcortical white matter, thalamus and hippocampus following TBI (Figs. 2-5). Little to no SBDP145-immunoreactivity was observed in sham animals, whereas in TBI animals at 3 hours post-injury there was detectable SBDP145 immunoreactivity in the neocortex, subcortical white matter and, to a lesser extent, the thalamus and hippocampus. More robust staining was seen within these brain regions by 24 hours following injury, with the immunoreactivity peaking at 24 to 48 hours post-injury. In the neocortex, SBDP145-immunoreactivity was diminished by 7 days post-injury (Fig. 2), whereas it persisted in the thalamus and, to a lesser extent, the hippocampus at 7 days post-injury, albeit at decreased levels (Figs. 3, 4). SDBP145-immunoreactivity was also markedly enhanced in white matter tracts throughout the brain of diffusely injured animals, including the corpus callosum, internal and external capsules, cingulum, fimbria, cerebellum and brainstem (Fig. 5 and data not shown). Comparatively low SBDP145 immunoreactivity was observed in the striatum, caudate putamen and globus pallidus at all time points assessed, suggesting that there are either regional differences in the vulnerability of the brain to injury-induced calpain-mediated spectrin proteolysis (CMSP) or that varying levels of force are applied to different brain regions in this diffuse/widespread injury model.
Figure 2.

Immunohistochemical detection of calpain-mediated alpha-II-spectrin proteolysis (CMSP) in the neocortex following diffuse brain injury. Using an antibody that targets the 145-kDa calpain-cleavage product of alpha-II-spectrin, there are a marked increases in calpain-mediated spectrin proteolysis is identified in the neocortex following diffuse brain injury. (A) Little to no SBDP145 immunoreactivity is observed in the cortex of sham animals. (B) SBDP145 immunoreactivity is detectable by 3 hours post-injury, particularly at the gray-white matter interface. (C, D) Levels of calpain-mediated spectrin proteolysis appear to peak at 24 hours (C) to 48 hours (D) post-injury; they were diminished to sham levels by 7 days post-injury (not shown). The majority of SBDP145-immunoreactivity appears to be localized to axons and axonal debris in the neocortex and subcortical white matter, but scattered somatic expression is also observed (B, inset). Scale bar = 20 μm.
Figure 5.

Calpain-mediated alpha-II-spectrin proteolysis (CMSP) in white matter tracts following diffuse brain injury. (A-D) SBDP145 immunocytochemistry reveals extensive immunoreactivity localized to axons and axonal debris in the internal capsule (A), external capsule (B), brainstem white matter tracts (C) and cingulum (D) at 24 hours post-injury. Scale bars = 50 μm for (D) and 20 μm for (A-C).
Figure 3.

Prolonged calpain-mediated alpha-II-spectrin proteolysis (CMSP) in the thalamus following diffuse brain injury. Immunocytochemistry demonstrates increased levels of SBD145 in the thalamus. (A) Relatively little SBDP145-immunoreactivity is observed in the thalamus of sham animals. (B, C) There is robust SBDP145 staining throughout the thalamus by 24 hours (B) and 48 hours (C) post-injury. (D) CMSP staining remained elevated albeit at apparently decreased levels from peak at 7 days post-injury. The majority of SBDP145 immunoreactivity appears localized to axons within the thalamus. (E-G) Neuronal somatic immunostaining, with particularly intense somatic staining in neurons of the thalamic reticular nucleus is also observed at 3 hours (E), 24 hours (F) and 7 days (G) post-injury. Scale bar = 50 μm.
Figure 4.

