

Abstract
To investigate the evolutionary origins of proteins encoded by the Poxviridae family of viruses, we examined all poxvirus protein coding genes using a method of characterizing and visualizing the similarity between these proteins and taxonomic subsets of proteins in GenBank. Our analysis divides poxvirus proteins into categories based on their relative degree of similarity to two different taxonomic subsets of proteins such as all eukaryote vs. all virus (except poxvirus) proteins. As an example, this allows us to identify, based on high similarity to only eukaryote proteins, poxvirus proteins that may have been obtained by horizontal transfer from their hosts. Although this method alone does not definitively prove horizontal gene transfer, it allows us to provide an assessment of the possibility of horizontal gene transfer for every poxvirus protein. Potential candidates can then be individually studied in more detail during subsequent investigation.
Results of our analysis demonstrate that in general, proteins encoded by members of the subfamily Chordopoxvirinae exhibit greater similarity to eukaryote proteins than to proteins of other virus families. In addition, our results reiterate the important role played by host gene capture in poxvirus evolution; highlight the functions of many genes poxviruses share with their hosts; and illustrate which host-like genes are present uniquely in poxviruses and which are also present in other virus families.
Keywords: Poxvirus, Comparative genomics, Bioinformatics, Horizontal gene transfer
1. Introduction
1.1. Poxviruses
The Poxviridae are a large family of double stranded DNA viruses whose members have a linear genome of 130–300 kbp, and replicate in the cytoplasm of eukaryote cells. The poxvirus family is composed of two subfamilies: the Entomopoxvirinae, comprised of viruses that infect insects, and the Chordopoxvirinae, comprised of viruses that infect vertebrates. Both subfamilies are further divided into genera, groups of viral species with genetic and antigenic similarity to one another. Chordopoxviruses are categorized into 9 genera: Avipoxvirus, Capripoxvirus, Cervidpoxvirus, Leporipoxvirus, Molluscipoxvirus, Orthopoxvirus, Parapoxvirus, Suipoxvirus, and Yatapoxvirus. Entomopoxviruses are categorized into the genera Alphaentomopoxvirus, Betaentomopoxvirus, and Gammaentomopoxvirus. The genus Orthopoxvirus is the most well characterized, and contains the species Variola virus, isolates of which are the causative agent of smallpox, as well as the species Vaccinia virus, containing less virulent but better studied viruses.
The poxvirus family is postulated to have a common evolutionary origin with four other families of large eukaryotic DNA viruses, collectively referred to as nucleocytoplasmic large DNA viruses (NCLDV) (Iyer et al., 2001). These families include Asfarviridae, containing viruses which infect both pig and parasitic arthropods, Iridoviridae, whose members infect invertebrates, fish or amphibians, Phycodnaviridae, containing viruses which infect eukaryotic algae, and the recently discovered Mimiviridae, whose members are only known to infect amoeba.
1.2. Poxvirus replication cycle
Poxvirus virions are ovoid or brick-shaped, and consist of an envelope surrounding an outer membrane, which itself surrounds a densely packed and membrane bound core containing a double-stranded DNA genome, enzymes, and transcription factors. Unlike most viruses, poxvirus virions do not rely on particular cell surface receptors, but are capable of binding and penetrating the outer membrane of nearly any cell type. Virion cores are released into the cytoplasm where they immediately synthesize early mRNAs that are translated into growth factors, cell signaling and immune defense molecules, enzymes, and other factors necessary for DNA replication and intermediate transcription. Uncoating of the core next allows the DNA genome to be replicated to form concatemeric molecules along with the transcription of intermediate genes that when translated, provide late transcription factors. Subsequent transcription and translation of the late genes produces virion structural proteins, enzymes, and early transcription factors for packaging into virions (Moss, 2001). During virion formation, the concatemeric DNA genomes are resolved into individual genomes, packaged into the core membranes, and mature within the cytoplasm to form infectious mature virions (MV). These are subsequently wrapped in modified Golgi membranes and transported to the periphery of the cell via attached actin filaments. Fusion of the wrapped virions with the plasma membrane results in release of enveloped virus (EV) (Condit et al., 2006).
1.3. Viral evolution
DNA viruses have much lower mutation rates and genetic variability than RNA viruses, with nucleotide substitution rates closer to those of their hosts, on the order of 10−7 to 10−9 mutations per site per round of replication (Drake and Hwang, 2005, Duffy et al., 2008). Their resulting genetic stability, together with their high levels of host specificity have led in part to the hypothesis that many DNA viruses cospeciate with their hosts (DeFilippis and Villarreal, 2001). Intricate relationships with their hosts are evidenced by the many immunological and cellular factors these viruses have obtained through host gene capture via recombination between viral and host DNA. Such acquisition of new coding information by poxviruses may contribute to the ability of the virus to manipulate the host immune response and other cellular machinery to provide a selective advantage for virus replication.
Evidence suggests many orthopoxviruses occasionally cross into other mammals from rodent reservoir populations, either as zoonotic infections of humans or via mutations that allow colonization of new host species (Esposito and Fenner, 2001, Li et al., 2007, Likos et al., 2005). Understanding species crossing events of both types is essential to understanding the threats poxviruses pose today. Investigation of evolutionary clues buried within the genome sequence of the virus, such as captured host genes left by such historical host interactions, may help us to better understand mechanisms of zoonotic infection, viral tropism, evolutionary adaptation, and pathogenesis.
1.4. Poxvirus recombination
In general, recombination may occur via homologous recombination, site-specific recombination, or non-homologous end joining, and may be between viral genomes or between a viral genome and some other genetic entity, such as the genome or cDNA of the viral host, or a co-infecting parasite or a plasmid. Recombination may be an important source of genetic variation among viruses, where it is often associated with rapid evolutionary divergence, due to the potential of providing a selective advantage much more quickly than through the accumulation of point mutations. Recombination has been detected in both DNA and RNA viruses including species of the families Caulimoviridae, Flaviviridae, Herpesviridae, Papillomaviridae, Picornaviridae, Potyviridae, Poxviridae, Polyomaviridae, Retroviridae, the genus Tobamovirus, and bacteriophages in the order Caudovirales (DeFilippis and Villarreal, 2001). Such evidence has led to the “modular” theory of virus evolution, whereby many viral genomes represent mosaics of genetic material obtained through multiple recombination events (Botstein, 1980, Shackelton and Holmes, 2004).
1.5. Roles of host-acquired proteins
Acquisition of host genes and apparent selection for maintenance of those genes has been documented in many virus families (Iyer et al., 2001, McFadden and Murphy, 2000). Many of the apparently host-derived genes fall into one of two well-defined categories of gene function: immunomodulatory genes and genes involved in nucleic acid metabolism. Viral proteins that are very similar to host genes are documented to interfere with a variety of host immune defense mechanisms including antigen display, cytokines and their receptors, cytoplasmic signaling resulting from immune activation, and genes involved in resistance of cells to oxidative stress and apoptosis (Hughes and Friedman, 2005, Shackelton and Holmes, 2004). Many DNA viruses encode genes involved in nucleic acid metabolism, with which they redirect the host nucleotide precursor pool to viral DNA synthesis (Iyer et al., 2006). These enzymes are often clearly similar to host enzymes, and are often very highly conserved, probably due to functional restraints on structure and biochemical properties. There are many additional, possibly host-derived genes, whose functions have not yet been fully explored, but based at least on similarity to other proteins, seem to manipulate various intracellular processes to facilitate steps in the viral life cycle. Examples of these are genes involved in signaling pathways, lipid and carbohydrate metabolism, vesicle transport, and protein–protein interactions (Afonso et al., 2000, Geserick et al., 2004, Laidlaw et al., 1998, Werden and McFadden, 2008).
1.6. Detection and analysis of horizontally transferred proteins
Several methods are available to detect genes that may have been horizontally transferred into virus genomes from hosts or other sources. These include phylogenetic inference, compositional features such as codon and nucleotide bias, and patterns of presence and absence of genes within genomes. A well accepted and widely used method to detect horizontal gene transfer (HGT) is demonstration of phylogenetic clustering of the gene of interest with taxa unrelated to the current genome in which the gene is found, to the exclusion of taxa more closely related to the current genome. This method provides information about the potential donor and recipient organisms, but its potential caveats include limited phylogenetic samples, undetected presence of paralogs, and unequal rates of evolution between lineages (Katz, 2002). Compositional features that may be used to detect recently horizontally acquired genes include nucleotide composition, oligonucleotide frequencies, and codon usage (Koonin and Wolf, 2008), but these methods work only for very recent HGT because the anomalous signatures of such genes decay rapidly due to continued evolution of the host genome (Katz, 2002, Koonin and Wolf, 2008, Monier et al., 2007), and these methods do not give information about the donor lineage (Katz, 2002). Presence of a gene within only a related subset of a taxonomic group is a possible indicator of HGT if apparent orthologs of the gene are present in unrelated taxa.
Sequence similarity alone is not accepted as a definitive demonstration of HGT or of close evolutionary relationship, since, for example, such results may be dependent on sampling biases present in the search databases used (Koski and Golding, 2001). However, sequence similarity measures can be a powerful tool for scanning very large amounts of data to find promising individual protein candidates for further analysis. Such sequence similarity analyses may also provide evidence for possible large-scale evolutionary trends across an entire virus taxon. This report therefore presents our effort to assess overall trends in HGT for members of the family Poxviridae and to identify individual poxvirus genes that show evidence of HGT for more detailed, subsequent studies.
2. Materials and methods
Protein databases for various taxonomic groups were assembled and searched using BLASTP for best matches to query sets of viral proteins. Results were processed with perl scripts, and displayed in two-dimensional taxonomic group plots. This straightforward method of visually comparing two sets of BLAST scores for a set of proteins has been utilized previously to compare proteins of a single genome to the proteins of two other genomes (NCBI, 2006, Rasko et al., 2005), and to compare proteins of one taxonomically grouped set of genomes to the proteins of two other taxonomically grouped sets of genomes (Lefkowitz et al., 2006).
2.1. Taxonomic group plots
-
•
Protein sequences were downloaded from GenBank (Benson et al., 2008) and from the Viral Bioinformatics Resource Center (Lefkowitz et al., 2005, VBRC, 2008) and were sorted by taxonomic divisions into BLAST-formatted databases. Datasets were downloaded in June 2007 and August 2007. These datasets from 2007 were the primary datasets used for the analyses presented in this report. Datasets were again downloaded in March 2009 and the analyses repeated to detect any significant changes that might occur in the results due to additions and changes in GenBank sequences. No significant differences were obtained. (Table S1 provides a list of downloaded protein groups used for analysis.)
-
•
The set of all proteins predicted to be encoded by poxviruses, and subsets of this set, were used to query individual taxonomic databases using NCBI command line blast version 2.2.15.
-
•
The single best BLASTP hit (based on BLAST bit score) for each query protein against each database was identified. Query proteins that gave low scores against each taxonomic database and would therefore be plotted, along with every other insignificant hit, near the origin of the graph, were removed from the analysis. The threshold for exclusion from the analysis was usually a bitscore of less than 60 with an E value of 10−5 or larger.
-
•
Results for each query protein were plotted on two-dimensional graphs with axes corresponding to mutually exclusive taxonomic databases. This allowed visual comparison of the query set's relationship to one taxonomic group to its relationship to another taxonomic group. This was done with a custom Java applet.
-
•
Normalization of bitscores using alignment length, or use of other measures such as percent identity or similarity, did not significantly change the results.
2.2. Interpretation of results
When the two taxonomic group databases being compared yield similar scores for a query protein, this indicates that each group contains at least one protein with about the same degree of pairwise similarity to the query protein. A set of query proteins with scores that are similar between the two databases creates a diagonal between the two axes.
When a point lies closer to one axis than to the other, this indicates that its best blast hit in the taxonomic group represented by the nearer axis has a greater degree of pairwise similarity to the query protein than its best match in the taxonomic group representing the opposing axis.
Any tendency of a set of query proteins to skew towards a particular taxonomic group might suggest a common evolutionary origin for those sequences either through descent from a common ancestor, or through multiple horizontal gene transfer events.
