

Summary
Production and trafficking of proteins entering the secretory pathway of eukaryotic cells is coordinated at the endoplasmic reticulum (ER) in a process that begins with protein translocation via the membrane-embedded ER translocon. The same complex is also responsible for the co-translational integration of membrane proteins and orchestrates polypeptide modifications that are often essential for protein function. We now show that the previously identified inhibitor of ER-associated degradation (ERAD) eeyarestatin 1 (ESI) is a potent inhibitor of protein translocation. We have characterised this inhibition of ER translocation both in vivo and in vitro, and provide evidence that ESI targets a component of the Sec61 complex that forms the membrane pore of the ER translocon. Further analyses show that ESI acts by preventing the transfer of the nascent polypeptide from the co-translational targeting machinery to the Sec61 complex. These results identify a novel effect of ESI, and suggest that the drug can modulate canonical protein transport from the cytosol into the mammalian ER both in vitro and in vivo.
Keywords: Cross-linking, ERAD, Protein secretion, Small molecule inhibitor
Introduction
The endoplasmic reticulum (ER) is a major site of synthesis for both membrane integrated and secretory proteins. Typically, nascent polypeptides are translocated co-translationally into or across the ER membrane via a proteinaceous membrane complex termed the ER translocon (Rapoport, 2007). Proteins destined for the ER are initially targeted from the cytosol by virtue of a hydrophobic span of amino acid residues that functions as a signal sequence and is recognised and bound by the signal recognition particle (SRP) as it emerges from the ribosome (Cross et al., 2009). SRP delivers the ribosome together with the nascent polypeptide chain to the ER translocon via a GTP-dependent interaction with the membrane-bound SRP receptor (SR) (Pool, 2005). The ER translocon pore minimally comprises one Sec61α-β-γ heterotrimer, where the crystal structure of an archaeal homologue suggests a regulated complex, capable of opening laterally to allow transmembrane (TM) segment integration (Van den Berg et al., 2004). When active, the ER translocon appears highly dynamic and is likely to exist as a dimer of Sec61α-β-γ complexes (Rapoport, 2007) together with accessory proteins required for co-translocational processing of nascent polypeptide chains (Hegde and Kang, 2008).
Subtle modulation of protein homeostasis in diseased cells, for example using small molecule inhibitors, is a promising strategy for therapeutic intervention (Balch et al., 2008). Given its central role in secretory and membrane protein biogenesis, the ER translocon is an obvious target for such approaches, yet to date few inhibitors of ER translocation that function in vivo have been identified. Two related cyclodepsipeptides, cotransin and CAM741, have been shown to bind to and block the activity of the Sec61 translocon (Besemer et al., 2005; Garrison et al., 2005; Mackinnon et al., 2007). However, this inhibitory effect appears to be highly substrate specific and only a small subset of precursor proteins are prevented from entering the ER in the presence of CAM741 (Harant et al., 2006; Harant et al., 2007).
Eeyarestatin 1 and 2 (ESI and ESII) were identified as small molecules that inhibit one or more steps in the pathway for ER-associated degradation (ERAD) (Fiebiger et al., 2004; Wang et al., 2008), potentially via an effect on the ER-associated p97 ATPase, causing a downstream block of the deubiquitylation process that facilitates such degradation (Wang et al., 2008). More recently, it has been shown that ESI induces a rapid ER stress response in mammalian cells and may have an anti-cancer activity similar to that of the proteasome inhibitor bortezomib (Wang et al., 2009). We now show that ESI acts as a potent inhibitor of co-translational protein transport across the ER membrane. This effect is apparent both in vitro and in vivo and provides a compelling molecular basis for the previously reported ESI-mediated induction of ER stress (Wang et al., 2009). We have characterised the mechanism and show that ESI prevents the transfer of nascent proteins from the membrane-targeting complex to the ER translocation machinery, most probably by inactivating the Sec61 complex. These results shed light on the physiological consequences of treating cells with ESI and provide a basis from which to more fully understand the cellular impact of this potentially useful pharmacological agent.
Results
ESI modulates protein processing at the ER of cultured mammalian cells
ESI, ESII and a third related molecule, ESR35 (see Fig. 1A), were initially characterised for their effect on cultured mammalian cells by analysing the secretion of pulse-labelled glycoproteins from HepG2 cells treated with each of the compounds. A wide range of glycoproteins were recovered from the media of control treated cells using a lectin-binding assay and a similar profile was observed when cells were first incubated with 8 μM ESR35 or ESII (Fig. 1B, cf. lanes 3-5). Strikingly, when cells were incubated with 8 μM ESI, an almost complete loss of secretory glycoproteins in the media was observed (Fig. 1B, cf. lanes 3-6). Total labelled cellular proteins appeared unaffected by the treatment (Fig. 1C), suggesting a distinct effect on protein secretion is elicited by ESI. Immunoprecipitation of the model secretory protein, α1-antitrypsin (α1-AT), from the media of treated cells confirmed this effect of ESI (Fig. 1D), whereas ESR35 and ESII caused no observable decrease in the secretion of the protein. As expected, treatment of the cells with dithiothreitol (DTT) or brefeldin A (BFA) resulted in the complete loss or a substantial reduction of secretory glycoproteins from the media, respectively (Fig. 1B, lanes 1 and 2) (Alberini et al., 1990; Orci et al., 1991).
Fig. 1.

ESI inhibits protein secretion. (A) Structures of eeyarestatin 1 (ESI), eeyarestatin 2 (ESII) and an ESI-related compound (ESR35). (B) Upper panel; glycoproteins from the media of pulse-labelled HepG2 cells treated with 1 mM DTT, 5 μg/ml BFA, 8 μM ESR35, ESII, ESI or a solvent control (DMSO) were isolated by incubation with ConA conjugated to Sepharose and analysed directly. Lower panel; treated cells were harvested and analysed by immunoblotting using antibodies specific for α-tubulin. (C) Total labelling of proteins in treated cells was analysed directly (upper panel) or by immunoblotting for α-tubulin (lower panel). (D) Upper panel: α1-antitrypsin (α1-AT) was recovered from the media of treated cells by immunoprecipitation. Lower panel; treated cells were harvested and analysed by immunoblotting using antibodies specific for α-tubulin.