Calpain-mediated alpha-II-spectrin (CMSP) proteolysis in the hippocampus following diffuse brain injury. Immunostaining for the 145-kDa breakdown product of alpha-II-spectrin demonstrates enhanced levels of spectrin proteolysis in the hippocampus. (A) Little to no SBDP145 immunoreactivity is observed in the hippocampus of sham animals. (B) Isolated neuronal somatic SBDP145-immunoreactivity is seen in the granule cell layer of the dentate gyrus at 3 hours post-injury. (C) Immunostaining is widespread throughout the hippocampus at 24 hours and later time points post-injury (not shown). Staining is primarily localized to axons and axonal debris. dg = dentate gyrus. Scale bar = 50 μm
In addition to providing a qualitative indication of the spatiotemporal pattern of CMSP following diffuse brain injury, LM analysis also revealed that the observed SBDP145 immunoreactivity was localized primarily to damaged axons and their associated swellings as well as the debris fields of degenerating axons (Fig. 6A, B). In addition to axonal labeling, isolated somatic SBDP145 labeling was also observed in the neocortex (Fig. 2B, insert), hippocampus (Fig. 4B) and thalamus, with particularly intense somatic immunoreactivity in neurons of the thalamic reticular nucleus (Fig. 3E-G). In contrast to the axonal labeling, however, the scattered neuronal somata staining positive for SBDP145 did not display any overt evidence of neuronal damage or death.
Figure 6.

Routine light microscopy (LM) and electron microscopy (EM) confirm axonal localization of calpain-mediated spectrin proteolysis (CMSP). (A, B) High magnification LM images of SBDP145 immunostaining in the thalamus suggest localization of CMSP to damaged axons and associated axonal swellings. The linear orientation of SBDP145 immunoreactivity in B suggests debris fields of degenerating axons. (C-E) SBDP145-immunostained sections processed for EM confirm localization of immunoreactivity in damaged axons and associated axonal swellings. Electron dense SBDP145-immunoreactivity (arrows) in a representative photomicrograph of a damaged thalamic axon exhibits a disrupted myelin sheath and cytoskeletal perturbation (C). Damaged axons display more mature bulb-like axon swellings characterized by pooling of intra-axonal organelles (D, E). Note the presence of SBDP145-immunoreactivity (arrows) within these swellings. Scale bar = 20 μm in A, B, 1 μm in C; 2 μm in D and E.
Ultrastructural Analysis
Ultrastructural analysis of SBDP145-immunostained sections provided confirmation that the immunoreactivity was localized primarily in damaged axons and their associated axonal swellings (Fig. 6). SBDP145-positive reaction product was observed in damaged axons throughout the neocortex and thalamus. These axons exhibited a disruption of their myelin sheath and/or cytoskeletal perturbation consistent with traumatically induced axonal damage as described in detail elsewhere (3). SBDP145 immunoreactivity was also observed in damaged axons displaying bulb-like axonal swellings characterized by pooling of intra-axonal organelles, a finding consistent with traumatically induced alteration (3). As at the LM level, SBDP145-labeled neuronal somata revealed no ultrastructural evidence of neuronal damage or death.
Western Blot Findings
Western blot analysis confirmed and extended the immunohistochemical findings by providing semiquantitative information on the spatiotemporal pattern of CMSP in the diffusely injured brain. Using antibodies to either intact alpha-II-spectrin or its 145-kDa calpain cleavage product, significant increases in calpain-mediated spectrin proteolysis were observed following injury in the neocortex, thalamus and hippocampus (Fig. 7). Similar to immunohistochemical observations, significantly heightened levels of calpain-mediated proteolysis were observed in the neocortex at 3, 24 and 48 hours post-injury before returning to basal levels by 7 days. Significantly enhanced levels of SBDP145 were observed at 24 and 48 hours post-injury in both the thalamus and hippocampus and, in contrast to the cortex, these levels remained significantly elevated by 7 days post-injury.
Figure 7.

Spatiotemporal pattern of calpain-mediated alpha-II-spectrin proteolysis (CMSP) following diffuse brain injury. Western blot analyses using an anti = SBDP145 reveal significant increases in calpain-mediated spectrin proteolysis in the neocortex (A, D), thalamus (B, E) and hippocampus (C, F) following diffuse brain injury. Representative immunoblots and densitometric measurements of SBDP145 (normalized to both sham and cyclophilin B load control values) are shown graphically and reveal significantly heightened levels of CMSP in the neocortex at 3 hours, 24 hours and 48 hours post-injury before returning to basal levels by 7 days. Significantly enhanced levels of SBDP145 are also observed at 24 hours and 48 hours post-injury in both the thalamus and hippocampus. In contrast to the cortex, SBDP145 levels remained elevated by 7 days post-injury in the latter two regions. *p < 0.05 ± SEM.