Each point on the graph is plotted based on the score of its single highest scoring hit in each of the target databases. In many cases, the target database provides many hits with scores that are nearly as high as the score for the single best hit. So the identity of the protein with the single best hit is only one representative of the group of all proteins with hits that exhibit closely related scores. The BLASTP bitscore is one of several metrics that can provide an indication of the degree of similarity between two proteins. No pairwise sequence metric can definitively establish an evolutionary relationship between two protein sequences, but many, including bitscore, can give clues regarding protein similarities. The similarities can be collectively examined to gauge general trends in the similarity between proteomes, and individual similarities might be suggestive of evolutionary relationships between proteins, which may then be followed up using more rigorous methods of investigation, such as phylogenetic analyses, to assess the nature and likelihood of possible evolutionary relationships between individual proteins.
2.3. Phylogenetic analyses
Phylogenetic analyses were conducted on a small number of poxvirus proteins suggested by taxonomic group plots as having potentially interesting evolutionary histories. Each poxvirus protein was aligned with the protein providing its best BLASTP score as depicted in the plot, along with similar sequences from representative taxa. Sequences were aligned using the CLUSTALW algorithm (Thompson et al., 1994) implemented in MEGA version 4 (Tamura et al., 2007). Consensus phylogenetic trees were constructed by the Maximum Parsimony method using MEGA version 4, by the Maximum Likelihood method using Garli 0.96 (Zwickl, 2006), and by Bayesian inference using MrBayes 3.12 (Ronquist and Huelsenbeck, 2003).
3. Results
Our initial analysis was performed using as a query set, all proteins predicted to be encoded by all species and isolates in both the chordopoxvirus and entomopoxvirus subfamilies of the Poxviridae. This query set was used to probe three protein databases: all proteins encoded by eukaryotes, all proteins encoded by bacteria, and all virus-encoded proteins except those encoded by poxviruses. The results from the viral protein database were plotted against results from eukaryote proteins (Fig. 1A) and against results from bacterial proteins (Fig. 1B), and results from the eukaryote and bacterial protein databases were plotted against each other (Fig. 1C). Overall, the resulting plots show that chordopoxvirus proteins tend to exhibit greater similarity to eukaryotic proteins than to bacterial or viral proteins, suggesting that many poxvirus proteins may share a common evolutionary origin with proteins of their eukaryotic hosts. The plots in Fig. 1 distinguish between chordopoxvirus and entomopoxvirus subsets of poxvirus proteins. Although entomopoxviruses share several of the host-like genes present in chordopoxviruses, entomopoxvirus proteins do not show the same general skew towards greater similarity with eukaryotic proteins in comparison to other viral proteins. This could be due to the relative shortage of insect sequences in GenBank or to a bias of entomopoxviruses towards trading genes with other insect viruses over acquiring them from hosts.
Fig. 1.
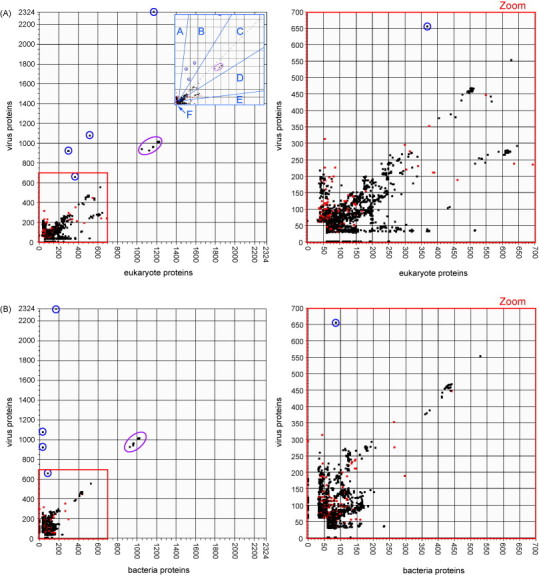
(A) Best BLASTP scores of poxvirus proteins against all eukaryote proteins vs. against proteins of all viruses except poxviruses. Inset shows regions described in the text. (B) Best BLASTP scores of poxvirus proteins against all bacteria proteins vs. against proteins of all viruses except poxviruses. (C) Best BLASTP scores of poxvirus proteins against all eukaryote proteins vs. against all bacteria proteins. Plotted chordopoxvirus proteins (6443 points) are represented by black squares, and plotted entomopoxvirus proteins (278 points) are represented by red squares. Notable points mentioned in the text are circled. Points circled in blue are proteins of an avian retrovirus integrated into the genome of fowlpox virus. The cluster of points circled in purple are the large subunit of ribonucleotide reductase.
3.1. Poxvirus proteins: eukaryote vs. virus axes
The prominent very high scoring proteins that skew towards the virus protein axis in both plots (Fig. 1A and B) are encoded by the copy of the avian retrovirus, reticuloendotheliosis virus, which has integrated into the genome of fowlpox virus, of the Avipoxvirus genus (Hertig et al., 1997). These proteins score very high against the virus database because they are identical to those encoded by reticuloendotheliosis virus. Their closest cousins among eukaryote-encoded proteins, also providing high blast bitscores, are those coded for by endogenous retroviruses of pig, koala and possum.
Many proteins lie in clusters, which are nearly always made up of orthologous proteins from various poxvirus species. Because of slight sequence variations between orthologs, the proteins in a cluster get slightly different scores against the target database proteins, but still close enough to form an orthologous cluster. One example is the cluster of ribonucleotide reductase large subunit (RNR1) poxvirus orthologs that is circled in Fig. 1A–C.
The proteins in Fig. 1A segregate into six categories based on their location on the plot: (A) along, or very close to the virus axis; (B) in the region between the diagonal and the virus axis; (C) on the diagonal between the two axes; (D) in the region between the diagonal and the eukaryote axis; (E) along or close to the eukaryote axis; and (F) proteins that fall near the origin and therefore do not exhibit significant sequence similarity to any proteins from other virus families or from eukaryotic species. Each category has its own range of values for the ratio of virus similarity to eukaryote similarity. For example, proteins in category A have recognizable sequence similarity to proteins of other viruses, as compared to their insignificant levels of similarity to proteins of eukaryotes, while category E is just the inverse, with a high eukaryote-to-virus sequence similarity ratio. Category C proteins have relatively equal levels of sequence similarity to proteins of both viruses and eukaryotes, and regions B and D on either side of the diagonal have recognizable similarity to proteins in both the eukaryote and virus databases, but get a higher score to one database than to the other. Poxvirus proteins plotted in the same region may have similar scores or similar score ratios, but they are not necessarily similar to one another in any other way, either by sequence similarity, by sequence length, by distribution among poxviruses, or by the species in either searched database which provide their closest match. Region F contains the majority of points, with 77% of points. A breakdown by numbers and percentages of points in each region of the plots in Fig. 1 is shown in Table 1 . A table of poxvirus proteins present in each region of Fig. 1A is available as supplemental Table S2. Table S2 identifies the taxonomic subset of poxviruses that encode each protein, the eukaryotic and/or virus species that exhibit the best scores to the poxvirus protein(s) and what is known about the function of the protein. This approach to evolutionary classification of poxvirus proteins is similar to that used to classify proteins of molluscum contagiosum virus (Senkevich et al., 1997).
Table 1.
Number and percentage of poxvirus points in each region of Fig. 1.
| Region | No. of points | % of points |
|---|---|---|
| Fig. 1A: X axis eukaryote proteins, Y axis proteins of all viruses except poxviruses | ||
| A, near Y axis | 657 | 2.3% |
| B, between Y axis and diagonal | 65 | 0.2% |
| C, near diagonal | 1,657 | 5.7% |
| D, between diagonal and X axis | 1,377 | 4.8% |
| E, near X axis | 2,965 | 10.3% |
| F, near origin | 22,190 | 76.8% |
| Fig. 1B: X axis bacteria proteins, Y axis proteins of all viruses except poxviruses | ||
| A, near Y axis | 1,695 | 5.8% |
| B, between Y axis and diagonal | 619 | 2.1% |
| C, near diagonal | 1,065 | 3.7% |
| D, between diagonal and X axis | 622 | 2.1% |
| E, near X axis | 1,307 | 4.5% |
| F, near origin | 23,783 | 81.8% |
| Fig. 1C: X axis eukaryote proteins, Y axis bacteria proteins | ||
| A, near Y axis | 258 | 0.9% |
| B, between Y axis and diagonal | 45 | 0.2% |
| C, near diagonal | 1,000 | 3.4% |
| D, between diagonal and X axis | 1,882 | 6.5% |
| E, near X axis | 3,371 | 11.6% |
| F, near origin | 22,535 | 77.5% |
The following sections outline representative poxvirus proteins from each category identified in Fig. 1A, identifying and discussing them in terms of their similarity to proteins from other virus families and/or eukaryotic species, their degree of distribution among poxvirus species, and their general category of function or putative function.
3.1.1. Region A: points near the virus axis
Poxvirus proteins that fall in this region of the plot have significant levels of sequence similarity to proteins of viruses in other virus families, but have no similarity to proteins of eukaryotes. For each poxvirus protein and its high scoring non-poxvirus protein or proteins, this high level of similarity could be due to a shared evolutionary origin, or to convergent evolution of proteins serving the same role in viruses with similar evolutionary niches.
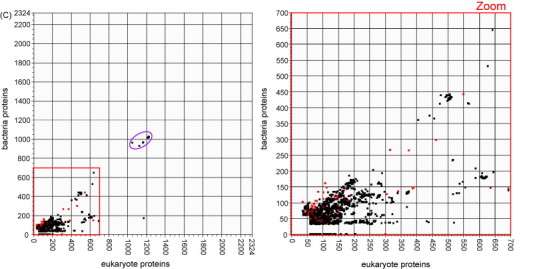
The highest scoring chordopoxvirus proteins along the virus axis are the large group of homologues of the variola virus protein B22R, whose high scores against the virus database result from a single possible relative of this protein present in cyprinid herpesvirus 3 (CyHV-3), a recently discovered member of the family Alloherpesviridae which is notable for having several genes with unexpected high levels of similarity to poxvirus genes (Ilouzea et al., 2006). B22R is present in every chordopoxvirus genus except parapoxvirus, and is the largest protein encoded by poxviruses. While its function is still unknown, it is predicted to contain carboxyl-terminal transmembrane domains and cysteine residues which may mediate disulfide bond formation (Tulman et al., 2006). The position of this protein in a sparsely populated area of the plot, and its potential for relationship to a protein in a herpesvirus makes it a good candidate for further investigation by phylogenetic analysis. The consensus tree of the high scoring sequence from CyHV-3 and representative poxvirus sequences in Fig. 2A shows that a horizontal transfer event may have occurred between virus predecessors of crocodile poxvirus and CyHV-3.
Fig. 2.
Phylogenetic reconstructions to investigate evolutionary histories of three poxvirus proteins appearing in different regions of the plot in Fig. 1A. All pictured trees were constructed by the method of Bayesian inference using MrBayes. The resulting topology for each tree agrees exactly with topology produced from the same alignment by the Maximum Likelihood method using Garli, and either agrees exactly or is very similar to topology produced by the Maximum Parsimony method using MEGA. MrBayes simulations for all three alignments were run with the GTR nucleotide substitution model and gamma distributed rate variation with an estimated proportion of invariable sites. The legend below each tree shows the scale for branch lengths as measured in expected nucleotide substitutions per site. The number to the right of each taxon name is the protein GI number for that sequence. (A) Variola virus B22R (plotted in region A near the virus axis) is a large surface glycoprotein and appears outside the poxvirus family only in the carp herpesvirus CyHV-3. (B) The interleukin-10 inhibitory cytokine (plotted on the diagonal) is evidently of eukaryote origin but has several apparent homologs in diverse virus genomes, potentially acquired in distinct gene transfer events. (C) Monoglyceride lipase (plotted in region E near the eukaryote axis) is an enzyme which may facilitate use of cellular fatty acids, and may have been acquired from a fish or reptilian host by a poxvirus ancestral to the orthopoxvirus and yatapoxvirus genera.
Nucleoside triphosphatase I (NPH-1) transcription termination factor is the only protein appearing along the virus axis that is encoded by both entomopoxviruses and chordopoxviruses. This protein is found in most chordopoxvirus genera as well as in Melanoplus sanguinipes entomopoxvirus (MSEV), and all versions get best scores against viruses of the NCLDV group.