This novel and unexpected observation of an ESI-induced inhibition of protein secretion from cultured cells appears more pronounced than that of the well-characterised inhibitor of Golgi to ER trafficking, BFA (Fig. 1B and C, cf. lanes 2-6), and was initially difficult to rationalise with the previously reported effects of ESI on ERAD (Fiebiger et al., 2004; Wang et al., 2008). We therefore undertook a more directed approach and analysed the effect of the compound on the biosynthesis of model proteins in a second cell line. To this end, HeLa cells transiently expressing the membrane glycoprotein TCRα (T-cell receptor α-subunit) were treated with ESI, ES2 and ESR35 before pulse labelling and immunoprecipitation. We found that the levels of the N-glycosylated population of TCRα were substantially reduced in cells treated with 8 μM ESI, whereas no effect was observed in cells treated with ESR35, ESII or a solvent control (Fig. 2A and quantification in Fig. 2B). The loss of N-glycosylated TCRα was accompanied by an increase in the level of non-glycosylated protein and a similar effect was also observed for a second model membrane protein, opsin (data not shown). Since the addition of N-linked oligosaccharides to proteins is a process coordinated in the lumen of the ER, these observations are indicative of an inhibition of protein processing at the ER. Alternatively, since ERAD typically progresses via deglycosylation of the substrate protein by cytosolic peptide N-glycanase prior to degradation by the proteasome, these results could also indicate an accumulation of retrotranslocated TCRα resulting from perturbation of the ERAD pathway (Fiebiger et al., 2004). Therefore, to differentiate between these two possibilities, HeLa cells were first incubated with the 8 μM ESI, ES2 or ESR35 for 1 hour before harvesting, semi-permeabilising with low concentrations of digitonin, and inclusion in an in vitro translation reaction (Wilson et al., 1995). This system limits any interference that may result from a potential effect of ESI on ERAD, which is not recapitulated in these experiments (Wilson et al., 2000), allowing a direct analysis of the effect of the compounds on protein biogenesis. A reproducible inhibition of the N-glycosylation of a third model membrane protein, a glycosylated variant of the proteolipid protein (gPLP), was observed in ESI-treated cells (Fig. 2C and quantification in Fig. 2D). Translation of luciferase in the same reaction as the gPLP further confirmed that this ESI treatment has no effect on protein synthesis per se (Fig. 2C, see luciferase).
Fig. 2.

ESI inhibits ER processing in vivo. (A) HeLa cells transiently transfected with FLAG-tagged T-cell receptor α-subunit (TCRα) or a mock transfection (lane 1; untreated) were treated with 8 μM ESR35, ESII, ESI or a solvent control (DMSO) as indicated for 1 hour before pulse-labelling and immunoprecipitation with antibodies specific for the FLAG tag. N-glycosylated TCRα is labelled as TCRαψ. (B) Quantification of the N-glycosylation population of transiently expressed, pulse-labelled TCRα following treatment with ESR35, ESII or ESI in HeLa cells as indicated (mean ± s.e.m.; n=3). (C) HeLa cells were treated with 8 μM eeyarestatins as indicated for 1 hour at 37°C before preparation for in vitro translocation assay by semi-permeabilisation with digitonin. Semi-intact cells were then used as a source of ER membrane in a co-translation reaction of a glycosylated variant of proteolipid protein (gPLP) and luciferase supplemented with [35S]Met/Cys and commercial rabbit reticulocyte lysate containing haemin. N-glycosylated gPLP is shown by gPLPψ. (D) Quantification of the N-glycosylated fraction of gPLP in the presence of the eeyarestatins-treated semi-intact cells (mean ± s.e.m.; n=4). (E) HeLa cells were treated with 8 μM eeyarestatins as indicated or with 2 mM DTT or 10 μg/ml tunicamycin (Tm) in culture for 10 minutes (upper panel) or 1 hour (lower panel) before harvesting and preparation by semi-permeabilisation. In vitro-derived mRNA for preprolactin (pPL) was then translated in the presence of these cells as a source of in vivo drug-treated ER membrane. Alternatively pPL was translated in the presence of canine pancreatic microsomes (RM) or in the absence of ER membrane to distinguish pPL from the processed prolactin (PL; lanes 1 and 2). Quantification was used to determine the relative processing of pPL (RP–pPL) for each treatment, and is shown below each lane.
To further distinguish the observed effect of ESI on protein biogenesis from any inhibition of the ERAD process, we used a second readout for ER processing that cannot be reversed as a consequence of ERAD, namely the proteolytic removal of N-terminal signal sequences following translocation into the ER lumen. When solvent-treated HeLa cells were harvested, semi-permeabilised and included in an in vitro preprolactin (pPL) translation reaction, the signal sequence of pPL was efficiently cleaved to yield prolactin (PL; Fig. 2E, lower panel, lanes 1-3). By contrast, when cells were first treated with 8 μM ESI in culture for 1 hour, signal sequence cleavage was significantly inhibited (Fig. 2E, lower panel, cf. lanes 3-6). A partial effect of ESI on this process was even seen after just a 10-minute incubation of the cells with ESI in culture (Fig. 2E, upper panel, cf. lanes 3-6), whereas ESII had a less pronounced effect and ESR35 was inactive (Fig. 2E, lower panel).
The treatment of cultured cells with ESI has been shown to induce signalling via the inositol-requiring protein-1 (IRE1) and protein-kinase RNA (PKR)-like ER kinase (PERK) branches of the unfolded protein response (UPR) (Wang et al., 2009). However, these pathways are activated after a prolonged treatment (>4 hours), and we observe no indication that translation is attenuated in HepG2 cells treated with ESI for 1 hour (see Fig. 1C). Nevertheless, we wished to address any potential role of ESI-induced UPR signalling, and distinguish between such an effect and a potentially novel and direct effect on ER processing or translocation. Since the endogenous cytosolic content is depleted from semi-permeabilised cells, UPR signalling is expected to be abolished in the subsequent translation reaction. Nevertheless, to more directly examine the possibility that the ER might be remodelled as a consequence of UPR signalling, HeLa cells were incubated with 2 mM DTT or 10 μg/ml tunicamycin for 1 hour, conditions known to elicit UPR signalling via all three branches of this process (DuRose et al., 2006), and the effect of these treatments compared with those of ESI. Neither DTT nor tunicamycin treatment resulted in any observable defect in pPL processing (Fig. 2E, lower panel), indicating that the effect of ESI is distinct from any consequences of UPR signalling caused by prolonged treatment of the cells with the compound (Wang et al., 2009). Furthermore, the rapidity of the effect of ESI treatment (Fig. 2E, upper panel) strongly suggests that it acts directly upon ER translocation or processing, and further distinguishes this effect from any longer-term consequences of blocking the ERAD pathway.
ESI inhibits protein translocation into the ER
Having established a direct and novel effect of ESI on protein biogenesis at the ER, we wished to understand the molecular basis for our observations. Hence, we prepared cell-free translation reactions using canine pancreatic ER microsomes preincubated with increasing concentrations of ESI to assess the effect of the compound in a well characterised reconstituted protein biogenesis system. Consistent with our previous results, we observed a dose-dependent inhibition of the N-glycosylation of an integral membrane protein, the P2X2 purinergic receptor (P2X2; Fig. 3A, upper panel, see P2X2ψ), whereas the synthesis of a cytosolic protein, SRP19 (19 kDa subunit of the signal recognition particle), translated in the same reaction as P2X2, was unaffected (Fig. 3A, lower panel, see SRP19). This assay was extended to carry out a second dose-response analysis, using another model membrane glycoprotein, the invariant chain of the class II major histocompatibility complex (MHC; Ii). Once again, a strong inhibition of N-glycosylation was seen with ESI (IC50 ∼70 μM; Fig. 3B), and to a lesser extent ESII (IC50 ∼200 μM; Fig. 3B). ESR35 had no inhibitory effect even at 1 mM (Fig. 3B). The effects of the compound on ER processing observed when treating cultured mammalian cells are therefore faithfully replicated in an in vitro system, although the IC50 for ESI treatment in vitro is higher than expected from the more potent effect observed in vivo.