SBDP145 CSF ELISA
We next sought to determine whether these observations would be reflected in the CSF of the diffusely injured rats. ELISA targeting the 145-kDa calpain-mediated degradation product of alpha-II-spectrin revealed significantly heightened levels of SBDP145 in the CSF of injured animals at 24 hours post-injury (Fig. 8). Sham animals exhibited negligible concentrations of SBDP145 in the CSF (1.44 ± 0.45 ng/mL), whereas animals subjected to diffuse brain injury had a slight but significant increase in CSF spectrin breakdown levels (6.16 ± 0.92 ng/mL), which coincided with the peak elevation in parenchymal SBDP 145 immunoreactivity at the 24 hours survival time point.
Figure 8.
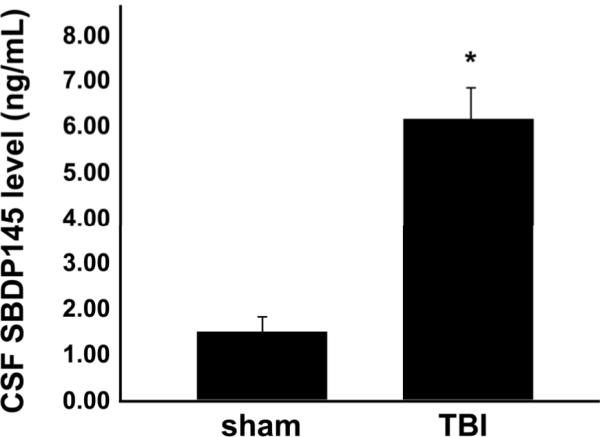
SBDP145 levels are increased in the CSF of diffusely injured animals. An ELISA targeting the 145-kDa calpain-mediated degradation product of alpha-II-spectrin reveals significantly enhanced SBDP145 levels in the CSF of injured animals (6.16 ± 0.92 ng/mL) compared to sham animals (1.44 ± 0.45 ng/mL) at 24 hours post-injury. *p < 0.05 ± SEM.
DISCUSSION
CMSP has been implicated as a key event in the pathological processes that follow TBI (6-13). The current study confirms these processes operate in an experimental model of diffuse/widespread brain injury uncomplicated by contusion and its related ischemic effects. We used several techniques to demonstrate CMSP throughout the brain parenchyma following moderate, diffuse TBI and assessed spectrin proteolytic cleavage products in the CSF of the injured animals. Our tissue sampling protocols allowed direct comparisons between regionally specific Western blot findings and immunohistochemical analyses that could then be compared to the more global ELISA evaluations of CSF SBDP145 levels. Taken together, our observations add to a growing body of experimental literature aimed at clarifying further the role of spectrin proteolysis in the diffusely injured brain (18, 30-32). They also indicate the potential utility of SBDPs as biomarkers for this form of brain injury, which is particularly relevant to human TBI, most of which involves mild to moderate insult with a significant diffuse injury component (1).
Increases in immunoreactivity for the 145-kDa spectrin breakdown product were observed following FPI in the neocortex, subcortical white matter, hippocampus and thalamus; the time course of spectrin proteolysis varied slightly among the regions. While CMSP peaked between 24 and 48 hours in the regions studied, there was a return to basal levels in the neocortex by day 7 post-injury; in contrast, heightened SBDP145 immunoreactivity persisted in the hippocampus and, to a greater extent, the thalamus up to 7 days post-injury. This pattern is consistent with previous findings in severely brain-injured animals manifesting significant contusional change (9, 14, 15, 22, 23, 33-35) as well as in investigations employing mild to moderate diffuse injury in which the animals exhibit more subtle and less frequent contusional change (18, 30-32). In these previous reports, brain injured animals demonstrated enhanced levels of CMSP as early as 15 minutes post-injury that persisted up to several weeks following injury; peak levels were typically observed between 24 and 48 hours post-insult. Despite similarities in the time course, however, the increased CMSP observed following brain injuries associated with contusion is more pronounced than that following a moderate, diffuse brain injury, a finding consistent with the magnitude of destruction caused by contusion injuries.