Among proteins along the virus axis with scores above 100 (approximate E values less than 10−22), there are 11 groups of orthologous proteins encoded by the entomopoxviruses, and 5 orthologous groups of proteins encoded by the chordopoxviruses. The highest scoring points include entomopoxvirus DNA and RNA repair enzymes, RNA ligase, and NAD+-dependent DNA ligase. Some entomopoxvirus proteins plotted in this region get their highest scores against proteins of viruses in the NCLDV group, but equally as many get high scores against proteins of viruses in the family Baculoviridae, where several, including the Fusolin/gp37 protein, and the Methionine-threonine-glycine (MTG) motif gene family member appear to enhance virus infectivity of the insect host (Dall et al., 2001). While many of the entomopoxvirus proteins plotted in this region score very high against proteins in other viruses, they are of unknown function, and contain no characterized domains.
3.1.2. Region B: points between the virus axis and the diagonal
Proteins plotted in this region have relatively high sequence similarity to proteins of other viruses as compared to their levels of similarity to eukaryote proteins. These poxvirus proteins may have a shared evolutionary origin with both virus and eukaryote ancestors, with greater similarity between the virus homologs due to similar evolutionary selection pressure and functional constraints on the virus genes, in contrast to the selection pressure on the eukaryotic versions of the protein. Poxvirus proteins in this category may share only one or a few protein domains with similar eukaryotic proteins, while best hits with proteins from other virus families exhibit similarity across the entire protein sequence.
Besides the proteins encoded by the reticuloendotheliosis virus integrated into fowlpox virus, the only points with scores above 100 which fall into region B, between the virus axis and the diagonal are encoded by members of the species Canarypox virus. CNPV153 has closest match with the viral replication protein, Rep, of members of the family Circoviridae, and the CNPV227 N1R/p28-like protein has closest match to acanthamoeba polyphaga mimivirus, of the family Mimiviridae.
3.1.3. Region C: points near the diagonal
Region C, surrounding the diagonal, contains proteins whose sequences are globally conserved throughout most DNA viruses and eukaryotes, but it also contains proteins which get a high score against sequences present in only one or a few members of the eukaryote or virus kingdom. Poxvirus proteins plotted in this region find best scores in the virus kingdom among possibly distantly related viruses, i.e. members of the NCLDV, as well as among species of other families including Herpesviridae and Adenoviridae. Many of these proteins are universally highly conserved, function in the synthesis and maintenance of DNA and RNA, and are present in many members of both the poxvirus family and the virus family in which the highest score is obtained, as well as in most eukaryotes. The ultimate origin of these proteins is uncertain, and their entries into the virus lineages may have occurred concurrently with the inception of the first ancestors of these viruses, or at many different times during the evolution of the different virus families. Other proteins plotted in this region are apparently of eukaryote origin, have functions involving immune response and intracellular processes, and seem likely to have been transferred horizontally from hosts into the corresponding virus families.
The highest scoring proteins along the diagonal are the large and small subunits of ribonucleotide reductase (RNR) (class 1A), an enzyme that controls the cellular concentration of deoxyribonucleotides. Although there are three classes of RNR, only class 1, subclass A is found in eukaryote-infecting viruses. RNR class 1 is made up of large (RNR1) and small (RNR2) subunits, with two of each subunit required to associate into a heterotetramer to form a functioning enzyme (Stubbe, 1990). Both subunits are very well conserved in all major taxonomic groups in which RNR type I appears: eukaryotes, eubacteria, bacteriophages and eukaryotic viruses. The large subunit of RNR (RNR1) is present in orthopoxviruses and suipoxviruses, while the small subunit of RNR (RNR2) is present in most chordopoxviruses. For both subunits, the percent identities between queries and highest scoring hits are between 80% and 90% percent similarity, with such high levels of sequence conservation likely due to the stringent structural requirements the enzyme must maintain in order to function (Torrents et al., 2002). Although many chordopoxvirus species encode only the small subunit, it is probably functioning in association with host-encoded RNR1, based on the finding that even RNR subunits from vastly different species can associate to form heterotetramers (Hamann et al., 1998).
In addition to the very high scoring RNR proteins, many other enzymes involved in nucleotide synthesis and metabolism are high on the diagonal, including deoxyuridine-triphosphatase (dUTPase), thymidine kinase (TK), thymidylate kinase (ThyK), deoxycytidine kinase, and the one example of thymidylate synthase present in poxviruses. All these enzymes catalyze steps in pyrimidine metabolism, in particular converting cellular pools of RNA components into nucleotides for synthesis of DNA. Also high on the diagonal are DNA polymerase, alpha and beta subunits of RNA polymerase, and DNA photolyase, a DNA repair enzyme well conserved in all branches of life, but notably missing from placental mammals. The poxvirus proteins in this category are widely, some even ubiquitously, distributed among poxviruses, and have most similar viral proteins outside poxviruses in a wide variety of double stranded DNA viruses, including members of the postulated NCLDV group of viruses such as the phycodnaviruses, iridoviruses and mimiviruses, as well as in viruses outside this group, such as adenoviruses and herpesviruses. Eukaryotic best hits come from an even wider range, spanning everything from fungi and plants to vertebrates and invertebrate animals. These types of proteins fulfill basic needs of DNA viruses and all organisms with DNA genomes, and both their omnipresence in nature and the high levels of sequence conservation can be confounding factors in attempts to phylogenetically trace their individual evolutionary lineages.
Many poxvirus proteins plotted on the diagonal have limited distribution among poxviruses and have best virus hits almost exclusively in putatively unrelated viruses, such as members of the baculovirus and herpesvirus families. Proteins in this category, which probably participate in downregulation of the host immune response, include interleukin-10 (IL-10) proteins and complement-control proteins. This category also includes semaphorins and c-type lectin-like proteins whose functions in poxviruses are unknown, but similar proteins in other organisms have roles in immunological pathways. Poxvirus encoded apoptosis-inhibiting proteins and copper/zinc superoxide dismutase protect infected cells against programmed cell death. These virally encoded proteins find highest scores among eukaryotes which seem likely to be hosts or closely related to hosts of the respective viruses, which make the viral proteins seem likely to be the products of independent horizontal gene transfer events from hosts. Although actual assessments of such potential gene transfers may be provided only by further analysis of each gene group, notably by phylogenetic inference, the locations of the points on these plots and the identities of the highest scoring proteins on each axis suggest candidates for study, and provide clues as to which proteins may yield the most interesting results. A good candidate for further study is IL-10, presumably of eukaryotic origin, but with several apparently homologous proteins among poxviruses and herpesviruses. A phylogenetic reconstruction of several viral and host IL-10 sequences is provided in Fig. 2B. Analysis of the phylogenetic relationship between these proteins suggests the possibility of several independent IL-10 HGT events between hosts and infecting viruses. Three HGT events are suggested into different lineages of herpesviruses, and two separate HGT events are suggested for poxviruses, with one each into the capripoxvirus and parapoxvirus lineages. It is notable that for many of these HGT events, the most closely related eukaryote IL-10 protein to a specific virus IL-10 protein is between the particular host species and the virus that infects that host.
A few orthologous groups of proteins plotted in this region have functions unrelated to DNA/RNA/nucleotide synthesis and have closest viral hits in viruses of the NCLDV group. The eukaryote species providing the highest scores to these proteins seem unlikely to be hosts of the respective poxviruses. 3-Beta-hydroxysteroid dehydrogenase proteins are widely distributed among poxvirus genera, are likely used to suppress the host inflammatory response, and find most similar virus proteins in fish-infecting iridoviruses. Orthopoxvirus and entomopoxvirus species encode a protein called vaccinia-related ser/thr kinase, which is widely distributed in the animal kingdom and seems to participate in regulation of cell cycle (Kang et al., 2008) and has closest virus relatives in iridovirus species. A few proteins with very limited poxvirus distribution have unknown functions and highest pairwise similarity to proteins of NCLDV member species. The ultimate evolutionary origins of these proteins are unknown.
3.1.4. Region D: points midway between the diagonal and the eukaryote axis
This region of the plot is one of the most densely populated, with many poxvirus proteins that show significant hits to eukaryotic proteins and lower scores to homologs in other viruses. As with region C, region D contains proteins whose high scores seem due to universal sequence conservation, as well as proteins of presumably eukaryote origin, whose high scores on both axes most likely reflect historical transfer of these genes by separate routes into poxviruses and other virus families.
Several poxvirus protein families have points in both regions C and D, including the vaccinia-related kinase family, the c-type lectin-like proteins, the TK enzymes, and ankyrin repeat proteins. As with their orthologs in region C, best virus matches for these are found both among NCLDV and non-NCLDV DNA viruses. All these are encoded by viruses in many poxvirus genera, and get best eukaryote hits among a variety of animals.
The best scoring protein sequences in region D of the plot are the ATP-dependent DNA ligases encoded by several poxvirus genera. These all have higher pairwise identity to proteins of various mammals than to their best virus hits, which are all among putatively unrelated nucleopolyhedrovirus (NPV) species, a group of viruses in the baculovirus family. This is the only DNA-related enzyme unique to this region of the plot.
Two additional region D proteins with wide distribution among poxvirus genera may have functions modulating host immune response. These are G protein-coupled receptors (GPCR) with significant similarity to known CC chemokine receptors, and proteins in the serpin superfamily of proteinase inhibitors, which are implicated in the regulation of tumor progression, of inflammation, and of cell death (Silverman et al., 2001, Viswanathan et al., 2009). Various mammals provide the best eukaryotic blast scores for most of these sequences, but some avipoxvirus proteins score best to a chicken protein. Herpesviruses provide best virus scores to most of the GPCR proteins, while mimivirus gives the best score fort all the serpins, as it has the only known viral serpin outside the poxvirus family. A third set of proteins, the soluble tumor necrosis factor receptor (TNFR) II homologs, and has slightly less widespread poxvirus distribution. These putatively protect infected cells from TNF-mediated cell death, and get highest scores to proteins of various mammals, and to viruses of the herpesvirus and iridovirus families.
Proteins with very limited distribution among poxvirus genera include a protein similar to eukaryotic initiation factor-4a (eIF-4a) and a protein possibly functioning as an oligoribonuclease, both encoded by diachasmimorpha longicaudata entomopoxvirus, the first known symbiotic entomopoxvirus, which infects a parasitic wasp. The best eukaryotic scores for these proteins come from potentially host-like species and both find best virus scores against NCLDV members. A possible dual specificity protein phosphatase, encoded by canarypox virus, and a protein similar to human MHC Class I, encoded by squirrel poxvirus, are plotted in this region for blast scores against proteins of vertebrates and non-NCLDV viruses. MHC Class I-like proteins encoded by poxviruses of several other genera are plotted very close to the origin due to low pairwise sequence identity to their best matches on both axes. MSEV and squirrel poxvirus each encode a sequence of unknown function, and which, although they do not share high identity with one another, may both be chromosome segregation ATPases.
3.1.5. Region E: points near the eukaryote axis
Region E contains poxvirus proteins that get notable scores against eukaryotic proteins and essentially insignificant scores against viral proteins. Poxvirus proteins that appear in this region are most likely of eukaryotic origin and have been transferred into poxviruses or ancestors of poxviruses, but have not been transferred or at least not maintained in sequenced viruses of any other present day virus family. Members of the poxvirus family may be the only virus species carrying these eukaryotic genes simply because poxviruses are more effective at capturing or maintaining host genes than other viruses. Alternatively, many of these genes may be absent from other viruses since they confer little or no selective advantage to these viruses, but do confer selective advantage to poxviruses due to unique aspects of their biology.
Poxvirus proteins plotted in this region include enzymes involved in lipid and carbohydrate metabolism, nucleotide metabolism, protection against oxidative damage, and intracellular processes including signaling, cell cycle control and apoptosis. The few proteins in this region which have wide distribution among poxvirus genera are kelch proteins and tyrosine protein kinase-like proteins. These proteins have unknown functions, and get best scores to proteins in a variety of vertebrates.
Orthologous groups of proteins from avipoxviruses appear more often in this region than proteins from any other genus. Several of these proteins have unknown function, but the functional characterizations of the others span the whole range of functions attributed to proteins of region E. Each orthologous group of avipoxvirus proteins gets best scores against a variety of eukaryotes, mostly vertebrates. Again, assessments of potential horizontal gene transfers may be provided only by detailed phylogenetic analysis of each gene group, but the wide range of vertebrates providing highest scores for each orthologous group is notable, and preliminary phylogenetic analyses (data not shown) may indicate that, although they score very well against vertebrate proteins, many of these avipoxvirus proteins may have begun diverging from the original host-acquired proteins in the ancient past.
