

Abstract
Background
Hypertrophic cardiomyopathy (HCM), the most common cause of sudden cardiac death in the young, is characterized by cardiac hypertrophy, myocyte disarray, and interstitial fibrosis. We propose that hypertrophy and fibrosis are secondary to the activation of trophic and mitotic factors and, thus, potentially reversible. We determined whether the blockade of angiotensin II, a known cardiotrophic factor, could reverse or attenuate interstitial fibrosis in a transgenic mouse model of human HCM.
Methods and Results
We randomized 24 adult cardiac troponin T (cTnT-Q92) mice, which exhibit myocyte disarray and interstitial fibrosis, to treatment with losartan or placebo and included 12 nontransgenic mice as controls. The mean dose of losartan and the mean duration of therapy were 14.2±5.3 mg · kg−1 · d−1 and 42±9.6 days, respectively. Mean age, number of males and females, and heart/body weight ratio were similar in the groups. Collagen volume fraction and extent of myocyte disarray were increased in the cTnT-Q92 mice (placebo group) compared with nontransgenic mice (9.9±6.8% versus 4.5±2.2%, P=0.01, and 27.6±10.6% versus 3.9±2.3%, P<0.001, respectively). Treatment with losartan reduced collagen volume fraction by 49% to 4.9±2.9%. The expression of collagen 1α (I) and transforming growth factor-β1, a mediator of angiotensin II profibrotic effect, were also reduced by 50%. Losartan had no effect on myocyte disarray.
Conclusions
Treatment with losartan reversed interstitial fibrosis and the expression of collagen 1α (I) and transforming growth factor-β1 in the hearts of cTnT-Q92 mice. These findings suggest that losartan has the potential to reverse or attenuate interstitial fibrosis, a major predictor of sudden cardiac death, in human patients with HCM.
Keywords: cardiomyopathy, fibrosis, collagen, death-sudden
Hypertrophic cardiomyopathy (HCM), the most common cause of sudden cardiac death (SCD) in the young,1 is caused by mutations in sarcomeric proteins.2 It is clinically diagnosed by unexplained cardiac hypertrophy and pathologically by myocyte hypertrophy, disarray, and interstitial fibrosis.3 Hypertrophy and fibrosis are the major determinants of mortality, morbidity, and SCD4,5 in HCM and in all acquired forms of cardiac diseases.
The genetic basis of HCM has been elucidated, and research efforts are being directed to decipher its molecular pathogenesis and to determine the reversibility of the phenotypes. We previously proposed that interstitial fibrosis, like cardiac hypertrophy, occurs “secondary” to the activation of trophic and mitotic factors in the heart6 and, thus, is potentially reversible by blocking cardiotrophic factors such as angiotensin II (Ang II). However, despite the well-established role of Ang II blockers in the attenuation of cardiac hypertrophy and fibrosis in acquired cardiac diseases, they are not conventionally used in the treatment of patients with HCM, a genetic paradigm of cardiac hypertrophy and fibrosis. We determined the effects of blocking Ang II on the interstitial collagen content of transgenic mice expressing mutant cardiac troponin T (cTnT)-Q92 protein,7 which causes HCM in humans.2
Methods
cTnT-Q92 Transgenic Mice Model
A full description of the cTnT-Q92 transgenic mice model has been published previously.7 In brief, the cardiac-restricted expression of cTnT-Q92 leads to myocyte disarray and interstitial fibrosis in two-thirds of 3- to 12-month-old mice but not to significant cardiac hypertrophy.7
Losartan Therapy
Age- and sex-matched adult cTnT-Q92 mice were randomized to treatment with either placebo (n=12) or losartan (n=12). Twelve nontransgenic mice, matched for age and sex with the cTnT-Q92 mice, were included as controls. Losartan was dissolved in daily drinking water at a concentration of 0.166 mg/mL, to provide ≈10 to 20 μg · kg−1 · d−1 of losartan per mouse. This dose has been shown to reduce myocardial fibrosis in a variety of experimental models without significantly affecting blood pressure or heart rate.8 Mice were weighed and water consumption was measured to calculate the precise daily intake of losartan.
Detection and Quantification of Fibrillar Collagen
Two complementary methods of Northern blotting of collagen α1 (I) mRNA and picrosirius red staining of collagen protein were used. To quantify the collagen volume fraction (CVF), 5-μm-thin ventricular sections were cut parallel to the atrioventricular groove and stained with collagen-specific Sirius red F3BA. Morphometric analysis was performed by an investigator who was blinded to randomization in 10 randomly selected fields per section, in 10 sections per mouse, and in 12 mice per group in a random fashion by computerized planimetry. Perimysial and endomysial collagens were included, but perivascular and epimysial collagens were excluded. CVF was calculated as the sum of all areas stained positive for Sirius red divided by the sum of all myocardial areas in each mouse.