Previous investigations into calpain activation and subsequent spectrin proteolysis in the injured brain (including both contusional and non-contusional domains) have largely associated these proteolytic processes with overt somatic damage and/or necrotic cell death of neurons in the contusional and peri-contusional regions (9, 14, 22, 23). By contrast, in the current study we found more evidence of axonal injury; results that are consistent with a growing body of experimental literature in which greater emphasis is placed on nerve fiber involvement (9, 13, 30, 36-38). With the exception of observing rare somatic localization, we found predominant axonal localization of SBDP145 immunoreactivity that was specifically related to damaged axons, their associated swellings and fields of axonal debris. These observations are in agreement with our previous findings of CMSP in damaged brain stem axons following diffuse brain injury as well as in human TBI cases that have shown SBDPs in damaged axons of the corpus callosum (13, 16). Despite the scattered somatic SBDP145 immunoreactivity in the current study, routine LM and EM analyses of the positively labeled neurons revealed no overt evidence of neuronal damage/death. Similarly, we previously reported CMSP-positive neocortical neurons that exhibited limited ultrastructural change and no evidence of necrotic damage in a different experimental model of diffuse brain injury (39). Taken together, these observations highlight the need for caution in interpreting the finding of elevated CMSP and its overall implications for neuronal injury and death. Further, the current study points toward a more significant role for calpain-mediated spectrin proteolysis in traumatic axonal injury and warrants further investigation of activated calpain as a potential therapeutic target in diffuse TBI (31, 38, 40).
We sought to determine whether parallel alterations in SBDP145 levels could be observed in the CSF of the brain injured animals. The increase of SBDP145 in the CSF of diffusely injured rats paralleled its increased expression in the brain parenchyma at 24 hours post-injury; this is consistent with previous studies in both contusion-based and diffuse experimental brain injury models (18, 41, 42). While alterations in SBDP levels in the CSF following contusive brain injury are more pronounced than those following a diffuse brain injury (41, 42 and unpublished observations), the current observation points toward the potential utility of SBDPs as biomarkers for detecting TBI of varying severity. In human studies, calpain-mediated SBDP levels in the CSF have correlated with initial severity of injury and clinical outcome (20). In addition to the experimental and clinical evidence, the potential utility of SBDPs as biomarkers is also supported by the fact that CSF samples are objectively quantifiable and alpha-II-spectrin expression is specific to the brain, therefore eliminating the potential confound of blood contamination (41). The levels and types of SBDPs (i.e. calpain vs. caspase-mediated) in the CSF may also provide insight into injury severity, type of injury (i.e. focal vs. diffuse), and the biochemical cascades/underlying pathological mechanisms, (i.e. apoptosis vs. necrosis). Thus, the information provided by assessing SBDPs in the CSF could guide treatment strategies and contribute to prediction of outcome. The potential utility of surrogate biomarkers of TBI is particularly relevant in cases of diffuse brain injury in which conventional imaging techniques often fail to reveal the full extent of diffuse axonal injury (43). To this end, ongoing studies in our lab are aimed at further investigating the effects of diffuse brain injury on SPDP levels in the CSF and working to develop serum-based assays sufficiently sensitive to detect SBDPs in the serum of injured experimental animals and humans.
Collectively, the current findings confirm the widespread occurrence of calpain-mediated proteolysis following diffuse brain injury uncomplicated by contusion. Moreover, these results strongly support the potential utility of spectrin proteolytic cleavage products as surrogate markers in the clinical monitoring of patients with diffuse brain injury.
ACKNOWLEDGMENTS
The authors gratefully acknowledge Lynn Davis, Susan Walker and Judy Williamson for their skilled technical support. KKW and RLH hold equity in Banyan Biomarkers, Inc., a company commercializing technology for detecting brain injury biomarkers.