Glutathione peroxidase protects against oxidative damage, and is the only avipoxvirus protein in region E that is also encoded by another poxvirus genus. Molluscum contagiosum virus encodes an ortholog of glutathione peroxidase that gets its highest blastp score against a similar protein in macaque, while the avipoxvirus sequences get highest scores against insect versions of the protein.
As with the avipoxvirus proteins, many of these proteins may have been transferred into the poxvirus lineage in the relatively distant past, from early vertebrates. The phylogenetic tree of the enzyme monoglyceride lipase (Fig. 2C), which appears in this region of the plot, provides evidence that the origin of the poxvirus homolog may represent a more ancient gene transfer into a poxvirus ancestor from an unknown host.
Many orthologous groups of proteins in this region have best blast scores scattered over a wide range of vertebrates, rather than among a narrowly defined group of species related to a potential HGT source. However, with only two exceptions both encoded by avipoxvirus species, all proteins find best scores against vertebrates, rather than against the wide variety of metazoa which provide the best scores for many of the potentially more universally conserved proteins plotted closer to the diagonal.
3.1.6. Region F: points near the origin
Approximately 77% of poxvirus proteins fall very close to the origin of this plot. These include genes that may be unique to the poxvirus family, as well as genes that in poxviruses have primary sequences too divergent to achieve high blastp scores against potentially orthologous proteins outside poxviruses. Examples of the former, poxvirus-specific genes include a DNA-binding phosphoprotein (Cop-F17R) and a structural protein (Cop-A12L). Examples of the latter, sequence-diverged genes, are a putative ATPase (Cop-A32L) and a capsid protein (Cop-D13L), both postulated to have orthologs in all members of NCLDV and included in the originally proposed core NCLDV genes (Iyer et al., 2001).
3.2. Poxviruses: bacteria vs. virus axes
In addition to the comparison of poxvirus proteins to proteins of eukaryotes and other viruses, we also compared the similarity of poxvirus proteins to proteins of bacteria and other viruses (Fig. 1B). For almost all poxvirus proteins, bacteria provide lower pairwise scores than eukaryotes. Notably, most of the large groups of proteins that lie on and below the diagonal in Fig. 1A skew in Fig. 1B towards the virus axis due to the absence of similar proteins in the bacterial kingdom.
With the exception of one entomopoxvirus protein, all proteins between the virus axis and the diagonal got higher scores against eukaryotes (Fig. 1A) than they get against bacteria. The exception is NAD+-dependent DNA ligase, encoded by MSEV and amsacta moorei entomopoxvirus (AMEV), which gets slightly higher scores against a sulfur-oxidizing bacterium and a fish-infecting mycoplasma than against its eukaryote best hits in amoeba. The unique status of this point on the plot marks it as a potentially interesting candidate for additional investigation. Preliminary analyses (data not shown) indicate that while apparent homologs of this gene are found predominately in bacterial genomes, a few are also found among species of bacteriophage and NCLDV, indicating a potential for interesting horizontal gene transfer events. These and all other such suggested relationships must of course be rigorously tested by phylogenetic analysis to provide the most reliable assessment of gene transfer pathways.
As in Fig. 1A, the diagonal in Fig. 1B contains several proteins highly conserved throughout nature. Fig. 1B also contains several proteins that in Fig. 1A were below the diagonal, showing high similarity to eukaryote proteins, but with scores against bacteria proteins more comparable to other virus proteins thus shifting them to the diagonal in Fig. 1B.
Nearly all points below the diagonal in Fig. 1B exhibit high bitscores against both bacterial and eukaryote proteins, although the eukaryote scores are usually higher. Among these proteins, all proteins with significant scores against virus proteins have mimivirus proteins as their best virus scores—possibly not surprising considering the many bacteria-like features of the mimivirus genome.
Poxvirus proteins plotted near the bacterial axis have similar scores with their best eukaryote protein hits. This region contains more avipoxvirus genes than genes from any other poxvirus genus. The only proteins in this region to get better bacterial than eukaryote scores come from the entomopoxvirus subfamily. There are the two different leucine-rich repeat (LRR) proteins encoded by AMEV, which get moderately good scores against eukaryotes yeast and plants, but get somewhat better scores against both a gram-negative anaerobic bacterium and a symbiotic green sulfur bacterium.
3.3. Poxvirus proteome subset
Although individual poxviruses usually contain more than 150 genes, only 49 of these are present in all of the fully sequenced poxviruses, with larger subsets being shared among members of each genus (Lefkowitz et al., 2006). In poxvirus genomes, the conserved “core” genes are involved in key functions such as replication, transcription and virion assembly, and tend to cluster in the central region of the linear genome, while genes that are unique to specific genera or species are distributed towards the two ends of the genome. Many of these peripheral genes encode proteins that manipulate host immune response and cellular processes, including apoptosis, antigen presentation and recognition, interferon functions and immune signaling processes.
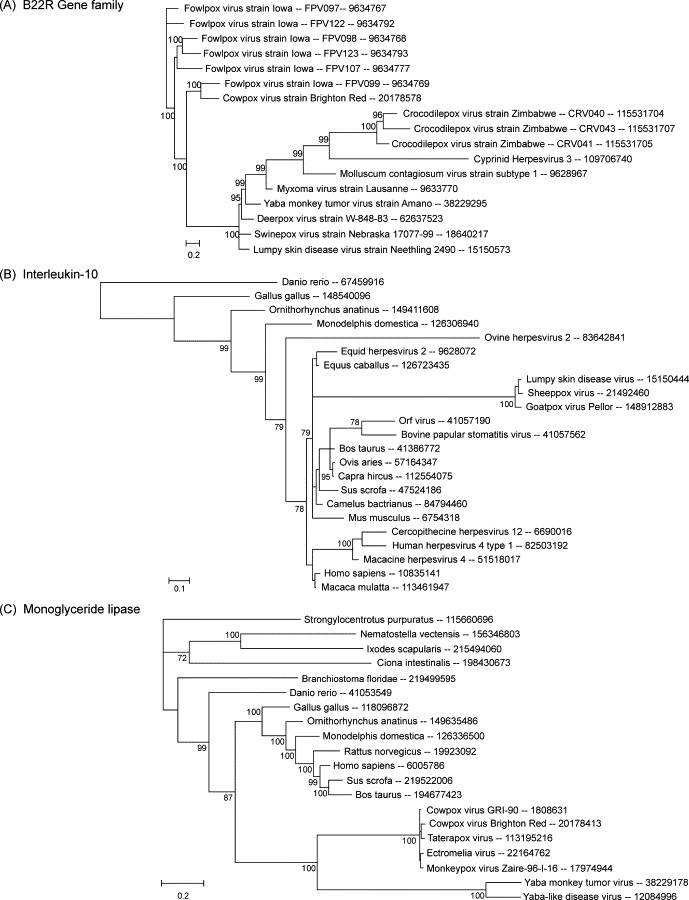
Cowpox virus strain Gri-90 has one of the largest genomes among orthopoxviruses, and contains essentially all genes found in other members of the genus. For this reason, it serves well as an archetypical orthopoxvirus genome for the purpose of orthopoxvirus gene analysis. All proteins of this strain were analyzed by taxonomic group plots, to compare the relationships of core and non-core protein subsets with eukaryotes and with viruses outside the poxvirus family (Fig. 3 ). In Fig. 3A, proteins were classified according to genomic location, as located centrally (red points) or non-centrally (black points), where the central region of the genome is defined as all genes from G13L to A47L. In Fig. 3B, proteins were classified according to the number of poxvirus species with conserved orthologs, with red points representing the most widely conserved proteins among poxviruses, and proteins of most limited distribution in black.
Fig. 3.
All proteins of cowpox strain GRI-90 were analyzed by taxonomic group plots, to compare the relationships of core and non-core protein subsets with proteins of eukaryotes and with proteins of viruses outside the poxvirus family. Panel (A) represents proteins classified according to genomic locus, as non-centrally located (black squares, 99 points) or centrally located (red squares, 115 points). Panel (B) represents proteins classified according to the number of poxvirus species with conserved orthologs, with genes in only 1–10 species in black (20 points), genes in 11–20 species in purple (59 points), genes in 21–30 species in blue (28 points), genes in 31–35 species in green (21 points), and genes in 36–40 species in red (86 points).
Results show that the diagonal contains universally conserved as well as species-specific genes (Fig. 3B), and contains proteins with both central and peripheral locations (Fig. 3A). However, the proteins that lie to the eukaryote side of the diagonal are predominantly non-centrally located and appear in a very limited number of species. Presence of these genes in only one or a few genera or species strongly suggests the genes were acquired by the cowpox virus lineage subsequent to its divergence (or the divergence of the most recent orthopoxvirus ancestor) from the other poxvirus genera. High scores with eukaryotic proteins may also indicate relatively recent transfer of the genes from eukaryotes, and/or strong selection for sequence identity with host proteins. The sparsely populated area near the virus axis has only proteins widely conserved among poxviruses, and these are almost exclusively centrally located, with the one exception being the poxvirus B22R protein. B22R is a surface glycoprotein that is conserved in every chordopoxvirus genus, and as mentioned above, has only one possible homolog outside the poxvirus family, in CyHV-3.
A genome map of cowpox virus strain Gri-90 (Shchelkunov et al., 1998) (GenBank accession no. X94355) in Fig. 4 depicts all cowpox virus genes color coded according to the degree of similarity of each cowpox virus protein to its best hit when compared against all virus (non-poxvirus) or all eukaryotic proteins. Genes and their descriptions are provided in Table 2 . Genes are labeled by their restriction fragment name and are colored according to the highest blastp bitscore obtained by the encoded poxvirus protein when searched against the respective taxonomy database. Bitscores are normalized by dividing by the highest possible bitscore the query protein could achieve, i.e. the bitscore it receives when compared to itself. Therefore the highest possible score for each comparison is 1. The map demonstrates the higher levels of similarity poxvirus proteins have to eukaryote proteins in comparison to virus proteins outside the poxvirus family. In addition, it is apparent that with only a few exceptions, poxvirus proteins with high levels of sequence identity to proteins of other organisms tend to lie towards the edges of the linear genome. Exceptions include S2R: thymidine kinase, L4L: ribonucleotide reductase large subunit, R2L: glutaredoxin 1, and E8L: carbonic anhydrase (virion protein).
Fig. 4.

A genome map of cowpox strain Gri-90 is color coded (see legend) according to the degree of similarity of each cowpox protein to its best hit when compared against all virus (non-pox) or eukaryote proteins.
Table 2.