Fig. 3.

Dose-dependent effect of ESI. (A) The P2X2 purinergic receptor (P2X2) was co-translated in vitro with the 19 kDa subunit of the signal recognition particle (SRP19) in the presence of [35S]Met/Cys and ER microsomes incubated with increasing concentrations of ESI, and the total translation products analysed directly. Fully glycosylated P2X2 is indicated by P2X2ψ. (B) Sigmoidal dose-response curves of the effect of ESR35, ESII and ESI on the glycosylation of the invariant chain of the major histocompatability (MHC) class II complex (Ii; normalised to DMSO control; mean ± s.e.m.; n=3). The IC50 for ESI is approximately 70 μM compared with approximately 200 μM for ESII.
When taken together with the inhibition of protein secretion in cultured cells elicited by ESI, the pronounced reduction of protein N-glycosylation observed in vitro suggests that either the translocation of proteins into the ER or the processing capacity of the ER is compromised in the presence of the drug. To distinguish between these two possibilities, we used a pPL protease protection assay. When pPL was synthesised in the presence of ER-derived microsomes, a fraction of the protein was translocated across the microsomal membrane with concomitant cleavage of the N-terminal signal sequence (PL; Fig. 4, cf. lanes 1 and 2; pPL and PL). Only the processed form of the protein is protected from proteolysis by proteinase K, indicating that it has been translocated across the ER membrane (Fig. 4, lane 3). Pre-incubation of the microsomal membranes with ESR35 had no effect upon the formation of the signal-cleaved, protease resistant form of pPL (Fig. 4, lanes 5 and 6), whereas ESII treatment lead to a modest loss of signal sequence processing (Fig. 4, lanes 8 and 9). In ESI-treated membranes, pPL signal sequence cleavage was almost undetectable (Fig. 4, lane 11). Since the unprocessed form is not protected from digestion with proteinase K (Fig. 4, lane 12), we conclude that ESI prevents the translocation of pPL into the ER lumen where it would be inaccessible to the protease. Importantly, ESI has no general effect on ER membrane integrity, since in a control reaction where Ii was first integrated into ER microsomes before treatment with the ESI, no effect upon the membrane-dependent protease-protected fragment was observed (supplementary material Fig. S1). Thus, inhibition of both signal sequence cleavage and N-glycosylation observed following ESI treatment of cultured mammalian cells and ER microsomes reflects a defect in protein translocation into the ER. ESII appears to have a minor effect on protein translocation in vitro, whereas the ESI analogue, ESR35, is inactive.
Fig. 4.
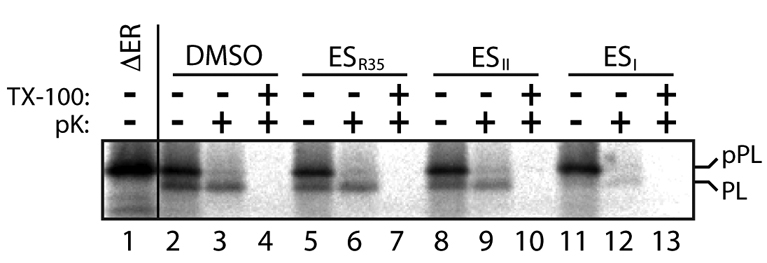
Protease-protection analysis of ESI-induced inhibition of ER translocation. pPL was synthesised in vitro in the presence of [35S]Met/Cys and ER microsomes treated with 250 μM ESR35, ESII, ESI or a solvent control (DMSO) as indicated. Samples were then digested with proteinase K (pK) in the presence or absence of detergent (TX-100) before solubilisation in sample buffer for direct analysis of the total translation products. Alternatively, pPL was translated in the absence of any ER membrane (lane 1) to distinguish the migration of the signal-cleaved pPL generated upon translocation into the ER (PL).
ESI inhibits Sec61-mediated co-translational translocation
To further explore the generality of the effect on ER translocation, we used N-glycosylation as a readout to analyse the effect of the drugs on the biogenesis of a range of membrane proteins, with different topologies and complexities. Hence, we analysed seven representative membrane proteins that utilise the classical co-translational pathway for their biogenesis. In each case we found that pretreatment of microsomes with 250 μM ESI had a pronounced inhibitory effect upon translocation at the ER (Fig. 5; see gPLP to US11). The effect of 250 μM ESII treatment was also always apparent, although less marked (Fig. 5), consistent with the higher IC50 of this compound (cf. Fig. 3).
Fig. 5.

ESI elicits a wide-ranging inhibition of co-translational translocation. Upper panel: the effect of 250 μM ESR35, ESII, and ESI on the translocation of multiple protein classes, normalised to DMSO controls (mean ± s.e.m.; n=3). ASGPr, asialoglycoprotein receptor; SPP, signal peptide peptidase; US11, unique short hCMV glycoprotein 11; gCytB5, glycosylated variant of cytochrome b5. All other abbreviations are as previously defined. In the case of gCytB5, analysis was of its Sec61-independent, post-translational integration. Lower panel: overview of topology and number of transmembrane spans for membrane proteins studied in the upper panel.
In mammalian cells, co-translational ER translocation of both membrane and secretory proteins is coordinated by the Sec61 complex, which both acts as a docking site for the translating ribosome following the SRP-dependent delivery process and forms a protein-conducting pore through the ER membrane. Given the wide-ranging effect on co-translational translocation that we observed following ESI treatment, the Sec61 complex is an obvious candidate for the site of action of the compound. In contrast to the classical precursors analysed previously, cytochrome b5 is a tail-anchored protein that is able to integrate spontaneously into the ER membrane via a pathway that is independent of the Sec61 complex (Brambillasca et al., 2005). We therefore analysed the effect of ESI on the integration of this unusual protein, using glycosylation as a reporter for membrane translocation (Rabu et al., 2008). We found that neither 250 μM ESI nor 250 μM ESII had any significant effect on the spontaneous integration of an N-glycosylated variant of cytochrome b5 (Fig. 5, see gCytB5). These data demonstrate that ESI has no effect on the activity of the ER luminal N-glycosylation machinery per se, because the modified cytochrome b5 was efficiently N-glycosylated even in the presence of the compound. Thus, the inhibition of ER translocation elicited by ESI appears to be limited to those proteins that are dependent upon the Sec61 translocon, strongly implicating this complex, or trans-acting components, as a target of the compound.