This work was supported by National Institute of Health grants 5R01HD055813-25 (JTP), 5P30NS047463-05 (JTP) and R01 NS049175-01 A1 (KKW) and Department of Defense grants DAMD17-03-1-0066 (RLH) and DAMD17-01-1-0765 (RLH).
REFERENCE
- 1.Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: A brief overview. J Head Trauma Rehabil. 2006;21:375–78. doi: 10.1097/00001199-200609000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Bramlett HM, Dietrich WD. Progressive damage after brain and spinal cord injury: Pathomechanisms and treatment strategies. Prog Brain Res. 2007;161:125–41. doi: 10.1016/S0079-6123(06)61009-1. [DOI] [PubMed] [Google Scholar]
- 3.Buki A, Povlishock JT. All roads lead to disconnection?--Traumatic axonal injury revisited. Acta Neurochir (Wien) 2006;148:181–93. doi: 10.1007/s00701-005-0674-4. [DOI] [PubMed] [Google Scholar]
- 4.Farkas O, Povlishock JT. Cellular and subcellular change evoked by diffuse traumatic brain injury: A complex web of change extending far beyond focal damage. Prog Brain Res. 2007;161:43–59. doi: 10.1016/S0079-6123(06)61004-2. [DOI] [PubMed] [Google Scholar]
- 5.Povlishock JT, Katz DI. Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil. 2005;20:76–94. doi: 10.1097/00001199-200501000-00008. [DOI] [PubMed] [Google Scholar]
- 6.Vanderklish PW, Bahr BA. The pathogenic activation of calpain: A marker and mediator of cellular toxicity and disease states. Int J Exp Pathol. 2000;81:323–39. doi: 10.1111/j.1365-2613.2000.00169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Czogalla A, Sikorski AF. Spectrin and calpain: A `target' and a `sniper' in the pathology of neuronal cells. Cell Mol Life Sci. 2005;62:1913–24. doi: 10.1007/s00018-005-5097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu J, Liu MC, Wang KK. Calpain in the CNS: From synaptic function to neurotoxicity. Sci Signal. 2008;1:tr3. doi: 10.1126/stke.114re1. [DOI] [PubMed] [Google Scholar]
- 9.Saatman KE, Bozyczko-Coyne D, Marcy V, et al. Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol. 1996;55:850–60. doi: 10.1097/00005072-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 10.Siman R, Baudry M, Lynch G. Brain fodrin: Substrate for calpain I, an endogenous calcium-activated protease. Proc Natl Acad Sci USA. 1984;81:3572–76. doi: 10.1073/pnas.81.11.3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siman R, Noszek JC. Excitatory amino acids activate calpain I and induce structural protein breakdown in vivo. Neuron. 1988;1:279–87. doi: 10.1016/0896-6273(88)90076-1. [DOI] [PubMed] [Google Scholar]
- 12.Roberts-Lewis JM, Siman R. Spectrin proteolysis in the hippocampus: A biochemical marker for neuronal injury and neuroprotection. Ann NY Acad Sci. 1993;679:78–86. doi: 10.1111/j.1749-6632.1993.tb18290.x. [DOI] [PubMed] [Google Scholar]
- 13.Buki A, Siman R, Trojanowski JQ, et al. The role of calpain-mediated spectrin proteolysis in traumatically induced axonal injury. J Neuropathol Exp Neurol. 1999;58:365–75. doi: 10.1097/00005072-199904000-00007. [DOI] [PubMed] [Google Scholar]
- 14.Newcomb JK, Kampfl A, Posmantur RM, et al. Immunohistochemical study of calpain-mediated breakdown products to alpha-spectrin following controlled cortical impact injury in the rat. J Neurotrauma. 1997;14:369–83. doi: 10.1089/neu.1997.14.369. [DOI] [PubMed] [Google Scholar]
- 15.Pike BR, Zhao X, Newcomb JK, et al. Regional calpain and caspase-3 proteolysis of alpha-spectrin after traumatic brain injury. Neuroreport. 1998;9:2437–42. doi: 10.1097/00001756-199808030-00002. [DOI] [PubMed] [Google Scholar]