Similarity of each cowpox virus Gri-90 protein to the best blastp hit in the eukaryote and virus (non-pox) protein datasets.
| Region | CPXV protein | Protein description | Eukaryote with best score | Euk. Bitscore | Virus with best score | Virus Bitscore |
|---|---|---|---|---|---|---|
| A | B22R | Surface glycoprotein | Strongylocentrotus purpuratus | 51 | Cyprinid herpesvirus 3 | 156 |
| E11L | NPH-I/Helicase, virion | Ciona intestinalis | 60 | Acanthamoeba polyphaga mimivirus | 135 | |
| B | J6R | Topoisomerase type I | Leishmania donovani infantum | 64 | Acanthamoeba polyphaga mimivirus | 97 |
| E6R | Morph, VETF-s (early transcription factor small) | Kluyveromyces lactis | 67 | Lymphocystis disease virus 1 | 99 | |
| E5R | NTPase, DNA replication | Pichia stipitis CBS 6054 | 41 | Acanthamoeba polyphaga mimivirus | 60 | |
| C | A41R | Semaphorin/CD100 antigen | Homo sapiens | 135 | Ovine herpesvirus 2 | 146 |
| B4R | Complement control/CD46/EEV | Pan troglodytes | 89 | Macaca mulatta rhadinovirus 17577 | 91 | |
| D9L | C-type lectin | Rattus norvegicus | 86 | Rat cytomegalovirus | 86 | |
| O4R | RNA pol (RPO147) | Mus musculus | 201 | Acanthamoeba polyphaga mimivirus | 186 | |
| A51R | Thymidylate kinase | Aedes aegypti | 167 | Chilo iridescent virus | 151 | |
| G4L | Ribonucleotide Reductase small subunit | Danio rerio | 506 | Lymantria dispar nucleopolyhedrovirus | 457 | |
| F9L | DNA-directed DNA polymerase | Tetrahymena thermophila SB210 | 113 | Human herpesvirus 7 | 102 | |
| A47L | Hydroxysteroid dehydrogenase | Bos taurus | 279 | Rana grylio virus 9506 | 247 | |
| A42R | Lectin homolog | Homo sapiens | 62 | African swine fever virus | 54 | |
| C17L | Complement binding (secreted) | Bos taurus | 190 | Macaca mulatta rhadinovirus 17577 | 166 | |
| J1L | Tyr/Ser phosphatase | Gallus gallus | 69 | Chilo iridescent virus | 61 | |
| C3L | Ankyrin | Trichomonas vaginalis G3 | 88 | Acanthamoeba polyphaga mimivirus | 76 | |
| A25R | RNA pol 132 (RPO132) | Aspergillus niger | 239 | Aedes taeniorhynchus iridescent virus | 207 | |
| G2L | DeoxyUTP pyrophosphatase (dUTPase) | Macaca mulatta | 164 | Spodoptera litura granulovirus | 138 | |
| L4L | Ribonucleotide reductase large subunit | Mus musculus | 1226 | Spodoptera litura nucleopolyhedrovirus | 1011 | |
| B1R | Ser/Thr kinase | Danio rerio | 246 | Chilo iridescent virus | 194 | |
| D | B11R | Ser/Thr kinase | Bos taurus | 154 | Chilo iridescent virus | 120 |
| K1R | Ankyrin | Trichomonas vaginalis G3 | 84 | Acanthamoeba polyphaga mimivirus | 62 | |
| B18R | Ankyrin | Trichomonas vaginalis G3 | 79 | Paramecium bursaria Chlorella virus 1 | 58 | |
| B3R | Ankyrin | Trichomonas vaginalis G3 | 112 | Acanthamoeba polyphaga mimivirus | 82 | |
| C7R | Ubiquitin Ligase/host defense modulator | Homo sapiens | 71 | Rock bream iridovirus | 52 | |
| M1L | Ankyrin/NFkB inhib | Trichomonas vaginalis G3 | 71 | Paramecium bursaria Chlorella virus 1 | 51 | |
| L8R | RNA helicase/NPH-II | Caenorhabditis elegans | 70 | Acanthamoeba polyphaga mimivirus | 50 | |
| C1L | Ankyrin | Strongylocentrotus purpuratus | 64 | Acanthamoeba polyphaga mimivirus | 45 | |
| A56R | TNF receptor (CrmC) | Pan troglodytes | 108 | Grouper iridovirus | 74 | |
| D14L | Ankyrin | Strongylocentrotus purpuratus | 87 | Acanthamoeba polyphaga mimivirus | 59 | |
| B16R | Ankyrin | Trichomonas vaginalis G3 | 91 | Acanthamoeba polyphaga mimivirus | 62 | |
| C11L | Ankyrin | Trichomonas vaginalis G3 | 68 | Acanthamoeba polyphaga mimivirus | 45 | |
| P1L | Ankyrin | Trichomonas vaginalis G3 | 94 | Acanthamoeba polyphaga mimivirus | 61 | |
| K3R | TNF-a receptor/CD27 cysteine-rich region | Rattus rattus | 136 | Singapore grouper iridovirus | 87 | |
| K2R | TNF receptor (CrmD) | Canis familiaris | 120 | Singapore grouper iridovirus | 72 | |
| S2R | Thymidine kinase | Homo sapiens | 248 | Cyprinid herpesvirus 3 | 147 | |
| D2L, I4R | TNF-α receptor II (CrmB) | Bos taurus | 149 | Grouper iridovirus | 87 | |
| M2L | Proteinase inhibitor I4, serpin | Monodelphis domestica | 160 | Acanthamoeba polyphaga mimivirus | 92 | |
| D3L, I3R | Ankyrin | Trichomonas vaginalis G3 | 97 | Acanthamoeba polyphaga mimivirus | 55 | |
| B12R | Serpin | Monodelphis domestica | 196 | Acanthamoeba polyphaga mimivirus | 102 | |
| D13L | Unknown | Mus musculus | 84 | Lymphocystis disease virus—isolate China | 43 | |
| D7L | Kelch-like | Rattus norvegicus | 71 | Clanis bilineata nucleopolyhedrosis virus | 34 | |
| A26L | A type inclusion protein | Trichomonas vaginalis G3 | 134 | Gryllus bimaculatus nudivirus | 64 | |
| O1R | Poly(A) polymerase-small (VP39) | Paramecium tetraurelia | 66 | Vibrio phage CTX | 31 | |
| B20R | Serpin | Bos taurus | 202 | Acanthamoeba polyphaga mimivirus | 94 | |
| C5R | Epidermal growth factor | Rattus norvegicus | 63 | Crimean-Congo hemorrhagic fever virus | 29 | |
| A53R | DNA ligase | Canis familiaris | 622 | Lymantria dispar nucleopolyhedrovirus | 274 | |
| C18L | Kelch-like | Canis familiaris | 111 | Pseudomonas phage phiEL | 48 | |
| A44R | Profilin homolog | Homo sapiens | 72 | Bacteriophage phi-MhaA1-PHL101 | 30 | |
| B19R | Kelch-like (EV-M-167) | Drosophila pseudoobscura | 107 | Acanthamoeba polyphaga mimivirus | 45 | |
| A57R | Kelch-like | Macaca mulatta | 202 | Human papillomavirus type 68 | 71 | |
| D11L | Kelch-like | Canis familiaris | 138 | Pseudomonas phage phiKZ | 43 | |
| E | R2L | Glutaredoxin 1 | Rattus norvegicus | 105 | Ectocarpus siliculosus virus | 33 |
| B9R | Kelch-like | Monodelphis domestica | 115 | Pseudomonas phage phiKZ | 35 | |
| G3L | Kelch-like | Mus musculus | 119 | Pseudomonas phage phiKZ | 35 | |
| A40L | CD47-like | Bos taurus | 119 | Ranid herpesvirus 2 | 34 | |
| G13L | Phospholipase EEV | Canis familiaris | 118 | Heliothis zea virus 1 | 32 | |
| B14R | IL-1 beta receptor | Rattus norvegicus | 144 | Enterobacteria phage RB69 | 35 | |
| E8L | Carbonic anhydrase/Virion | Homo sapiens | 143 | Acanthamoeba polyphaga mimivirus | 32 | |
| B2R | Schlafen | Mus musculus | 231 | Choristoneura fumiferana MNPV | 47 | |
| A59R | Guanylate kinase | Mus musculus | 202 | Sapovirus SaKaeo-15/Thailand | 35 | |
| M5L | Putative monoglyceride lipase | Rattus norvegicus | 280 | Paramecium bursaria Chlorella virus 1 | 42 | |
| T1R | NMDA receptor-like protein | Bos taurus | 256 | Chimpanzee cytomegalovirus | 32 | |
| M4L | Nicking-joining enzyme | Rattus norvegicus | 368 | Bombyx mori nuclear polyhedrosis virus | 32 | |
| F | E10R | mutT motif/NPH-PPH/RNA levels regulator | Tetrahymena thermophila SB210 | 40 | Aedes taeniorhynchus iridescent virus | 53 |
| A19R | DNA Helicase, transcription | Ashbya gossypii ATCC 10895 | 48 | Ectocarpus siliculosus virus | 59 | |
| D10L | CPV-B-012 | Rattus norvegicus | 49 | Rat cytomegalovirus | 51 | |
| E1R | Large capping enzyme | Trichomonas vaginalis G3 | 58 | Acanthamoeba polyphaga mimivirus | 52 | |
| D4L, I2R | Ankyrin | Aedes aegypti | 59 | Acanthamoeba polyphaga mimivirus | 46 | |
| A48R | Superoxide dismutase-like | Lasius niger | 57 | Mamestra configurata nucleopolyhedrovirus B | 44 | |
| C9L | Ankyrin/host range | Trichomonas vaginalis G3 | 59 | Acanthamoeba polyphaga mimivirus | 45 | |
| D8L | Ankyrin | Trichomonas vaginalis G3 | 59 | Ectocarpus siliculosus virus | 42 | |
| A22R | DNA processivity factor | Plasmodium falciparum 3D7 | 52 | Bacteriophage 85 | 35 | |
| C15L | Unknown | Monodelphis domestica | 55 | Acanthamoeba polyphaga mimivirus | 36 | |
| F3L | IFN resistance/PKR inhibitor (Z-DNA binding) | Rattus norvegicus | 52 | Paramecium bursaria Chlorella virus 1 | 33 | |
| B17R | IFN-alpha/beta receptor | Pan troglodytes | 56 | Listeria phage A118 | 32 | |
| B7R | IFN-gamma receptor | Canis familiaris | 57 | Choristoneura occidentalis granulovirus | 31 | |
| C10L | Unknown | Trypanosoma rangeli | 20 | Enterobacteria phage K1-5 | 19 | |
| A39R | Unknown | Entamoeba histolytica HM-1:IMSS | 24 | Rabies virus | 23 | |
| H6R | RNA pol | Debaryomyces hansenii CBS767 | 27 | Acanthamoeba polyphaga mimivirus | 24 | |
| N2R | Unknown | Entamoeba histolytica HM-1:IMSS | 27 | Pseudomonas phage D3 | 25 | |
| A14L | Virion maturation | Pichia stipitis CBS 6054 | 29 | Equid herpesvirus 2 | 26 | |
| A31L | Virion morphogenesis | Paramecium tetraurelia | 29 | Plum pox virus | 25 | |
| NULL | Unknown | Musca domestica | 29 | Rice tungro bacilliform virus | 25 | |
| G8L | Cytoplasmic protein | Aspergillus oryzae | 28 | Cryptophlebia leucotreta granulovirus | 27 | |
| G14L | Unknown | Cosmospora coccinea | 29 | Acanthamoeba polyphaga mimivirus | 27 | |
| B10R | Unknown | Paramecium tetraurelia | 29 | Tomato chlorosis virus | 27 | |
| A46R | Unknown | Plasmodium falciparum 3D7 | 30 | Human immunodeficiency virus 1 | 26 | |
| G17R | DNA-binding phosphoprotein | Plasmodium vivax | 30 | Murid herpesvirus 1 | 26 | |
| NULL | Unknown | Dictyostelium discoideum AX4 | 31 | Influenza A virus (A/seal/Massachusetts/1/80(H7N7)) | 27 | |
| L5L | IMV protein VP13 | Mustela vison | 32 | Staphylococcus phage Twort | 28 | |
| A13L | Structural protein | Cryptococcus neoformans var. neoformans B-3501A | 32 | Bacteriophage phBC6A51 | 28 | |
| A10L | Membrane protein | Pichia stipitis CBS 6054 | 34 | Influenza A virus (A/Hong Kong/481/97(H5N1)) | 26 | |
| A3L | Thioredoxin-like | Caenorhabditis elegans | 31 | Impatiens necrotic spot virus | 30 | |
| A18L | IMV MP PO4 | Plasmodium vivax | 35 | Mycoreovirus 3 | 27 | |
| L2L | Unknown | Aspergillus niger | 30 | Cyanophage phage S-PM2 | 31 | |
| A15L | IMV PO4 MP | Tetrahymena thermophila SB210 | 32 | Gryllus bimaculatus nudivirus | 30 | |
| C2L | MPV-Z-N3R | Xenopus tropicalis | 32 | Avian infectious bronchitis virus | 30 | |
| N5R | Entry and fusion IMV protein | Cryptosporidium parvum Iowa II | 33 | Chrysodeixis chalcites nucleopolyhedrovirus | 29 | |
| G15L | Unknown conserved | Paramecium tetraurelia | 34 | Influenza A Virus (A/Fujian/555/2003(H3N2)) | 28 | |
| B13R | Unknown | Aspergillus nidulans FGSC A4 | 34 | American plum line pattern virus | 29 | |
| G7L | Unknown | Plasmodium yoelii yoelii | 37 | Lactococcus lactis bacteriophage Q30 | 26 | |
| F10R | Disulfide bond formation | Plasmodium berghei | 31 | African swine fever virus | 32 | |
| H2L | Unknown | Drosophila melanogaster | 34 | Plutella xylostella multiple nucleopolyhedrovirus | 30 | |
| A16L | Unknown | Plasmodium berghei | 34 | Rice stripe virus | 30 | |
| B5R | Unknown | Leishmania braziliensis | 34 | Little cherry virus 1 | 29 | |
| J2R | Entry and cell–cell fusion | Canis familiaris | 34 | Acanthamoeba polyphaga mimivirus | 29 | |
| E7R | RNA pol 18(RPO18) | Tetrahymena thermophila SB210 | 34 | Glypta fumiferanae ichnovirus | 30 | |
| A5L | Core protein | Monodelphis domestica | 34 | Hibiscus latent Fort Pierce virus | 30 | |