Expression of collagen α1 (I) mRNA, the major collagen in the heart,9 was detected by Northern blotting. In brief, 10-μg aliquots of cardiac total RNA extracts were loaded onto a formaldehyde-agarose gel, electrophoresed, and transferred to nylon membranes. A 194-bp of fragment of the coding sequence of the COL1A1 gene (accession number S67530) was amplified by polymerase chain reaction (primers: forward, 5′tccctgaggtcagctgcgtac3′; reverse, 5′cgtattcttccgggcagaaag3′), labeled with [32P]dCTP, and hybridized to membranes in the presence of Denhardt’s reagent in hybridization solution for 48 hours. Membranes were washed and exposed to an x-ray film.
Detection and Quantification of Transforming Growth Factor-β1 Expression
To detect and quantify the expression of transforming growth factor-β1 (TGF-β1), thin myocardial sections were incubated with goat anti–TGF-β1 polyclonal antibody at a concentration of 1 μg/mL, followed by incubation with a biotinylated anti-goat secondary antibody, also at a concentration of 1 μg/mL. Signals were detected by peroxidase reaction. Areas stained positive for the TGF-β1 expression, excluding perivascular and epimysial areas, were quantified in 6 fields per section, in 10 sections per mouse, and in 12 mice per group and expressed as percent of total myocardial area.
Detection and Quantification of Myocyte Disarray
Myocyte disarray was detected and quantified as previously described.7 Myocyte disarray was defined as bundles of myocytes that were aligned perpendicularly or obliquely to each other or were interspersed in different directions. Minor variations and areas of myocardium at the junctions of interventricular septum with the ventricles, near the blood vessels, and trabeculations were excluded. Extent of myocyte disarray was quantified in 6 fields per section, in 10 sections per mouse, and in 12 mice per group.
Statistical Methods
Differences in variables among the groups were compared by ANOVA, followed by Bartlett’s test for the homogeneity of variances. Variables with unequal SDs were compared by the nonparametric Kruskal-Wallis test.
Results
Study Groups
No mice died during the experiments. Baseline parameters among the 3 groups, including mean age (447±69, 409±53, and 403±49.4 days in the losartan, placebo, and nontransgenic groups, respectively; P=0.31), male/female ratio (6/6 in each group), and heart weight/body weight ratio (4.1±0.5, 3.9±0.5, and 3.9±0.7 mg/g in the losartan, placebo, and nontransgenic groups, respectively; P=0.73) were not significantly different. The mean daily dose of losartan was 14.2±5.3 mg · kg−1 · d−1 (range, 8.8 to 23.6 mg · kg−1 · d−1). The mean duration of therapy was 42±9.6 days (range, 34 to 56 days).
Nontransgenic and Placebo Groups
Consistent with the previous observations,7 the mean CVF was increased in the cTnT-Q92 mice in the placebo group compared with the mice in the nontransgenic group (9.8±6.8% versus 4.5±2.2%; P=0.028; Figure 1A). TGF-β1 was predominantly localized to perivascular and epimysial areas in the nontransgenic mice (Figure 1B), whereas in the cTnT-Q92 mice, TGF-β1 was also expressed in the perimysial and endomysial regions. As observed in human patients with HCM,10 the expression of TGF-β1 was increased by 2-fold in the cTnT-Q92 mice (1.1±0.38% versus 0.66±0.34% in nontransgenic mice; P=0.008). Myocyte disarray comprised 27.6±10.6% of the myocardium in the cTnT-Q92 mice treated with placebo compared with 3.9±2.3% in the nontransgenic mice (P<0.001; Figure 1C). There was a strong correlation between CVF and percent of myocyte disarray (Pearson correlation, 0.81; P=0.001) in the cTnT-Q92 group.
Figure 1.
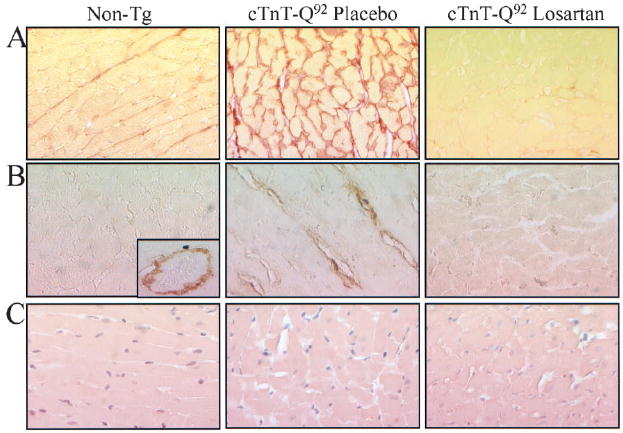
Interstitial fibrosis, expression of TGF-β1, and myocyte disarray in nontransgenic (non-tg) and cTnT-Q92 transgenic mice treated with placebo or losartan. A, B, and C show representative photomicrographs of thin myocardial sections stained with Sirius red F3BA, anti-TGF-β1 antibody, and hematoxylin and eosin, respectively, in experimental groups. Inset in B shows expression of TGF-β1 in nontransgenic group, which was localized to perivascular area.