ESI prevents nascent chain transfer from SRP to the Sec61 complex
We next analysed the effect of the drug upon the interactions of specific cellular components known to mediate the SRP-dependent delivery of proteins to the ER translocon in order to better understand how ESI blocks ER translocation. We first addressed the possibility that ESI might prevent the binding of the SRP targeting complex to the SRP receptor (SR), a critical step for precursor delivery to the Sec61 complex (Cross et al., 2009). Ribosome-nascent chain-SRP complexes were first generated by translation of a truncated pPL mRNA transcript in vitro, in the absence of any ER membrane. The exclusion of a stop codon from these transcripts causes the polypeptide to remain associated with the ribosome, which can then be purified, together with any additional associated components, by ultracentrifugation. The interaction between the bound SRP and purified, recombinant SR was then probed directly by cross-linking (Pool et al., 2002). Under control conditions, the α-subunit of SR (SRα) can be cross-linked to SRP54 in the presence of the non-hydrolysable GTP analogue GMP-PNP (β, γ-imidoguanosine 5′-triphosphate; Fig. 6A, cf. lanes 1, 2 and 5, see SRP54×SRα). Replacing GMP-PNP with GDP in the reaction, which precludes the SRP-SR interaction, prevents the formation of this adduct (Fig. 6A, lane 8). This assay therefore accurately reports the GTP-dependent interaction of SRP and SR (Pool et al., 2002). When recombinant SR was pre-incubated with either ESI or ESR35, no inhibition of the binding of SR to SRP was detected (Fig. 6A, cf. lanes 5, 6 and 7, see SRP54×SRα). We conclude that ESI acts to block a step occurring after the delivery of ribosome-nascent chain complexes (RNCs) to SR at the ER membrane.
Fig. 6.

ESI targets the Sec61 complex. (A) pPL86-RNC-SRP complexes were incubated either alone (lanes 1-4) or with recombinant SR (0.25 μM; lanes 5-8) which had been pre-treated with 250 μM ESR35, ESII, and ESI as indicated in the presence of either GDP or a non-hydrolysable GTP analogue, GMP-PNP. Samples were cross-linked with DSS and analysed by immunoblotting with anti-SRP54 serum. Cross-linking adducts between SRP54 and ribosomal proteins L23a and L35 and SRα are indicated. (B) pPL86-RNCs synthesised in a wheat germ extract system were added to PKRMs treated with 250 μM ESR35, ESII, or ESI as indicated and incubated to facilitate targeting before addition of puromycin. SRP-independent translocation was competitively inhibited by saturation of the targeting reaction with wheat germ ribosomes (lane 3). Some of the non-translocated, puromycin-released pPL86 is lost to high molecular weight complexes in the presence of ESI (not shown). Numbers show the relative processing of the pPL86 into PL56 (RP-PL56), where a co-migrating band (see lane 2; white circle) is subtracted as background. (C) PreprocecropinA (pPCec) was translated in vitro in the absence of membranes before being treated with ESR35, ESII, or ESI (250 μM; as indicated) ER microsomes were added, ensuring strictly post-translational delivery to the ER membrane. Translocation across the ER membrane is reported by the cleavage of the signal sequence to yield procecropinA (PCec). (D) Quantification of the ER processing of pPCec (mean ± s.e.m.; n=5).
Following the interaction of SRP and SR at the membrane of the ER, classical co-translational translocation proceeds by transfer of the ribosome-nascent chain complex to the Sec61 complex. In order to more closely examine the selectivity of ESI-induced inhibition and test the effect of the compound on the Sec61 complex, we exploited an SRP-independent translocation reaction in which only the Sec61 complex is required to support translocation (Jungnickel and Rapoport, 1995). In this assay, a short pPL nascent chain tethered to the ribosome can be translocated without the requirement for SRP by virtue of the affinity of the pPL signal sequence for the Sec61 complex (Jungnickel and Rapoport, 1995). Thus, 86-residue ribosome-bound pPL nascent chains (pPL86-RNCs) were generated using a wheat germ system containing SRP that is incompatible with the SR of mammalian ER-derived microsomes (Jungnickel and Rapoport, 1995). Incubation of these pPL86-RNCs with SRP-stripped microsomes (PKRMs) followed by treatment of the reaction with puromycin results in signal sequence cleavage of the translocon-associated pPL86 chains, providing a readout for their translocation. Thus, in the absence of PKRMs, uncleaved pPL86 was observed following puromycin treatment (Fig. 6B, lanes 1 and 2), whereas in the presence of solvent-treated membranes, the majority of the protein was found in the cleaved, translocated form following puromycin treatment (Fig. 6B, cf. lanes 4 and 5, see PL56). This SRP-independent translocation was competitively inhibited by an excess of wheat germ ribosomes (Fig. 6B, cf. lanes 3 and 5), confirming the specificity of the reaction (Neuhof et al., 1998). Incubation of SRP-stripped membranes with ESR35 had little effect on pPL86 translocation, whereas ESII caused a slight decrease and ESI a substantial reduction in polypeptide transport across the ER membrane (Fig. 6B, cf. lanes 2, 5, 7, 9 and 11; see also quantification of RP-pPL86). Treatment of these translocation intermediates with proteinase K indicated that in the presence of ESI, the ribosome-nascent chain complex fails to establish a protease-resistant complex with the Sec61 complex (supplementary material Fig. S2), consistent with a direct effect of ESI on Sec61-mediated translocation. The short insect secretory protein preprocecropinA (pPCec) is delivered to the ER membrane via an SRP- and ribosome-independent post-translational mechanism, and translocated via the Sec61 complex (Erdmann et al., 2009; Klappa et al., 1994). We therefore exploited this unusual translocation event to further examine the effect of ESI on SRP-independent translocation at the ER membrane. To this end, pPCec was first synthesised in vitro before the addition of control or ESI-treated ER microsomes to facilitate post-translational translocation. On exposure to the ER lumen pPCec is cleaved by the signal peptidase complex yielding PCec (Fig. 6C, cf. lanes 1 and 2). This process is effectively inhibited in the presence of ESI-treated membranes (Fig. 6C, cf. lanes 2 and 5 and quantification in Fig. 6D). Thus, ESI-mediated inhibition persists even for substrates that are delivered to the ER membrane independently of the ribosome or the SRP targeting machinery, and we conclude that ESI most probably affects the Sec61 complex directly.