| G6L | Unknown | Plasmodium chabaudi | 35 | Bacteriophage 933W | 29 | |
| D6L | Alpha-amanitin sensitivity | Plasmodium falciparum 3D7 | 34 | Bacillus thuringiensis phage MZTP02 | 31 | |
| A36R | Unknown | Schizosaccharomyces pombe | 34 | Acanthamoeba polyphaga mimivirus | 30 | |
| G16L | Unknown | Trichomonas vaginalis G3 | 34 | Maize dwarf mosaic virus | 30 | |
| H7R | Unknown | Tetrahymena thermophila SB210 | 35 | Streptococcus thermophilus bacteriophage Sfi19 | 29 | |
| A32R | Unknown | Dictyostelium discoideum | 35 | Avian infectious bronchitis virus | 30 | |
| M6R | Unknown | Aspergillus terreus NIH2624 | 34 | Feline calicivirus | 31 | |
| A34R | EEV Glycoprotein | Caenorhabditis elegans | 35 | Feline leukemia virus (strain Sarma) | 30 | |
| F7R | Soluble/Myristyl EEV | Plasmodium chabaudi | 35 | Chilo iridescent virus | 29 | |
| J5R | VLTF-4 (late transcription factor 4) | Canis familiaris | 35 | Measles virus | 30 | |
| H9R | VLTF-1 | Apis mellifera | 35 | Acanthamoeba polyphaga mimivirus | 31 | |
| P2L | NFkB inh | Arabidopsis thaliana | 35 | Lymphocystis disease virus 1 | 31 | |
| B21R | Unknown | Paramecium tetraurelia | 36 | Human adenovirus type 13 | 30 | |
| B8R | Virulence factor | Babesia bovis | 34 | Simian immunodeficiency virus | 32 | |
| A50L | Unknown | Candida albicans SC5314 | 35 | Paramecium bursaria Chlorella virus 1 | 31 | |
| H4L | Glutaredoxin 2 | Gibberella zeae PH-1 | 37 | Ecotropis obliqua NPV | 30 | |
| L6L | Telomere-binding protein | Trypanosoma cruzi | 35 | Staphylococcus phage Twort | 32 | |
| Q1L | Virokine/NFkB inh/Str resemblence to apoptotic reg | Dictyostelium discoideum | 34 | Neodiprion abietis nucleopolyhedrovirus | 33 | |
| A37R | IEV-specific | Tetrahymena thermophila SB210 | 35 | Influenza A virus (A/Chicken/NY/29878/91 (H2N2)) | 32 | |
| C12L | Unknown | Theileria parva | 34 | Acanthamoeba polyphaga mimivirus | 34 | |
| N1R | Myristylated MP IMV | Tetrahymena thermophila SB210 | 35 | Lymphocystis disease virus 1 | 33 | |
| A23R | Holliday junction resolvase | Rattus norvegicus | 35 | Trichoplusia ni ascovirus 2c | 32 | |
| G11L | Unknown | Entamoeba histolytica HM-1:IMSS | 35 | Human enterovirus 94 | 32 | |
| F11L | Virion core protein | Dictyostelium discoideum AX4 | 35 | Acanthamoeba polyphaga mimivirus | 32 | |
| C4L | Unknown | Tetrahymena thermophila SB210 | 37 | Acanthamoeba polyphaga mimivirus | 31 | |
| C8L | IL-18 BP | Macaca mulatta | 37 | Mamestra configurata nucleopolyhedrovirus B | 31 | |
| D12L | TNF receptor (CrmB) | Candida albicans SC5314 | 35 | Ilesha virus | 33 | |
| A1L | VLTF-2 (late transcription factor 2) | Mus musculus | 36 | KI polyomavirus Stockholm 60 | 32 | |
| B6R | Virulence, ER resident | Plasmodium berghei | 36 | Human papillomavirus type 50 | 32 | |
| S1R | Virion morph | Entamoeba histolytica | 38 | Cherry chlorotic rusty spot associated totiviral-like dsRNA 3 | 30 | |
| A20L | Unknown | Mus musculus | 37 | Mycobacteriophage Halo | 32 | |
| A29L | IMV MP/virus entry | Plasmodium berghei | 35 | Porcine epidemic diarrhea virus | 33 | |
| A21L | Entry and cell–cell Fusion | Medicago truncatula | 39 | Human immunodeficiency virus type 1 | 30 | |
| A55R | Intracellular TLR and IL-1 signaling inhibitor | Caenorhabditis briggsae | 34 | Bacteriophage 2638A | 35 | |
| E2L | Virion core | Rhipicephalus evertsi | 34 | Bovine enteric calicivirus | 34 | |
| J3L | IMV heparin binding surface protein | Neosartorya fischeri NRRL 181 | 37 | Enterobacteria phage JS98 | 32 | |
| J7R | Unknown | Theileria annulata | 37 | Rachiplusia ou multiple nucleopolyhedrovirus | 32 | |
| H10R | Entry-fusion complex protein | Plasmodium falciparum 3D7 | 38 | Epiphyas postvittana nucleopolyhedrovirus | 32 | |
| C14L | Unknown | Plasmodium falciparum 3D7 | 37 | Bluetongue virus 22 | 32 | |
| H8L | Virion assembly protein | Paramecium tetraurelia | 37 | Leucania separata nuclear polyhedrosis virus | 32 | |
| A30L | RNA pol 35(RPO35) | Trichomonas vaginalis G3 | 37 | Fiji disease virus | 33 | |
| A6R | RNA pol 19 (RPO19) | Trichomonas vaginalis G3 | 38 | Emiliania huxleyi virus 86 | 32 | |
| E9R | mutT motif/NTP-PPH | Tetrahymena thermophila SB210 | 38 | Chilo iridescent virus | 32 | |
| A43L | Virulence/secreted | Plasmodium falciparum 3D7 | 37 | Clostridium phage c-st | 34 | |
| F5R | Virosome component | Monodelphis domestica | 38 | Citrus tristeza virus | 32 | |
| D5L, I1R | Unknown | Plasmodium chabaudi | 35 | Lymphocystis disease virus 1 | 35 | |
| A38R | Unknown | Theileria annulata | 38 | Murid herpesvirus 4 | 32 | |
| A28L | Fusion protein | Tribolium castaneum | 40 | Lymphocystis disease virus—isolate China | 31 | |
| A54R | Unknown | Plasmodium berghei | 38 | Taura syndrome virus | 33 | |
| A12R | Viral membrane formation | Paramecium tetraurelia | 38 | Staphylococcus aureus prophage phiPV83 | 33 | |
| D1L, I5R | Chemokine binding protein | Dictyostelium discoideum AX4 | 39 | Lactobacillus plantarum bacteriophage LP65 | 32 | |
| E4R | Uracil-DNA glycosylase | Plasmodium berghei | 35 | Gallid herpesvirus 1 | 37 | |
| O2R | RNA pol (RPO22) | Entamoeba histolytica HM-1:IMSS | 41 | Choristoneura fumiferana MNPV | 30 | |
| F8R | ER-localized MP | Hordeum vulgare | 38 | Oryctes rhinoceros virus | 34 | |
| A9R | VITF-3 34kda subunit | Paramecium tetraurelia | 39 | Adoxophyes orana granulovirus | 34 | |
| N3L | Internal virion protein | Plasmodium falciparum 3D7 | 40 | Adeno-associated virus | 32 | |
| G12L | IEV associated | Danio rerio | 37 | Bat coronavirus (BtCoV/133/2005) | 35 | |
| G1L | Apoptosis inhibitor (mitochondrial-associated) | Plasmodium falciparum 3D7 | 39 | Spodoptera litura granulovirus | 34 | |
| L3L | DNA-binding phosphoprotein | Tetrahymena thermophila SB210 | 40 | Acanthamoeba polyphaga mimivirus | 32 | |
| L7L | Virion core protease | Plasmodium falciparum 3D7 | 39 | Lactococcus phage Q54 | 34 | |
| E3R | Virion core | Plasmodium falciparum 3D7 | 38 | Agrotis segetum granulovirus | 35 | |
| G9L | Disulfide bond formation | Gallus gallus | 40 | Human immunodeficiency virus 1 | 33 | |
| A17L | Myristylated entry/cell–cell fusion protein | Danio rerio | 40 | Lymphocystis disease virus—isolate China | 33 | |
| O3L | Unknown MP | Dictyostelium discoideum AX4 | 34 | Lymphocystis disease virus 1 | 39 | |
| F6R | Unknown | Tetrahymena thermophila SB210 | 39 | Human immunodeficiency virus 1 | 35 | |
| A24R | VITF-3 45kda subunit | Oryza sativa (japonica cultivar-group) | 40 | Autographa californica nucleopolyhedrovirus | 34 | |
| A27L | P4c precursor | Plasmodium yoelii yoelii | 41 | Bacteriophage RM 378 | 32 | |
| C19L | Unknown | Tetrahymena thermophila SB210 | 39 | Bacteriophage 66 | 35 | |
| C6L | IL-1 receptor antagonist | Plasmodium yoelii yoelii | 42 | Human papillomavirus type 14D | 32 | |
| G10L | Ser/Thr kinase Morph | Plasmodium vivax | 41 | Acanthamoeba polyphaga mimivirus | 34 | |
| Q2L | Alpha-amanitin sensitivity | Trichomonas vaginalis G3 | 41 | Xestia c-nigrum granulovirus | 33 | |
| G5L | 36 kDa major membrane protein | Danio rerio | 39 | Cyanophage phage S-PM2 | 36 | |
| A45R | Membrane glycoprotein-class I | Plasmodium falciparum 3D7 | 37 | Maize dwarf mosaic virus | 38 | |
| A52R | Putative Phosphotransferase/anion transport protein | Plasmodium chabaudi | 40 | Chilo iridescent virus | 35 | |
| E12L | Small capping enzyme | Tetrahymena thermophila SB210 | 39 | Porcine rotavirus | 37 | |
| N4R | Core package/transcription | Plasmodium falciparum | 41 | Staphylococcusphage CNPH82 | 34 | |
| L1L | DNA-binding protein | Strongylocentrotus purpuratus | 42 | Helicoverpa armigera nuclear polyhedrosis virus | 34 | |
| A49R | IL-1 signaling inhibitor | Cryptosporidium parvum Iowa II | 45 | Aedes taeniorhynchus iridescent virus | 30 | |
| C13L | Host range virulence factor | Plasmodium falciparum 3D7 | 41 | Plutella xylostella granulovirus | 35 | |
| H3R | VLTF (late transcription elongation factor) | Trichomonas vaginalis G3 | 41 | Chilo iridescent virus | 35 | |
| H5R | Unknown | Caenorhabditis elegans | 41 | Acanthamoeba polyphaga mimivirus | 36 | |
| B15L | Unknown | Dictyostelium discoideum AX4 | 42 | Tomato leaf curl Madagascar virus | 35 | |
| E13L | Trimeric virion coat protein (rifampicin res) | Bigelowiella natans | 42 | Neodiprion sertifer nucleopolyhedrovirus | 35 | |
| F1L | Poly (A) polymerase-large (VP55) | Strongylocentrotus purpuratus | 42 | Staphylococcus phage 187 | 35 | |
| H1L | Predicted metallo-protease | Dictyostelium discoideum | 39 | Acanthamoeba polyphaga mimivirus | 39 | |
| A11L | P4a precursor | Tetraodon nigroviridis | 43 | Acanthamoeba polyphaga mimivirus | 35 | |
| C16L | IL-1 receptor antagonist | Plasmodium falciparum 3D7 | 42 | Acanthamoeba polyphaga mimivirus | 37 | |
| A35R | C-type lectin-like EEV protein | Caenorhabditis briggsae | 44 | African swine fever virus | 35 | |
| A33L | ATPase/DNA packaging protein | Trichomonas vaginalis G3 | 43 | Cotesia congregata bracovirus | 36 | |
| A58R | Hemagglutinin | Anas platyrhynchos | 48 | Heliothis zea virus 1 | 33 | |
| F4L | RNA pol (RPO30) | Homo sapiens | 41 | African swine fever virus | 41 | |
| J4L | RAP94 (RNA pol assoc protein) | Plasmodium berghei | 45 | Trichoplusia ni SNPV | 37 | |
| F2L | Unknown | Entamoeba histolytica HM-1:IMSS | 44 | Staphylococcus aureus phage phiP68 | 39 | |
| R1L | Unknown | Pichia stipitis CBS 6054 | 41 | Gryllus bimaculatus nudivirus | 42 | |
| M3L | IFN resistance/eIF2 alpha-like PKR inhibitor | Anopheles gambiae str. PEST | 40 | Silurus glanis ranavirus | 44 | |
| A2L | VLTF-3 (late transcription factor 3) | Paramecium tetraurelia | 48 | Acanthamoeba polyphaga mimivirus | 37 | |
| A7L | Virion morphogenesis | Plasmodium reichenowi | 48 | Acanthamoeba polyphaga mimivirus | 40 | |
| A8L | VETF-L (early transcription factor large) | Plasmodium yoelii yoelii | 45 | Acanthamoeba polyphaga mimivirus | 44 | |
| A4L | P4b precursor | Tetrahymena thermophila SB210 | 40 | Acanthamoeba polyphaga mimivirus | 49 | |
4. Discussion and conclusions
Protein coding genes of poxviruses have been the subject of much research. Poxvirus immunomodulatory genes, those both with and without host homologs, have been extensively examined (Finlay and McFadden, 2006, Iyer et al., 2006, McFadden and Murphy, 2000, Monier et al., 2007, Seet et al., 2003, Stanford et al., 2007) as have the gene content and gene families present in poxvirus species, and evolutionary relationships based on phylogenies of those genes (Bratke and McLysaght, 2008, Gubser et al., 2004, Iyer et al., 2001, Iyer et al., 2006, Lefkowitz et al., 2006, McLysaght et al., 2003, Upton et al., 2003, Xing et al., 2006). It is apparent that many genes have entered poxvirus genomes via horizontal transfer both from their hosts and also possibly from other viruses.
