

Summary
The Rho family of GTPases regulates many aspects of cellular behavior through alterations to the actin cytoskeleton [1-6]. The majority of the Rho family proteins function as molecular switches cycling between the active, GTP-bound, and the inactive, GDP-bound, conformations [1-6]. Unlike typical Rho family proteins, the Rnd subfamily members, including Rnd1, Rnd2, RhoE/Rnd3, and RhoH, are GTPase deficient, and thus expected to be constitutively active [7-10]. Here, we identify an unexpected role for RhoE/Rnd3 in the regulation of the p53-mediated stress response. We show that RhoE is a transcriptional p53 target gene, and that genotoxic stress triggers actin depolymerization, resulting in actin-stress fiber disassembly through p53-dependent RhoE induction. Silencing of RhoE induction in response to genotoxic stress maintains stress fiber formation and strikingly increases apoptosis, implying an antagonistic role for RhoE in p53-dependent apoptosis. We found that RhoE inhibits ROCK I (Rho associated kinase I) activity during genotoxic stress thereby suppressing apoptosis. We demonstrate that the p53-mediated induction of RhoE in response to DNA damage favors cell survival partly through inhibition of ROCK I-mediated apoptosis. Thus, RhoE is anticipated to function by regulating ROCK I signaling to control the balance between cell survival and cell death in response to genotoxic stress.
Results and Discussion
We identified RhoE as a p53 responsive gene through a DNA chip expression array that compared gene expression in the presence or absence of p53. To confirm this data, we looked for upregulation of RhoE mRNA and protein in the p53 inducible EJ cell line and through adenoviral (Ad) expression of p53 in p53 null Saos2 cells (Figure 1A, Figure S1). Since p53 is important in the DNA damage response, the levels of RhoE mRNA and protein (Figure 1B, Figure S1) were studied in several p53 positive and negative cell lines after treatment with various DNA damaging agents: mitomycin C (MMC), etoposide (ETO), camptothecin (CPT). RhoE was induced in response to p53 or DNA damage in cells containing wild-type p53 with kinetics similar to those of the p53-dependent gene, p21 (Figure 1A). Inhibition of p53 activation in response to DNA damage, using p53 siRNA, abrogated RhoE induction (Figure 1B). We next investigated whether RhoE could be a direct p53 transcription target by mapping the transcriptional start site in a 5’ RACE experiment and found three potential p53 binding sites (p53-BS) in the RhoE promoter, which matched the consensus p53 binding sequence by 80-90% and contained the required conserved nucleotide sequence [11] (Figure 1C). To test the p53 response of these sites on the RhoE promoter, we linked the promoter, containing the three potential binding sites at positions −249, −580, and −2220, to a luciferase reporter gene. Co-transfection of the construct with a wt-p53 expression plasmid into p53 null Saos2 cells, increased luciferase activity significantly, while co-transfection with vector alone (Figure 1C) or mutant p53 (V143A) failed to do so (data not shown). We next generated point mutations at several consensus sequences of the three potential p53-BS in the RhoE promoter. A luciferase construct mutated at the -2220 position (designated M-1) significantly decreased luciferase activity in response to wt-p53, while mutations in either the −249 or −580 sites had little effect on luciferase activity (Figure 1C). U2OS cells, containing wt-p53, were transfected with the luciferase constructs, then treated with CPT for 24 hr in order to test the ability of endogenous p53 to activate the putative p53-BS in the RhoE promoter. These results confirm the data obtained in Saos2 cells that transcriptional activity is only affected upon mutation of the −2200 p53-BS (Figure 1C). To determine if p53 could bind to the RhoE promoter in vivo, a chromatin immunoprecipitation (ChIP) assay was carried out in Saos2 cells infected with Ad-p53 or Ad-GFP. The RhoE genomic fragment containing the -2200 p53-BS was specifically immunoprecipitated as a p53 protein-DNA complex with an anti-p53 antibody but not with anti-HA antibody (Figure 1D, Figure S1). PCR amplification of a region containing a p53-binding site in the p21 promoter served as a positive control (Figure 1D). In order to examine whether endogenous p53 could bind to the RhoE promoter in vivo after DNA damage, a ChIP assay was performed in CPT treated U2OS cells. The same genomic fragments were amplified from complexes immunoprecipitated with a p53-specific antibody, but not with control IgG in DNA damaged U2OS cells (Figure 1D). Although the fragments were present in both DMSO and CPT treated U2OS cells, there was a significant increase in the amount of DNA amplified from the complexes of the DNA damaged cells. ChIP assays performed using primers specific to either the −249 or −580 p53-BS in the RhoE promoter demonstrated no significant binding (data not shown). These results indicate that RhoE is a transcriptional target of p53, and that the −2220 p53-BS plays a role in p53-mediated RhoE transcription.