Since components of the SRP-dependent targeting pathway appear to be unaffected by ESI, a cross-linking analysis was exploited to monitor the transfer of the nascent chain from SRP to the Sec61 complex and determine the molecular basis for the effect of ESI (Cross and High, 2009). P2X2-RNCs were first synthesised in vitro by truncating the mRNA transcript at a site selected such that the first P2X2 TM segment, which functions as a hydrophobic ER targeting signal (Cross and High, 2009), is predicted to have just emerged from the ribosome exit tunnel. A single cysteine was introduced into the TM segment so that the molecular environment of P2X2 TM1 could be monitored by thiol-dependent cross-linking. In the absence of ER membranes, TM1 was found directly adjacent to the SRP targeting machinery, resulting in the formation of a cross-linking adduct with the 54 kDa subunit of the SRP complex (SRP54; Fig. 7, lane 3, see SRP54×P2X2148). When ER-derived membranes and GTP were present, adducts representing P2X2 cross-linked to Sec61α were generated, indicating that P2X2-RNCs have been transferred from SRP and have engaged the ER translocon (Fig. 7, lane 8, see Sec61α×P2X2148). If ER membranes are treated with ESR35 before incubation with P2X2-RNCs, identical cross-linking adducts to the control reaction were observed (Fig. 7, cf. lanes 5-8 and lanes 9-12), consistent with the efficient integration of P2X2 seen in the presence of ESR35 (cf. Fig. 5). Treatment with ESII had an observable effect upon nascent chain transfer from SRP, and far fewer P2X2 polypeptides formed adducts with Sec61α (Fig. 7, lane 16). The effect of ESI is even more striking and no detectable cross-linking of P2X2 chains to Sec61α was observed in ESI-treated microsomes, whereas SRP54 adducts persisted (Fig. 7, lane 20). We conclude that ESI, and to a lesser extent ESII, inhibit the transfer of nascent polypeptides from SRP to the Sec61 complex. Inhibition of this process most likely proceeds by a direct effect on the Sec61 complex, causing potent attenuation of co-translational translocation both in vitro and in cultured mammalian cells.
Fig. 7.

ESI inhibits the transfer of the nascent chain from the targeting complex to the translocation complex. Truncated and HA-tagged P2X2 [Q56C] 148 ribosome-nascent chain (RNC) complexes were synthesised in rabbit reticulocyte lysate and added to ER membranes treated with a solvent control (DMSO) or 250 μM ESR35, ESII, or ESI as indicated, or analysed in the absence of membranes (ΔER). The reactions were supplemented with 1 mM GTP and incubated for a further 10 minutes to facilitate membrane targeting prior to cross-linking with bismaleimidohexane (BMH; +) or a solvent control (DMSO; –) and immunoprecipitation (IP) with anti-serum specific to the nascent chain (HA), Sec61α subunit (α) or SRP54 subunit (54). Adducts representing the nascent P2X2 chain cross-linked to Sec61α and SRP54 are indicated. An unknown membrane-independent adduct is indicated by an asterisk; secondary adducts between P2X2, Sec61α and Sec61β are indicated by white circles (Cross and High, 2009).
Discussion
We have identified and characterised a novel effect of the small molecule ESI. Using both in vivo and in vitro treatments combined with the analysis of signal sequence cleavage and protein N-glycosylation, we have shown that ESI inhibits the co-translational translocation of protein precursors across the ER membrane. This study made use of an analogue of ESI, ESR35, which provides both a valuable comparator for the effects of drug treatments and confirms the specificity of the ESI-induced inhibition of ER translocation. In cultured cells, inhibition of ER translocation was accompanied by a perturbation of the secretory pathway, consistent with a wide-ranging effect of ESI on protein export. Over the time periods used in our analysis, we found no evidence that protein synthesis was affected by the presence of ESI, either in vitro or in vivo. Likewise, the membrane targeting of newly synthesised proteins to the ER was not inhibited, and the interactions between the nascent chain, SRP and the SRP receptor were all unaffected by ESI. Instead, we found that ESI acts to prevent the transfer of the nascent polypeptide chain from the membrane targeting complex to the ER translocon, most probably by a direct effect on the Sec61 complex (Fig. 8).
Fig. 8.
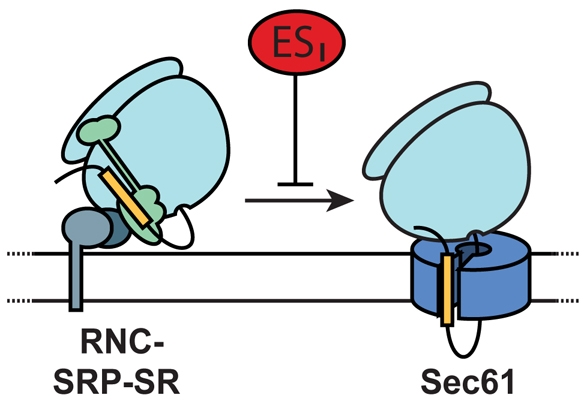
ESI inhibits productive interaction between RNCs and the ER translocon. ESI does not affect protein synthesis or membrane targeting of newly synthesised proteins. The transfer of the nascent chain from the targeting complex to the ER translocon appears to be abolished in the presence of ESI, most likely by a direct effect on the Sec61 complex.
The IC50 for the inhibitory effect of ESI in vitro is ∼70 μM, yet the treatment of cultured mammalian cells with only 8 μM ESI is sufficient to block ER translocation both in vivo, and when in-vivo-treated cells are subsequently semi-permeabilised and used in an in vitro translocation assay. This discrepancy may result from metabolism of ESI in vivo, as has been suggested previously (Wang et al., 2008). Interestingly, a similar requirement for an approximately tenfold higher in vitro concentration of the cyclodepsipeptides cotransin and CAM741 compared with that required for efficient in vivo inhibition has been reported (Besemer et al., 2005; Garrison et al., 2005). Moreover, it is well established that BFA is only active in vitro at concentrations of ∼150 μM (Orci et al., 1991), whereas in vivo it effectively inhibits protein secretion at sub-micromolar concentrations (Klausner et al., 1992). In this study we have examined the effect of ESI on two mammalian cell lines, and it should be noted that different cell lines might exhibit altered sensitivity to ESI treatment (Wang et al., 2009).
The wide-ranging effect of ESI is in contrast to the more substrate-specific translocation blockade observed with CAM741 and cotransin (Besemer et al., 2005; Garrison et al., 2005; Harant et al., 2007). These compounds bind Sec61α and inhibit the translocation of VCAM1 at a post-targeting stage, i.e. after the ribosome-bound nascent chain has engaged the ER translocon (Besemer et al., 2005; Garrison et al., 2005; Mackinnon et al., 2007). Thus, although ESI, CAM741 and cotransin all seem to target the Sec61 complex, we propose that ESI prevents the transfer of the nascent polypeptide chain from the membrane-bound SRP delivery complex to the ER translocon (see Fig. 8), and therefore appears to act upstream of the two cyclodepsipeptides. A comparable translocation defect can be elicited at the ER by artificially loading the membrane with cholesterol (Nilsson et al., 2001), although, in contrast to ESI, this sterol treatment is also known to inhibit the unassisted integration of cytochrome B5 (Brambillasca et al., 2005). Thus, the mechanism for ESI activity appears to be distinct from other inhibitors of ER translocation identified to date. The simplest model for the activity of ESI is that its effect on the Sec61 complex sterically perturbs the association of the SR-SRP-RNC complex with the ER translocon (see Fig. 8).