Effects of Losartan on Cardiac Phenotype
Losartan significantly reduced interstitial fibrillar collagen in the cTnT-Q92 mice (Figures 1A and 2A). Perimysial and endomysial CVF were reduced by 49% in the losartan group compared with the placebo group (4.9±2.9% versus 9.8±6.8%; P=0.040) and was similar to the values in the nontransgenic mice. Expression of collagen α1 (I), the predominant collagen in the heart,9 was also reduced by 50% in the losartan group (Figure 2B).
Figure 2.
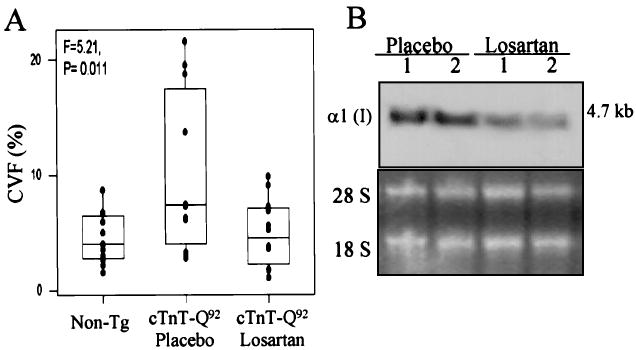
A, Box-and-whisker plot showing median (bar), first and third quartiles (box), lowest and highest values, and individual data points for CVF. Mean CVF of myocardium was 4.5±2.2% (range, 1.5% to 8.7%) in nontransgenic mice (non-tg; n=12), 9.8±6.8% (range, 2.76% to 21.6%) in placebo group (n=12), and 4.9±2.9% (range, 1.05% to 9.90%) in losartan group (n=12) (P=0.011, 2 df). B represents a Northern blot showing expression of collagen 1α (I) and 28S and 18S bands in experimental groups.
Expression of TGFβ-1, a known mediator of the profibrotic effect of Ang II, was also reduced in the losartan group compared with the placebo group (0.53±0.48% versus 1.1±0.38%, P=0.004; Figure 1B), and it was similar to that in the nontransgenic mice (0.66±0.34%). The extent of myocyte disarray was not significantly different between the losartan and placebo groups (23.4±9.6% versus 27.6±10.6%, P=0.32; Figure 1C).
Discussion
A transgenic mouse model of human HCM was established by cardiac-restricted expression of mutant cTnT-Q92 protein, which is known to cause HCM in humans.2 Adult cTnT-Q92 mice, which exhibit myocyte disarray and interstitial fibrosis,7 were matched for age and sex and treated with either losartan or placebo for 6 weeks. Interstitial fibrosis, which was detected by 2 complementary methods of Northern blotting for collagen α1 (I) mRNA and picrosirius red staining of collagen protein, was reduced by 49% in the losartan group. Expression of TGF-β1 protein was also reduced significantly, but the extent of myocyte disarray remained unchanged. These results in a genetic animal model of HCM support the hypothesis that interstitial fibrosis is a secondary and reversible phenotype. We propose that the inhibition of Ang II in human patients with HCM, which at the present time is considered unconventional, could have salutary effects by attenuating fibrosis, a major risk factor for SCD.4 We think this treatment warrants investigation.