From an evolutionary perspective, the genes poxviruses share with other viruses have been examined most notably in the context of exploring the hypothesis that the poxvirus family may share a common ancestor with several other families of large DNA viruses (the NCLDV). This hypothesis is based largely on the set of similar proteins these viruses share (at a sequence and/or functional level), which may have served as a “core” set of NCLDV genes. Poxviruses also code for genes with significant sequence similarity to genes from non-NCLDV virus family members, including virulence genes shared by entomopoxviruses, baculoviruses and iridoviruses (Dall et al., 2001, Means et al., 2007), host-interaction genes present in poxviruses and herpesviruses (Afonso et al., 2000, Iyer et al., 2006, McFadden and Murphy, 2000), and other poxvirus proteins with notable levels of similarity to genes of a recently discovered fish herpesvirus (Ilouze et al., 2006).
The potential for horizontal gene transfer into poxviruses has been examined using several methods, including phylogenetic reconstructions, gene synteny analysis, and anomalous base composition. Phylogenetic reconstructions of gene families with members in other viruses and their hosts have suggested that multiple horizontal gene transfer (HGT) events have taken place into poxvirus genomes from other viruses (Dall et al., 2001) and from their eukaryotic hosts (Bratke and McLysaght, 2008, Hughes, 2002, Hughes and Friedman, 2005, Monier et al., 2007). Anomalous base composition (DaSilva and Upton, 2005, Monier et al., 2007), and gene synteny analysis (Bratke and McLysaght, 2008, McLysaght et al., 2003) have found evidence for HGT from hosts to poxviruses, including multiple HGT events for some genes. All methods of analysis conclude that the presence of many genes is best explained by HGT, although the process may not be frequent and recent (Lefkowitz et al., 2006, Monier et al., 2007), and some genes with noted similarity to genes of other organisms are proposed to not have been obtained via HGT (Hughes and Friedman, 2005, Iyer et al., 2001).
The goals of our current analysis were to develop a method of measuring and visualizing the similarities of all proteins expressed by virus isolates belonging to the entire poxvirus family to various taxonomically distinct sets of proteins from other organisms. This analysis was designed to detect overall trends in gene similarity and to detect individual genes that may be of interest due to anomalous characteristics with regard to such levels of similarity. Each individual protein may then be further investigated with regard to its function, distribution in poxviruses and other organisms, and via phylogenetic analysis, to determine its most likely evolutionary history. Proteins identified as interesting candidates for follow-up research by this method may be further studied using more traditional phylogenetic methods as illustrated by our initial phylogenetic analyses of proteins in Fig. 2. Overall trends in sequence similarity of different subsets of poxvirus proteins, as well as information about individual proteins implicated by our analysis may contribute valuable information about the evolution of poxviruses and the mechanisms of host pathogenesis.
Overall, analysis by taxonomic group plots shows that chordopoxvirus proteins tend to exhibit greater similarity to eukaryotic proteins than to bacterial or viral proteins, suggesting that many poxvirus proteins may share a common evolutionary origin derived from proteins of their eukaryotic hosts. Although entomopoxviruses also contain host-like genes, both with and without homologs in chordopoxviruses, entomopoxvirus proteins do not show the same general skew towards similarity to eukaryotic proteins. However, entomopoxviruses encode quite a few proteins with notably greater similarity to proteins of other viruses than to bacterial or eukaryotic proteins. The relatively small sampling of insect proteins available in GenBank could partly account for the low scores of these proteins to the eukaryote database, with insects being represented by 799,971 proteins and 210 complete genomes, compared to a vertebrate collection of 1,787,682 proteins and 1559 complete genomes. However, with only 3 exceptions, all chordopoxvirus proteins which achieve similarly high scores to proteins of other viruses are proteins with sequences universally conserved throughout nature, such as ribonucleotide reductase, DNA photolyase and RNA polymerase. For viruses of both the entomopoxvirus and chordopoxvirus subfamilies, the most similar virus proteins outside the poxvirus family are found both among members of the postulated NCLDV group of viruses and among non-NCLDV members, with viruses of the families Baculoviridae, Herpesviridae and Iridoviridae most represented.
Inspection of the individual proteins represented on the plots reveals that many of the proteins are universally highly conserved. These function in the synthesis and maintenance of DNA and RNA, and are present in many virus species, as well as in most eukaryotes. All the poxvirus enzymes that convert cellular pools of nucleotides for RNA synthesis into deoxyribonucleotides for synthesis of DNA fall either on the diagonal or just below it. The ultimate origin of these proteins is uncertain, but those with greater similarity to eukaryote proteins may have been transferred more recently into the poxvirus lineage than into the other virus families in which they appear, or these proteins may have been constrained for functional purposes towards high sequence identity with host proteins.
Many other proteins highlighted by this analysis are apparently of eukaryote origin, and fall either on the diagonal, just below it, or near the eukaryote axis, depending on their degree of similarity to proteins presumably transferred into viruses outside the poxvirus family. These have functions involving immune response and intracellular processes, and seem likely to have been transferred horizontally from hosts into poxviruses as well as into the families of other, non-poxvirus viruses. The functions of these proteins are presumed to be advantageous to the biology of viruses in all families where these proteins appear.
Proteins near the eukaryotic axis in Fig. 1A are only present in viruses of the family Poxviridae. The majority of these proteins are involved in the manipulation of intracellular processes, including redox state, protein signaling cascades, and lipid and carbohydrate metabolism, as well as involved in the manipulation of the extracellular environment. Some of these proteins are of unknown function. The fact that these eukaryotic-like proteins are found only among viruses in the poxvirus family may be informative about what cellular processes and signaling cascades are unique to poxvirus infections. Finally, there are many proteins that are seemingly unique to poxviruses, with no significant sequence similarity to known proteins among other viruses, eukaryotes or bacteria.
Together these results give us a picture of the many different subsets of proteins present in poxviruses, and allow us to draw some conclusions about each subset based on where else in nature proteins of these types appear. Investigation of the similarities and origins of particular proteins may yield further insights into poxvirus evolution and pathogenesis. For example, the fact that the poxvirus versions of universally highly conserved enzymes such as RNR have significantly more sequence similarity to RNR of eukaryotes than to those of bacteria or other viruses may imply a need for interoperability of the poxvirus enzymes with host proteins. Another example is the presence of different clusters of poxvirus TK sequences, where TK encoded by entomopoxviruses and avipoxviruses cluster together on the plot in a different location from the cluster of TK proteins encoded by poxviruses in other genera, agreeing with previously published suggestions that the TK enzymes of avipoxvirus, entomopoxvirus and the other chordopoxvirus genera may have different origins (Bratke and McLysaght, 2008, Koonin and Senkevich, 1992).
Finally, by using taxonomic group plots to study the proteome of cowpox virus, we show that the most host-like genes tend to lie at the ends of the linear genome and have the most limited distributions among poxvirus species.
More explicit conclusions about individual proteins, including gene origins, relationships to proteins of other organisms, and details of potential horizontal gene transfer events, will require additional, more extensive analyses at the level of each individual gene. Such investigations will require phylogenetic reconstruction of individual protein families utilizing sequences obtained from accurate annotations of poxvirus genomes, with particular attention to providing an accurate gene prediction for each genome and to the presence or absence of particular genes in each genome.
In conclusion, using taxonomic group plots to analyze proteins of poxviruses confirms the presence of many eukaryotic-like proteins in the genomes of poxvirus species, underscoring the importance of the contribution of host gene capture in the evolution of these viruses. These results also provide an overview of the functional significance of many of the genes poxviruses share with their hosts, and expose which host genes are captured uniquely by poxviruses and which are captured by other virus families as well. Information yielded by more comprehensive phylogenetic analysis of poxvirus genes to genes of their hosts and other viruses, will illustrate details of molecular mechanisms of poxvirus adaptation and survival throughout the history of the virus family, giving a richer picture of the evolution of this once devastating and still dangerous group of viral pathogens.
Acknowledgements
We would like to thank the staff of the Viral Bioinformatics Research Center (www.vbrc.org) for invaluable contributions, support and guidance. This work was supported by NIH/NIAID Contract No. HHSN266200400036C to EJL.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.virusres.2009.05.006.