Treatment of cultured cells with ESI has been shown to be highly cytotoxic and, in human cancer cells, the upregulation of transcription factors ATF3 and ATF4, following the induction of UPR signalling by ESI, was suggested to have antitumour activity (Wang et al., 2009). In the present study, we have identified and characterised a rapid inhibition of ER translocation by ESI, and we suggest that this effect of the drug might contribute to the induction of ER stress by preventing the entry of newly synthesised enzymes and chaperones into the ER lumen. Furthermore, the accumulation of cytosolic proteins that would result from an inhibition of ER translocation might also contribute to the cytotoxicity of ESI and its potential anticancer properties (Rane et al., 2008; Wang et al., 2009). The inhibition of ER translocation by the cyclodepsipeptide apratoxinA has also been linked to an anticancer effect, in this case by preventing the biosynthesis of cancer-associated receptor tyrosine kinases (Liu et al., 2009). Thus, modulation of protein homeostasis by targeting fundamental cellular components such as the Sec61 complex appears to be a valid target for therapeutic intervention.
ESI was initially identified as an inhibitor of a virally induced ERAD of the class I heavy chain (HC) of the MHC (Fiebiger et al., 2004), and this effect was subsequently shown to extend to other ERAD substrates as evidenced by the accumulation of polyubiquitinated proteins destined for degradation via the proteasome (Wang et al., 2008). Our data indicate that discerning a direct effect of ESI on the ERAD pathway in vivo (Fiebiger et al., 2004; Wang et al., 2008) is complicated by the potential for upstream effects caused by an inhibition of protein translocation into the ER. This complexity is demonstrated by the capacity of ESI to inhibit the ER integration of the viral US11 protein (see Fig. 5), raising the possibility that the stabilising effect of ESI upon the US11-mediated ERAD of MHC class I HC is, at least in part, due to reduced levels of US11 in the ER (cf. Fiebiger et al., 2004). Likewise, several endogenous components of the ERAD machinery are rapidly turned over (Cali et al., 2008; Wu et al., 2007), and any ESI-mediated inhibition of their translocation into the ER would also most probably disrupt the native degradation pathway. Alternatively, the Sec61 complex has been strongly linked to the ERAD process by a number of studies (Romisch, 2005; Scott and Schekman, 2008; Willer et al., 2008), and it is possible that inactivation of the ER translocation machinery simultaneously blocks translocation and retrotranslocation. Although at present we are unable to exclude the possibility that ESI has multiple targets in cells, it provides a novel tool to further dissect the processes of ER translocation and ERAD.
Materials and Methods
HA-tagged SPP was from B. Martoglio (ETH, Switzerland), TCRα was a gift from R. Kopito (Stanford University, USA), ASGPr was from M. Speiss (University of Basel, Switzerland) and US11 was from E. Weirtz (Leiden University Medical Centre, Netherlands). Wild-type P2X2 was a gift from R. A. North (University of Manchester, UK). Purified SR-his-αβΔN was prepared in the laboratory of I. Sinning (University of Heidelberg, Germany). Antisera specific for Sec61β, SRP54 and SRβ were from B. Dobberstein (University of Heidelberg, Germany), anti-Sec61α and the pPCec construct were gifts from R. Zimmermann and S. Lang (University of the Saarland, Germany) and TAT1 anti-α–tubulin was from K. Gull (University of Oxford, UK). ESI, ESII and ESR35 were synthesised in house (C.M., M.P., A.C.C., S.H., S.L.F., R.W. et al., unpublished data), dissolved in 100% DMSO and stored at –80°C.
In vivo analysis
Confluent HepG2 cells were treated with the eeyarestatins, ESI, ESII or a third related molecule, ESR35 for 1 hour in culture before a 30-minute starvation in the presence of the compounds in methionine- and cystine-free medium supplemented with 2 mM glutamine. Cells were then metabolically labelled in the presence of the eeyarestatins by incubation in starvation medium containing 22 μCi/ml [35S]Met/Cys protein labelling mix (Perkin-Elmer) for 30 minutes. Cells were rinsed twice in PBS and chased for 90 minutes in serum-free medium (Invitrogen). The resulting medium was then removed, and precleared before incubation with ConA-Sepharose at 4°C overnight. Bound proteins were washed and solublised in SDS-PAGE sample buffer (0.2 M Tris-HCl pH 6.8, 10 mM EDTA, 1 M sucrose, 1% L-methionine, 0.02% Bromophenol Blue, 80 mM DTT, 6% SDS). For analysis of in vivo translocation, HeLa cells were transiently transfected with N-terminally FLAG-tagged TCRα overnight before treatment of the cells for 1 hour at 37°C with 8 μM ESI, ESII or ESR35, or a solvent control (DMSO). Cells were then pulse-labelled as described for the secretion analysis, TCRα recovered by immunoprecipitation using antibodies specific for the FLAG-tag epitope (Sigma-Aldrich), and translocation analysed by N-glycosylation.
In vitro translocation assays
Canine pancreatic rough microsomes were prepared as previously described (Walter and Blobel, 1983) and treated directly with ESI, ESII, ESR35 or an equal volume of DMSO and incubated on ice for 1 hour before inclusion in translocation analyses. Alternatively, HeLa cells were treated with 8 μM ESI, ESII, ESR35 and incubated at 37°C before harvesting and preparation as semi-intact cells (Wilson et al., 1995) for use in translocation assays. Cell-free translation was performed in rabbit reticulocyte lysate (Promega) supplemented with [35S]Met/Cys and an in vitro synthesised mRNA transcript. Membranes were used at 10% v/v in translation assays, with the concentration of the eeyarestatin in the final reaction being diluted pro rata. Total translation products were analysed by direct solublisation of the reaction in SDS-PAGE sample buffer and incubation for 10 minutes at 70°C. Where appropriate, the membrane-associated fraction of the reaction was isolated by centrifugation at 100,000 g through a high-salt sucrose cushion (HSC500; 25 mM Hepes-KOH pH 7.8, 650 mM potassium acetate, 10 mM magnesium acetate, 500 mM sucrose) prior to analysis. Protease protection studies of the membane-associated material used 500 μg/ml Proteinase K for 1 hour on ice in the presence or absence of 1% Triton X-100 and the reaction was stopped by the addition of 1 mM phenylmethyl sulphonyl fluoride (PMSF). For analysis of the spontaneous, post-translational integration of cytochrome b5, mRNA encoding cytB5 fused with an opsin glycosylation tag at the C-terminus (gCytB5) was prepared by in vitro transcription (Rabu et al., 2008). gCytB5-ribosome-nascent chain (RNC) complexes were then generated in the presence of [35S]methionine for 7 minutes at 25°C in a wheat germ extract system, purified by ultracentrifugation through a high-salt sucrose cushion (HSC1000; 25 mM Hepes pH 7.8, 650 mM potassium acetate, 10 mM magnesium acetate, 1 M sucrose) at 175,000 g for 20 minutes at 4°C before resuspension in RM buffer (50 mM potassium acetate, 2 mM magnesium acetate, 50 mM Hepes pH 7.8, 250 mM sucrose, 1 mM dithiothreitol). Release from the ribosome was induced by addition of 2 mM puromycin (Sigma, UK), 1 mM GTP and incubation at 37°C for 5 minutes. Ribosome-released nascent chains were then added to rabbit reticulocyte lysate and ER microsomes that had been pretreated with 250 μM ESI, ESII or ESR35 for 1 hour on ice and these samples incubated for 30 minutes at 37°C to facilitate spontaneous integration. For SRP-independent targeting assays (Jungnickel and Rapoport, 1995), pPL86-RNCs (Hauser et al., 1995) were generated in vitro in a wheat germ extract system supplemented with [35S]Met/Cys for 7 minutes at 25°C and purified by ultracentrifugation through HSC1000 at 175,000 g for 20 minutes at 4°C before resuspension in RM buffer. ER microsomes stripped of SRP and ribosomes by high-salt and puromycin treatment (PKRMs) (Hauser et al., 1995) were treated with a eeyarestatin for 1 hour on ice before incubation with the pPL86-RNCs for 10 minutes at 30°C. Translocation was then analysed by the addition of 2 mM puromycin, 1 mM GTP and incubation at 37°C for 10 minutes. Bound tRNAs were removed by RNaseA (Sigma, UK) treatment at 250 μg/ml for 5 minutes at 37°C. Samples were resolved by Tris-tricine or Tris-glycine SDS-PAGE and analysed by FLA-3000 phosphorimaging (Fuji, Japan). Quantitative analysis was done with AIDA v3.52 (Raytest Isotopenmessgerate, Germany) and statistical analyses with Prism v4.0 for Macintosh (GraphPad, USA).