We performed quantitative morphometric measurements in a blinded fashion, randomly, and in 1200 fields per group. To corroborate the reduction of CVF with losartan, we determined the amount of expression of collagen 1α (I) mRNA and TGF-β1 protein, which were also reduced. Our findings are also consistent with the well-established role of Ang II blockers in the downregulation of expression of collagen 1α (I) and TGF-β1 in models of secondary hypertrophy and fibrosis.11,12 We did not perform functional studies to determine whether a reduction in interstitial fibrosis led to improvement in cardiac function or electrophysiological properties. The anticipated effect is salutary, through either the reversal of interstitial fibrosis or the beneficial hemodynamic effects. The absence of functional data does not detract from our observation that treatment with losartan reverses interstitial fibrosis in a genetic mouse model of HCM, a finding that raises the possibility of new therapeutic option for human HCM. The cTnT-Q92 mice do not exhibit cardiac hypertrophy or increased incidence of SCD.7 Whether the blockade of Ang II could reverse or attenuate hypertrophy or fibrosis in the presence of hypertrophy or reduce the risk of SCD remains to be determined. The results of recent studies in human patients with hypertensive hypertrophic heart disease show that inhibiting Ang II could attenuate interstitial fibrosis, which would further enhance the potential salutary effects of Ang II blockers in HCM.13
In summary, blocking Ang II reversed and normalized interstitial fibrosis and reduced the expression of collagen 1α (I) and TGF-β1 in the heart of a mutant cTnT-Q92 mouse model of HCM. These findings support the hypothesis that interstitial fibrosis, a major predictor of SCD in HCM,1 is a secondary phenotype that could be reversed. We propose that inhibiting Ang II in human patients with HCM (the most common cause of SCD in the young1), which at the present time is considered unconventional, could have salutary effects and warrants investigation.
Acknowledgments
Supported by grants from the National Heart, Lung, and Blood Institute, Specialized Centers of Research (P50-HL42267 to 01), and an Established Investigator Award (9640133N) from the American Heart Association, National Center, Dallas, Texas.
References
- 1.Maron BJ, Shirani J, Poliac LC, et al. Sudden death in young competitive athletes: Clinical, demographic, and pathological profiles. JAMA. 1996;276:199–204. [PubMed] [Google Scholar]
- 2.Seidman CE, Seidman JG. Molecular genetic studies of familial hypertrophic cardiomyopathy. Basic Res Cardiol. 1998;93(suppl 3):13–16. doi: 10.1007/s003950050196. [DOI] [PubMed] [Google Scholar]
- 3.Maron BJ, Roberts WC. Quantitative analysis of cardiac muscle cell disorganization in the ventricular septum of patients with hypertrophic cardiomyopathy. Circulation. 1979;59:689–706. doi: 10.1161/01.cir.59.4.689. [DOI] [PubMed] [Google Scholar]
- 4.Shirani J, Pick R, Roberts WC, et al. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol. 2000;35:36–44. doi: 10.1016/s0735-1097(99)00492-1. [DOI] [PubMed] [Google Scholar]
- 5.Spirito P, Bellone P, Harris KM, et al. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med. 2000;342:1778–1785. doi: 10.1056/NEJM200006153422403. [DOI] [PubMed] [Google Scholar]
- 6.Marian AJ. Pathogenesis of diverse clinical and pathological phenotypes in hypertrophic cardiomyopathy. Lancet. 2000;355:58–60. doi: 10.1016/s0140-6736(99)06187-5. [DOI] [PubMed] [Google Scholar]
- 7.Oberst L, Zhao G, Park JT, et al. Dominant-negative effect of a mutant cardiac troponin T on cardiac structure and function in transgenic mice. J Clin Invest. 1998;102:1498–1505. doi: 10.1172/JCI4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Melo LG, Veress AT, Chong CK, et al. Salt-sensitive hypertension in ANP knockout mice is prevented by AT1 receptor antagonist losartan. Am J Physiol. 1999;277:R624–R630. doi: 10.1152/ajpregu.1999.277.3.R624. [DOI] [PubMed] [Google Scholar]
- 9.Weber KT. Targeting pathological remodeling: concepts of cardioprotection and reparation. Circulation. 2000;102:1342–1345. doi: 10.1161/01.cir.102.12.1342. [DOI] [PubMed] [Google Scholar]
- 10.Li RK, Li G, Mickle DA, et al. Overexpression of transforming growth factor-beta1 and insulin-like growth factor-I in patients with idiopathic hypertrophic cardiomyopathy. Circulation. 1997;96:874–881. doi: 10.1161/01.cir.96.3.874. [DOI] [PubMed] [Google Scholar]
- 11.Tomita H, Egashira K, Ohara Y, et al. Early induction of transforming growth factor-beta via angiotensin II type 1 receptors contributes to cardiac fibrosis induced by long-term blockade of nitric oxide synthesis in rats. Hypertension. 1998;32:273–279. doi: 10.1161/01.hyp.32.2.273. [DOI] [PubMed] [Google Scholar]
- 12.Kawano H, Do YS, Kawano Y, et al. Angiotensin II has multiple profibrotic effects in human cardiac fibroblasts. Circulation. 2000;101:1130–1137. doi: 10.1161/01.cir.101.10.1130. [DOI] [PubMed] [Google Scholar]
- 13.Brilla CG, Funck RC, Rupp H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation. 2000;102:1388–1393. doi: 10.1161/01.cir.102.12.1388. [DOI] [PubMed] [Google Scholar]