- 16.McCracken E, Hunter AJ, Patel S, et al. Calpain activation and cytoskeletal protein breakdown in the corpus callosum of head-injured patients. J Neurotrauma. 1999;16:749–61. doi: 10.1089/neu.1999.16.749. [DOI] [PubMed] [Google Scholar]
- 17.Pineda JA, Wang KK, Hayes RL. Biomarkers of proteolytic damage following traumatic brain injury. Brain Pathol. 2004;14:202–209. doi: 10.1111/j.1750-3639.2004.tb00054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siman R, McIntosh TK, Soltesz KM, et al. Proteins released from degenerating neurons are surrogate markers for acute brain damage. Neurobiol.Dis. 2004;16:311–20. doi: 10.1016/j.nbd.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 19.Farkas O, Polgar B, Szekeres-Bartho J, et al. Spectrin breakdown products in the cerebrospinal fluid in severe head injury--preliminary observations. Acta Neurochir (Wien) 2005;147:855–61. doi: 10.1007/s00701-005-0559-6. [DOI] [PubMed] [Google Scholar]
- 20.Pineda JA, Lewis SB, Valadka AB, et al. Clinical significance of alphaII-spectrin breakdown products in cerebrospinal fluid after severe traumatic brain injury. J Neurotrauma. 2007;24:354–366. doi: 10.1089/neu.2006.003789. [DOI] [PubMed] [Google Scholar]
- 21.Cardali S, Maugeri R. Detection of alphaII-spectrin and breakdown products in humans after severe traumatic brain injury. J Neurosurg Sci. 2006;50:25–31. [PubMed] [Google Scholar]
- 22.Deng Y, Thompson BM, Gao X, et al. Temporal relationship of peroxynitrite-induced oxidative damage, calpain-mediated cytoskeletal degradation and neurodegeneration after traumatic brain injury. Exp Neurol. 2007;205:154–65. doi: 10.1016/j.expneurol.2007.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hall ED, Sullivan PG, Gibson TR, et al. Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: More than a focal brain injury. J Neurotrauma. 2005;22:252–65. doi: 10.1089/neu.2005.22.252. [DOI] [PubMed] [Google Scholar]
- 24.Kelley BJ, Lifshitz J, Povlishock JT. Neuroinflammatory responses after experimental diffuse traumatic brain injury. J Neuropathol Exp Neurol. 2007;66:989–1001. doi: 10.1097/NEN.0b013e3181588245. [DOI] [PubMed] [Google Scholar]
- 25.Sullivan HG, Martinez J, Becker DP, et al. Fluid-percussion model of mechanical brain injury in the cat. J Neurosurg. 1976;45:521–34. [PubMed] [Google Scholar]
- 26.Dixon CE, Lyeth BG, Povlishock JT, et al. A fluid percussion model of experimental brain injury in the rat. J Neurosurg. 1987;67:110–19. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- 27.Nath R, Huggins M, Glantz SB, et al. Development and characterization of antibodies specific to caspase-3-produced alpha II-spectrin 120 kDa breakdown product: marker for neuronal apoptosis. Neurochem Int. 2000;37:351–61. doi: 10.1016/s0197-0186(00)00040-1. [DOI] [PubMed] [Google Scholar]
- 28.Wang KK. Calpain and caspase: Can you tell the difference? Trends Neurosci. 2000;23:20–26. doi: 10.1016/s0166-2236(99)01536-2. [DOI] [PubMed] [Google Scholar]
- 29.Stone JR, Walker SA, Povlishock JT. The visualization of a new class of traumatically injured axons through the use of a modified method of microwave antigen retrieval. Acta Neuropathol. 1999;97:335–45. doi: 10.1007/s004010050996. [DOI] [PubMed] [Google Scholar]
- 30.Huh JW, Widing AG, Raghupathi R. Basic science; repetitive mild non-contusive brain trauma in immature rats exacerbates traumatic axonal injury and axonal calpain activation: a preliminary report. J Neurotrauma. 2007;24:15–27. doi: 10.1089/neu.2006.0072. [DOI] [PubMed] [Google Scholar]