Appendix A. Supplementary data
References
- Afonso C.L., Tulman E.R., Lu Z., Zsak L., Kutish G.F., Rock D.L. The genome of Fowlpox virus. J. Virol. 2000;74(8):3815–3831. doi: 10.1128/jvi.74.8.3815-3831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson D.A., Karsch-Mizrachi I., Lipman D.J., Ostell J., Wheeler D.L. GenBank. Nucleic Acids Res. 2008;36(Database issue):D25–D30. doi: 10.1093/nar/gkm929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botstein D. A theory of modular evolution for bacteriophages. Ann. N. Y. Acad. Sci. 1980;354:484–490. doi: 10.1111/j.1749-6632.1980.tb27987.x. [DOI] [PubMed] [Google Scholar]
- Bratke K.A., McLysaght A. Identification of multiple independent horizontal gene transfers into poxviruses using a comparative genomics approach. BMC Evol. Biol. 2008;8:67. doi: 10.1186/1471-2148-8-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condit R.C., Moussatche N., Traktman P. In a nutshell: structure and assembly of the vaccinia virion. In: Maramorosch K., Shatkin A.J., editors. vol. 66. Academic Press; 2006. pp. 31–124. (Advances in Virus Research). [DOI] [PubMed] [Google Scholar]
- Dall D., Luque T., O’Reilly D. Insect–virus relationships: sifting by informatics. Bioessays. 2001;23:184–193. doi: 10.1002/1521-1878(200102)23:2<184::AID-BIES1026>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- DaSilva M., Upton C. Host-derived pathogenicity islands in poxviruses. Virol. J. 2005;2:30. doi: 10.1186/1743-422X-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFilippis V.R., Villarreal L.P. Virus evolution. In: Knipe D.M., Howley P.M., Griffin D.E., editors. Fields Virology. 4th ed. Lippincott Williams & Wilkins; Philadelphia, PA: 2001. [Google Scholar]
- Drake J., Hwang C. On the mutation rate of herpes simplex virus type 1. Genetics. 2005;170(2):969–970. doi: 10.1534/genetics.104.040410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S., Shackelton L., Holmes E. Rates of evolutionary change in viruses: patterns and determinants. Nat. Rev. Genet. 2008;9(4):267–276. doi: 10.1038/nrg2323. [DOI] [PubMed] [Google Scholar]
- Esposito J.J., Fenner F. Poxviruses. In: Knipe D.M., Howley P.M., Griffin D.E., editors. Fields Virology. 4th ed. Lippincott Williams & Wilkins; Philadelphia, PA: 2001. [Google Scholar]
- Finlay B.B., McFadden G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell. 2006;124:767–782. doi: 10.1016/j.cell.2006.01.034. [DOI] [PubMed] [Google Scholar]
- Geserick P., Kaiser F., Klemm U., Kaufmann S., Zerrahn J. Modulation of T cell development and activation by novel members of the Schlafen (slfn) gene family harbouring an RNA helicase-like motif. Int. Immunol. 2004;16(10):1535–1548. doi: 10.1093/intimm/dxh155. [DOI] [PubMed] [Google Scholar]
- Gubser C., Hué S.p., Kellam P., Smith G.L. Poxvirus genomes: a phylogenetic analysis. J. Gen. Virol. 2004;85:105–117. doi: 10.1099/vir.0.19565-0. [DOI] [PubMed] [Google Scholar]
- Hamann C., Lentainge S., Li L., Salem J., Yang F., Cooperman B. Chimeric small subunit inhibitors of mammalian ribonucleotide reductase: a dual function for the R2 C-terminus? Protein Eng. Des. Sel. 1998;11(3):219–224. doi: 10.1093/protein/11.3.219. [DOI] [PubMed] [Google Scholar]
- Hertig C., Coupar B.E.H., Gould A.R., Boyle D.B. Field and vaccine strains of Fowlpox virus carry integrated sequences from the avian retrovirus, reticuloendotheliosis virus1. Virology. 1997;235:367–376. doi: 10.1006/viro.1997.8691. [DOI] [PubMed] [Google Scholar]
- Hughes A.L. Origin and evolution of viral interleukin-10 and other DNA virus genes with vertebrate homologues. J Mol Evol. 2002;54:90–101. doi: 10.1007/s00239-001-0021-1. [DOI] [PubMed] [Google Scholar]
- Hughes A.L., Friedman R. Poxvirus genome evolution by gene gain and loss. Mol. Phylogenet. Evol. 2005;35:186–195. doi: 10.1016/j.ympev.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Ilouze M., Dishon A., Kahan T., Kotler M. Cyprinid herpes virus-3 CyHV-3 bears genes of genetically distant large DNA viruses. FEBS Lett. 2006;580:4473–4478. doi: 10.1016/j.febslet.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Ilouzea M., Dishona A., Kahanb T., Kotlera M. Cyprinid herpes virus-3 CyHV-3 bears genes of genetically distant large DNA viruses. FEBS Lett. 2006;580:4473–4478. doi: 10.1016/j.febslet.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Iyer L.M., Aravind L., Koonin E.V. Common origin of four diverse families of large Eukaryotic DNA viruses. J. Virol. 2001;75(23):11720–11734. doi: 10.1128/JVI.75.23.11720-11734.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer L.M., Balaji S., Koonin E.V., Aravind L. Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Res. 2006;117:156–184. doi: 10.1016/j.virusres.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Kang T.-H., Park D.-Y., Kim W., Kim K.-T. VRK1 phosphorylates CREB and mediates CCND1 expression. J. Cell Sci. 2008;121:3035–3041. doi: 10.1242/jcs.026757. [DOI] [PubMed] [Google Scholar]
- Katz L.A. Lateral gene transfers and the evolution of eukaryotes: theories and data. Int. J. Syst. Evol. Microbiol. 2002;52(Pt 5):1893–1900. doi: 10.1099/00207713-52-5-1893. [DOI] [PubMed] [Google Scholar]
- Koonin E.V., Senkevich T.G. Evolution of thymidine and thymidylate kinases: the possibility of independent capture of TK genes by different groups of viruses. Virus Genes. 1992;6:2. doi: 10.1007/BF01703067. [DOI] [PubMed] [Google Scholar]
- Koonin E.V., Wolf Y.I. Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 2008;36(21):6688–6719. doi: 10.1093/nar/gkn668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koski L.B., Golding G.B. The closest BLAST hit is often not the nearest neighbor. J. Mol. Evol. 2001;52(6):540–542. doi: 10.1007/s002390010184. [DOI] [PubMed] [Google Scholar]
- Laidlaw S.M., Anwar M.A., Thomas W., Green P., Shaw K., Skinner M.A. Fowlpox virus encodes nonessential homologs of cellular alpha-SNAP, PC-1, and an orphan human homolog of a secreted nematode protein. J. Virol. 1998;72(8):6742–6751. doi: 10.1128/jvi.72.8.6742-6751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz E.J., Upton C., Changayil S.S., Buck C., Traktman P., Buller R.M.L. Poxvirus bioinformatics resource center: a comprehensive Poxviridae informational and analytical resource. Nucleic Acids Res. 2005;33(Database issue):D311–D316. doi: 10.1093/nar/gki110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz E.J., Wang C., Upton C. Poxviruses: past, present and future. Virus Res. 2006;117:105–118. doi: 10.1016/j.virusres.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Li Y., Carroll D.S., Gardner S.N., Walsh M.C., Vitalis E.A., Damon I.K. On the origin of smallpox: correlating variola phylogenics with historical smallpox records. PNAS. 2007;104(40):15787–15792. doi: 10.1073/pnas.0609268104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likos A.M., Sammons S.A., Olson V.A., Frace A.M., Li Y., Olsen-Rasmussen M., Davidson W., Galloway R., Khristova M.L., Reynolds M.G., Zhao H., Carroll D.S., Curns A., Formenty P., Esposito J.J., Regnery R.L., Damon I.K. A tale of two clades: monkeypox viruses. J. Gen. Virol. 2005;86:2661–2672. doi: 10.1099/vir.0.81215-0. [DOI] [PubMed] [Google Scholar]
- McFadden G., Murphy P.M. Host-related immunomodulators encoded by poxviruses and herpesviruses. Curr. Opin. Microbiol. 2000;3:371–378. doi: 10.1016/s1369-5274(00)00107-7. [DOI] [PubMed] [Google Scholar]
- McLysaght A., Baldi P.F., Gaut B.S. Extensive gene gain associated with adaptive evolution of poxviruses. PNAS. 2003;100(26):15655–15660. doi: 10.1073/pnas.2136653100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Means J.C., Penabaz T., Clem R.J. Identification and functional characterization of AMVp33, a novel homolog of the baculovirus caspase inhibitor p35 found in Amsacta moorei entomopoxvirus. Virology. 2007;358:4376–4447. doi: 10.1016/j.virol.2006.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monier A., Claverie J.-M., Ogata H. Horizontal gene transfer and nucleotide compositional anomaly in large DNA viruses. BMC Genomics. 2007;8:456. doi: 10.1186/1471-2164-8-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B. Poxviridae: the viruses and their replication. In: Knipe D.M., Howley P.M., editors. Fields Virology. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 2849–2883. [Google Scholar]
- NCBI . National Center for Biotechnology Information (NCBI); 2006. TaxPlot. www.ncbi.nlm.nih.gov/sutils/taxik2.cgi/ [Google Scholar]
- Rasko D.A., Myers G.S.A., Ravel J. Visualization of comparative genomic analyses by BLAST score ratio. BMC Bioinform. 2005;6:2. doi: 10.1186/1471-2105-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F., Huelsenbeck J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19(12):1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Seet B.T., Johnston J., Brunetti C.R., Barrett J.W., Everett H., Cameron C., Sypula J., Nazarian S.H., Lucas A., McFadden G. Poxviruses and immune evasion. Annu. Rev. Immunol. 2003;21:377–423. doi: 10.1146/annurev.immunol.21.120601.141049. [DOI] [PubMed] [Google Scholar]
- Senkevich T.G., Koonin E.V., Bugert J.J., Darai G., Moss B. The genome of molluscum contagiosum virus: analysis and comparison with other poxviruses. Virology. 1997;233:19–42. doi: 10.1006/viro.1997.8607. [DOI] [PubMed] [Google Scholar]
- Shackelton L.A., Holmes E.C. The evolution of large DNA viruses: combining genomic information of viruses and their hosts. Trends Microbiol. 2004;12(10):458–465. doi: 10.1016/j.tim.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Shchelkunov S.N., Safronov P.F., Totmenin A.V., Petrov N.A., Ryazankina O.I., Gutorov V.V., Kotwal G.J. The genomic sequence analysis of the left and right species-specific terminal region of a Cowpox virus strain reveals unique sequences and a cluster of intact ORFs for immunomodulatory and host range proteins. Virology. 1998;243:432–460. doi: 10.1006/viro.1998.9039. [DOI] [PubMed] [Google Scholar]
- Silverman G., Bird P., Carrell R., Church F., Coughlin P., Gettins P., Irving J., Lomas D., Luke C., Moyer R., Pemberton P., Remold-O’Donnell E., Salvesen G., Travis J., Whisstock J. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J. Biol. Chem. 2001;276(36):33293–33296. doi: 10.1074/jbc.R100016200. [DOI] [PubMed] [Google Scholar]
- Stanford M.M., McFadden G., Karupiah G., Chaudhri G. Immunopathogenesis of poxvirus infections: forecasting the impending storm. Immunol. Cell Biol. 2007:1–10. doi: 10.1038/sj.icb.7100033. [DOI] [PubMed] [Google Scholar]
- Stubbe J. Ribonucleotide reductases. Adv. Enzymol. Relat. Areas Mol. Biol. 1990;63:349–419. doi: 10.1002/9780470123096.ch6. [DOI] [PubMed] [Google Scholar]
- Tamura K., Dudley J., Nei M., Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007:24. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrents E., Aloy P., Gibert I., Rodríguez-Trelles F. Ribonucleotide reductases: divergent evolution of an ancient enzyme. J. Mol. Evol. 2002;(55):138–152. doi: 10.1007/s00239-002-2311-7. [DOI] [PubMed] [Google Scholar]
- Tulman E.R., Delhon G., Afonso C.L., Lu Z., Zsak L., Sandybaev N.T., Kerembekova U.Z., Zaitsev V.L., Kutish G.F., Rock D.L. Genome of Horsepox virus. J. Virol. 2006;80(18):9244–9258. doi: 10.1128/JVI.00945-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton C., Slack S., Hunter A.L., Ehlers A., Roper R.L. Poxvirus orthologous clusters: toward defining the minimum essential Poxvirus genome. J. Virol. 2003;77(13):7590–7600. doi: 10.1128/JVI.77.13.7590-7600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viral Bioinformatics Resource Center (VBRC), 2008. www.vbrc.org.
- Viswanathan K., Richardson J., Togonu-Bickersteth B., Dai E., Liu L., Vatsya P., Sun Y.M., Yu J., Munuswamy-Ramanujam G., Baker H., Lucas A.R. Myxoma viral serpin, Serp-1, inhibits human monocyte adhesion through regulation of actin-binding protein filamin B. J. Leukoc. Biol. 2009;85(3):418–426. doi: 10.1189/jlb.0808506. [DOI] [PubMed] [Google Scholar]
- Werden S.J., McFadden G. The role of cell signaling in poxvirus tropism: the case of the M-T5 host range protein of myxoma virus. Biochim. Biophys. Acta. 2008;1784:228–237. doi: 10.1016/j.bbapap.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Xing K., Deng R., Wang J., Feng J., Huang M., Wang X. Genome-based phylogeny of poxvirus. Intervirology. 2006;49(4):207–214. doi: 10.1159/000090790. [DOI] [PubMed] [Google Scholar]
- Zwickl D.J. The University of Texas at Austin; 2006. Genetic Algorithm Approaches for the Phylogenetic Analysis of Large Biological Sequence Datasets under the Maximum Likelihood Criterion. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.