Figure 1. p53-dependent induction of RhoE.

(A) RhoE mRNA is induced after tetracycline (tet) removal in EJ-p53 cells (tet-off). Total RNA was prepared from these samples and northern blots were performed sequentially using 32P-labeled probe against RhoE, p21, and 36B4 (loading control). Total RNA was prepared from other p53 +/+ and -/- cell lines after mitomycin C (MMC, 1 μg/ml) or etoposide (ETO, 25 μM) treatment at the indicated times.
(B) U2OS and Saos2 cells were transfected with siRNA oligos targeting wt-p53 or luciferase control for 1 day, then treated with either 25 μM of ETO for 24 hr (U20S). or infected with adenovirus expressing p53 (Ad-p53) or Ad-GFP control (Saos2). Protein extracts were immunoblotted with RhoE, p21, p53 and β-actin antibodies.
(C) The p53 binding site at -2200 is responsible for RhoE transcription. Luciferase assay reporter gene constructs containing the putative p53-recognition sequences within the RhoE promoter were co-transfected into Saos2 cells with either wt-p53 or control expression constructs. U2OS cells were also transfected with the reporter plasmids, then treated with 600 nM camptothecin (CPT) for 24 hr. The reporter plasmids used were the: empty PGL3 vector, the full length (FL) promoter, six reporter constructs with p53-binding site mutations (M1-M6), and the p21 promoter was used as a control. The putative p53 recognition sequence within the RhoE promoter and the p53-consensus binding sequences (BS) are shown. Error bars represent the standard deviation from three independent experiments.
(D) Chromatin immunoprecipitation (ChIP) assay of p53 DNA binding to the RhoE promoter. Saos2 cells were infected with Ad-p53 or Ad-GFP for 24 hr, then ChIP was performed using either a p53 antibody or an HA-tag antibody (negative control). U2OS cells were also used for ChIP analysis of endogenous p53 activity upon DNA damage by 24 hr treatment with 600 nM CPT. Amplification products were 235 bp (RhoE) and 417 bp (p21), respectively.
To investigate whether RhoE induction in response to genotoxic stress contributes directly to p53-dependent stress responses, we used RhoE shRNA in the pBabe-U6-shRNA retroviral vector to knockdown the induction of endogenous RhoE in response to DNA damage. Transfection of RhoE shRNA resulted in the suppression of endogenous RhoE induced by CPT treatment of U2OS cells, while control shRNA (scrambled) had no effect (Figure 2A). To exclude off-target effects of shRNA, two independent RhoE shRNAs were used, both of which reduced RhoE expression (Figure 2A). In subsequent experiments, the data presented used the R1 RhoE shRNA and the results confirmed by transfection of R2.
Figure 2. RNAi ablation of endogenous RhoE induction in response to genotoxic stress restores stress fiber formation and enhances apoptosis.

(A) Expression of RhoE protein and mRNA in RhoE knockdown cells. RhoE expression was analyzed by northern and western blot analysis in U2OS cells transiently transfected with shRNA against RhoE (R1, R2) or control scrambled RhoE shRNA (C1, C2), then treated the following day with 600 nM CPT for the indicated times.
(B) RhoE effects actin stress fiber formation in response to genotoxic stress. U2OS cells stably transfected with RhoE shRNA (R1) or scrambled RhoE shRNA plasmids and were treated with CPT (600 nM) for 24 hr. Cells were stained for filamentous actin with rhodamine-conjugated phalloidin.