Cross-linking and immunoprecipitation
Truncated P2X2 [Q56C] 148 mRNA transcripts were synthesised from a PCR-generated template, translated for 7 minutes at 30°C and purified by centrifugation though HSC1000 at 175,000 g for 20 minutes at 4°C before resuspension in RM buffer and incubation with ER membranes (crude microsome preparation, including additional SRP). P2X2 [Q56C] 148 was subjected to cross-linking by addition of 1 mM bismaleimidohexane (BMH), prepared in DMSO, and incubation at 30°C for 10 minutes before quenching with 5 mM β-mercaptoethanol and RNaseA treatment. Following denaturation with 1% SDS, immunoprecipitation (IP) was achieved using a tenfold dilution of the sample in Triton IP buffer (10 mM Tris-HCl pH 7.6, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100); pre-clearing and IP with indicated antisera was as previously described (Ismail et al., 2008). Cross-linking analysis of SRP54 was performed as described elsewhere (Pool et al., 2002). Briefly, pPL86-RNCs were synthesised in the presence of purified canine SRP (Martoglio et al., 1997) in a rabbit reticulocyte lysate system, purified by centrifugation through HSC500 and resuspended (to 200 nM) in RNC buffer (25 mM Hepes, KOH pH 7.6, 2 mM magnesium acetate, 2 mM DTT, 1 mM cycloheximide, 120 mM potassium acetate), before addition of 250 nM purified SR-his-αβΔN (Fulga et al., 2001) which had been pre-treated with ESI, ESII or ESR35 and 200 μM GMP-PNP or GDP. Cross-linking was induced by addition of 40 μM disuccinimidyl suberate (DSS) and incubation at 30°C for 10 minutes.
Supplementary Material
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/122/23/4393/DC1
This work was supported by PhD and project grant funding from the BBSRC, including an award from the SCIBS initiative, and a Wellcome Trust PhD studentship. We acknowledge all those who have provided helpful comments and reagents used during this study and thank Blanche Schwappach, Stephen Taylor and Martin Lowe in particular for their advice during the preparation of this manuscript. Deposited in PMC for release after 6 months.
References
- Alberini, C. M., Bet, P., Milstein, C. and Sitia, R. (1990). Secretion of immunoglobulin M assembly intermediates in the presence of reducing agents. Nature 347, 485-487. [DOI] [PubMed] [Google Scholar]
- Balch, W. E., Morimoto, R. I., Dillin, A. and Kelly, J. W. (2008). Adapting proteostasis for disease intervention. Science 319, 916-919. [DOI] [PubMed] [Google Scholar]
- Besemer, J., Harant, H., Wang, S., Oberhauser, B., Marquardt, K., Foster, C. A., Schreiner, E. P., de Vries, J. E., Dascher-Nadel, C. and Lindley, I. J. (2005). Selective inhibition of cotranslational translocation of vascular cell adhesion molecule 1. Nature 436, 290-293. [DOI] [PubMed] [Google Scholar]
- Brambillasca, S., Yabal, M., Soffientini, P., Stefanovic, S., Makarow, M., Hegde, R. S. and Borgese, N. (2005). Transmembrane topogenesis of a tail-anchored protein is modulated by membrane lipid composition. EMBO J. 24, 2533-2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cali, T., Galli, C., Olivari, S. and Molinari, M. (2008). Segregation and rapid turnover of EDEM1 by an autophagy-like mechanism modulates standard ERAD and folding activities. Biochem. Biophys. Res. Commun. 371, 405-410. [DOI] [PubMed] [Google Scholar]
- Cross, B. C. S. and High, S. (2009). Dissecting the physiological role of selective transmembrane-segment retention at the ER translocon. J. Cell Sci. 122, 1768-1777. [DOI] [PubMed] [Google Scholar]
- Cross, B. C. S., Sinning, I., Luirink, J. and High, S. (2009). Delivering proteins for export from the cytosol. Nat. Rev. Mol. Cell. Biol. 10, 255-264. [DOI] [PubMed] [Google Scholar]
- DuRose, J. B., Tam, A. B. and Niwa, M. (2006). Intrinsic capacities of molecular sensors of the unfolded protein response to sense alternate forms of endoplasmic reticulum stress. Mol. Biol. Cell 17, 3095-3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann, F., Jung, M., Eyrisch, S., Lang, S., Helms, V., Wagner, R. and Zimmermann, R. (2009). Lanthanum ions inhibit the mammalian Sec61 complex in its channel dynamics and protein transport activity. FEBS Lett. 583, 2359-2364. [DOI] [PubMed] [Google Scholar]
- Fiebiger, E., Hirsch, C., Vyas, J. M., Gordon, E., Ploegh, H. L. and Tortorella, D. (2004). Dissection of the dislocation pathway for type I membrane proteins with a new small molecule inhibitor, eeyarestatin. Mol. Biol. Cell 15, 1635-1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulga, T. A., Sinning, I., Dobberstein, B. and Pool, M. R. (2001). SRbeta coordinates signal sequence release from SRP with ribosome binding to the translocon. EMBO J. 20, 2338-2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison, J. L., Kunkel, E. J., Hegde, R. S. and Taunton, J. (2005). A substrate-specific inhibitor of protein translocation into the endoplasmic reticulum. Nature 436, 285-289. [DOI] [PubMed] [Google Scholar]
- Harant, H., Lettner, N., Hofer, L., Oberhauser, B., de Vries, J. E. and Lindley, I. J. (2006). The translocation inhibitor CAM741 interferes with vascular cell adhesion molecule 1 signal peptide insertion at the translocon. J. Biol. Chem. 281, 30492-30502. [DOI] [PubMed] [Google Scholar]
- Harant, H., Wolff, B., Schreiner, E. P., Oberhauser, B., Hofer, L., Lettner, N., Maier, S., de Vries, J. E. and Lindley, I. J. (2007). Inhibition of vascular endothelial growth factor cotranslational translocation by the cyclopeptolide CAM741. Mol. Pharmacol. 71, 1657-1665. [DOI] [PubMed] [Google Scholar]