- 31.Kupina NC, Nath R, Bernath EE, et al. The novel calpain inhibitor SJA6017 improves functional outcome after delayed administration in a mouse model of diffuse brain injury. J Neurotrauma. 2001;18:1229–40. doi: 10.1089/089771501317095269. [DOI] [PubMed] [Google Scholar]
- 32.Kupina NC, Detloff MR, Bobrowski WF, et al. Cytoskeletal protein degradation and neurodegeneration evolves differently in males and females following experimental head injury. Exp Neurol. 2003;180:55–73. doi: 10.1016/s0014-4886(02)00048-1. [DOI] [PubMed] [Google Scholar]
- 33.Aikman J, O'Steen B, Silver X, et al. Alpha-II-spectrin after controlled cortical impact in the immature rat brain. Dev Neurosci. 2006;28:457–65. doi: 10.1159/000094171. [DOI] [PubMed] [Google Scholar]
- 34.Thompson SN, Gibson TR, Thompson BM, et al. Relationship of calpain-mediated proteolysis to the expression of axonal and synaptic plasticity markers following traumatic brain injury in mice. Exp Neurol. 2006;201:253–65. doi: 10.1016/j.expneurol.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 35.Huh JW, Franklin MA, Widing AG, et al. Regionally distinct patterns of calpain activation and traumatic axonal injury following contusive brain injury in immature rats. Dev Neurosci. 2006;28:466–76. doi: 10.1159/000094172. [DOI] [PubMed] [Google Scholar]
- 36.Serbest G, Burkhardt MF, Siman R, et al. Temporal profiles of cytoskeletal protein loss following traumatic axonal injury in mice. Neurochem Res. 2007;32:2006–14. doi: 10.1007/s11064-007-9318-9. [DOI] [PubMed] [Google Scholar]
- 37.Saatman KE, Abai B, Grosvenor A, et al. Traumatic axonal injury results in biphasic calpain activation and retrograde transport impairment in mice. J Cereb Blood Flow Metab. 2003;23:34–42. doi: 10.1097/01.WCB.0000035040.10031.B0. [DOI] [PubMed] [Google Scholar]
- 38.Ai J, Liu E, Wang J, et al. Calpain inhibitor MDL-28170 reduces the functional and structural deterioration of corpus callosum following fluid percussion injury. J Neurotrauma. 2007;24:960–78. doi: 10.1089/neu.2006.0224. [DOI] [PubMed] [Google Scholar]
- 39.Farkas O, Lifshitz J, Povlishock JT. Mechanoporation induced by diffuse traumatic brain injury: An irreversible or reversible response to injury? J Neurosci. 2006 Mar 22;26:3130–40. doi: 10.1523/JNEUROSCI.5119-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buki A, Farkas O, Doczi, et al. Preinjury administration of the calpain inhibitor MDL-28170 attenuates traumatically induced axonal injury. J Neurotrauma. 2003;20:261–68. doi: 10.1089/089771503321532842. [DOI] [PubMed] [Google Scholar]
- 41.Pike BR, Flint J, Dutta S, et al. Accumulation of non-erythroid alpha II-spectrin and calpain-cleaved alpha II-spectrin breakdown products in cerebrospinal fluid after traumatic brain injury in rats. J Neurochem. 2001;78:1297–1306. doi: 10.1046/j.1471-4159.2001.00510.x. [DOI] [PubMed] [Google Scholar]
- 42.Ringger NC, O'Steen BE, Brabham JG, et al. A novel marker for traumatic brain injury: CSF alphaII-spectrin breakdown product levels. J Neurotrauma. 2004;21:1443–56. doi: 10.1089/neu.2004.21.1443. [DOI] [PubMed] [Google Scholar]
- 43.Belanger HG, Vanderploeg RD, Curtiss G, et al. Recent neuroimaging techniques in mild traumatic brain injury. J Neuropsychiatry Clin Neurosci. 2007;19:5–20. doi: 10.1176/jnp.2007.19.1.5. [DOI] [PubMed] [Google Scholar]