- Hauser, S., Bacher, G., Dobberstein, B. and Lutcke, H. (1995). A complex of the signal sequence binding protein and the SRP RNA promotes translocation of nascent proteins. EMBO J. 14, 5485-5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde, R. S. and Kang, S. W. (2008). The concept of translocational regulation. J. Cell Biol. 182, 225-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail, N., Crawshaw, S. G., Cross, B. C. S., Haagsma, A. C. and High, S. (2008). Specific transmembrane segments are selectively delayed at the ER translocon during opsin biogenesis. Biochem. J. 411, 495-506. [DOI] [PubMed] [Google Scholar]
- Jungnickel, B. and Rapoport, T. A. (1995). A posttargeting signal sequence recognition event in the endoplasmic reticulum membrane. Cell 82, 261-270. [DOI] [PubMed] [Google Scholar]
- Klappa, P., Zimmermann, M. and Zimmermann, R. (1994). The membrane proteins TRAMp and sec61 alpha p may be involved in post-translational transport of presecretory proteins into mammalian microsomes. FEBS Lett. 341, 281-287. [DOI] [PubMed] [Google Scholar]
- Klausner, R. D., Donaldson, J. G. and Lippincott-Schwartz, J. (1992). Brefeldin A: insights into the control of membrane traffic and organelle structure. J. Cell Biol. 116, 1071-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y., Law, B. K. and Luesch, H. (2009). Apratoxin a reversibly inhibits the secretory pathway by preventing cotranslational translocation. Mol. Pharmacol. 76, 91-104. [DOI] [PubMed] [Google Scholar]
- Mackinnon, A. L., Garrison, J. L., Hegde, R. S. and Taunton, J. (2007). Photo-leucine incorporation reveals the target of a cyclodepsipeptide inhibitor of cotranslational translocation. J. Am. Chem. Soc. 129, 14560-14561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martoglio, B., Hauser, S. and Dobberstein, B. (1997). Cell Biology: A Laboratory Handbook (ed. J. C. Celis), pp. 265-273. San Diego, CA: Academic Press.
- Neuhof, A., Rolls, M. M., Jungnickel, B., Kalies, K. U. and Rapoport, T. A. (1998). Binding of signal recognition particle gives ribosome/nascent chain complexes a competitive advantage in endoplasmic reticulum membrane interaction. Mol. Biol. Cell 9, 103-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson, I., Ohvo-Rekila, H., Slotte, J. P., Johnson, A. E. and von Heijne, G. (2001). Inhibition of protein translocation across the endoplasmic reticulum membrane by sterols. J. Biol. Chem. 276, 41748-41754. [DOI] [PubMed] [Google Scholar]
- Orci, L., Tagaya, M., Amherdt, M., Perrelet, A., Donaldson, J. G., Lippincott-Schwartz, J., Klausner, R. D. and Rothman, J. E. (1991). Brefeldin A, a drug that blocks secretion, prevents the assembly of non-clathrin-coated buds on Golgi cisternae. Cell 64, 1183-1195. [DOI] [PubMed] [Google Scholar]
- Pool, M. R. (2005). Signal recognition particles in chloroplasts, bacteria, yeast and mammals (review). Mol. Membr. Biol. 22, 3-15. [DOI] [PubMed] [Google Scholar]
- Pool, M. R., Stumm, J., Fulga, T. A., Sinning, I. and Dobberstein, B. (2002). Distinct modes of signal recognition particle interaction with the ribosome. Science 297, 1345-1348. [DOI] [PubMed] [Google Scholar]
- Rabu, C., Wipf, P., Brodsky, J. L. and High, S. (2008). A precursor-specific role for Hsp40/Hsc70 during tail-anchored protein integration at the endoplasmic reticulum. J. Biol. Chem. 283, 27504-27513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane, N. S., Kang, S. W., Chakrabarti, O., Feigenbaum, L. and Hegde, R. S. (2008). Reduced translocation of nascent prion protein during ER stress contributes to neurodegeneration. Dev. Cell 15, 359-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport, T. A. (2007). Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 450, 663-669. [DOI] [PubMed] [Google Scholar]
- Romisch, K. (2005). Endoplasmic reticulum-associated degradation. Annu. Rev. Cell Dev. Biol. 21, 435-456. [DOI] [PubMed] [Google Scholar]
- Scott, D. C. and Schekman, R. (2008). Role of Sec61p in the ER-associated degradation of short-lived transmembrane proteins. J. Cell Biol. 181, 1095-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Berg, B., Clemons, W. M., Jr, Collinson, I., Modis, Y., Hartmann, E., Harrison, S. C. and Rapoport, T. A. (2004). X-ray structure of a protein-conducting channel. Nature 427, 36-44. [DOI] [PubMed] [Google Scholar]
- Walter, P. and Blobel, G. (1983). Preparation of microsomal membranes for cotranslational protein translocation. Methods Enzymol. 96, 84-93. [DOI] [PubMed] [Google Scholar]
- Wang, Q., Li, L. and Ye, Y. (2008). Inhibition of p97-dependent protein degradation by Eeyarestatin I. J. Biol. Chem. 283, 7445-7454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q., Mora-Jensen, H., Weniger, M. A., Perez-Galan, P., Wolford, C., Hai, T., Ron, D., Chen, W., Trenkle, W., Wiestner, A. et al. (2009). ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc. Natl. Acad. Sci. USA 106, 2200-2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer, M., Forte, G. M. and Stirling, C. J. (2008). Sec61p is required for ERAD-L: genetic dissection of the translocation and ERAD-L functions of Sec61P using novel derivatives of CPY. J. Biol. Chem. 283, 33883-33888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, C. M., Farmery, M. R. and Bulleid, N. J. (2000). Pivotal role of calnexin and mannose trimming in regulating the endoplasmic reticulum-associated degradation of major histocompatibility complex class I heavy chain. J. Biol. Chem. 275, 21224-21232. [DOI] [PubMed] [Google Scholar]
- Wilson, R., Allen, A. J., Oliver, J., Brookman, J. L., High, S. and Bulleid, N. J. (1995). The translocation, folding, assembly and redox-dependent degradation of secretory and membrane proteins in semi-permeabilized mammalian cells. Biochem. J. 307, 679-687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Y., Termine, D. J., Swulius, M. T., Moremen, K. W. and Sifers, R. N. (2007). Human endoplasmic reticulum mannosidase I is subject to regulated proteolysis. J. Biol. Chem. 282, 4841-4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}