(C) RhoE depletion increases genotoxic stress-induced apoptosis. U2OS cells stably transfected with RhoE shRNA or scrambled RhoE shRNA plasmids were exposed to CPT (600 nM) or ETO (25 μM) for 24 hr. The M1 cell population represents the percentage of sub-G1 cells from each sample as calculated from 3 independent experiments. The percentages in the right panel indicate the number of apoptotic cells, which stain Annexin V positive and PI negative.
(D) DNA damage-induced cell blebbing is increased by RhoE knockdown. U2OS cells expressing control (scrambled) or RhoE shRNA plasmids were cultured on cover glass and treated with 600 nM CPT or DMSO for 48 hr then imaged by phase-contrast microscopy. Arrows indicate cells with membrane-blebbing. The percentage of cells that bleb at 32 hr were quantified and represents the mean ± S.E.M. of 3 experiments.
RhoE is known to inhibit RhoA/ROCK signaling and block actin-stress fiber formation [7,8,10,12,13]. CPT treatment of U2OS cells induced a dramatic loss of actin stress fibers as indicated by F-actin staining, while DMSO-treated cells maintained actin stress fibers (Figure 2B). Depletion of RhoE by shRNA in CPT-treated U2OS cells caused a reproducible recovery of actin stress fibers, as compared to cells expressing control shRNA (Figure 2B), suggesting that RhoE induction was required for the DNA damage-mediated loss of actin stress fibers. This recovery of actin stress fibers upon ablation of RhoE is consistent with overexpression studies showing RhoE inhibits actin stress fiber formation [7,8,12]. Furthermore, suppression of RhoE in response to genotoxic insult resulted in a significant increase in apoptosis from ~20% to ~50% as measured by flow cytometry analysis of cells stained with propidium iodide (PI) or by Annexin V/PI co-staining (Figure 2C). To confirm the p53 dependence of RhoE’s effect on apoptosis and to demonstrate that this result is not limited to U2OS cells, these experiments were repeated in normal dermal fibroblasts (IMR90), p53 null Saos2 cells, and p53 +/+ and -/- HCT116 cells (Figure S2, Figure S3). p53 wild type cells transiently transfected with shRhoE were able to enhance CPT induced apoptosis, as measured by TUNEL and DNA fragmentation assays (Figure S2, Figure S3). However, there was no noticeable change in the DNA damage response upon knocking down RhoE in p53 null cells (Figure S2, Figure S3). While increased stress fiber formation was observed upon inhibition of RhoE and treatment with CPT (Figure 2B), at later time points we were able to see an increase in membrane blebbing, a hallmark of cellular apoptosis, compared to control cells (Figure 2D).
The concomitant increase in actin fibers and cell death in RhoE depleted cells treated by DNA damage suggests a link between RhoE mediated-inhibition of actin stress fiber formation and ROCK I mediated cell death. ROCK I is an emerging protein kinase that is responsible for membrane blebbing in apoptotic cells [14-19]. Owing to the fact that RhoE interacts with ROCK I, and RhoE overexpression inhibits ROCK I-induced myosin light chain (MLC) phosphorylation [12,19], we investigated whether RhoE induction by DNA damage inhibits ROCK I kinase activity. We determined the effect of RhoE depletion on endogenous ROCK I kinase activity by measuring the level of MYPT1 (regulatory subunit of MLC-phosphatase) phosphorylation. While, CPT treated U2OS cells increased ROCK I activation more than 2-fold, depletion of RhoE in CPT treated cells further increased ROCK I activity (Figure 3A). We also examined the effect of RhoE over-expression on ROCK I activity in CPT treated 293ET cells containing dysfunctional p53. CPT treatment increased ROCK I activity in 293ET cells, but RhoE expression was not induced due to the lack of p53. Overexpression of RhoE was able to inhibit ROCK I activation (Figure 3B). This data suggests that p53 induction of RhoE antagonizes ROCK I-mediated cellular outcomes through a direct inhibition of ROCK I activity.
Figure 3. Effect of RhoE on ROCK I kinase activity during genotoxic stress.
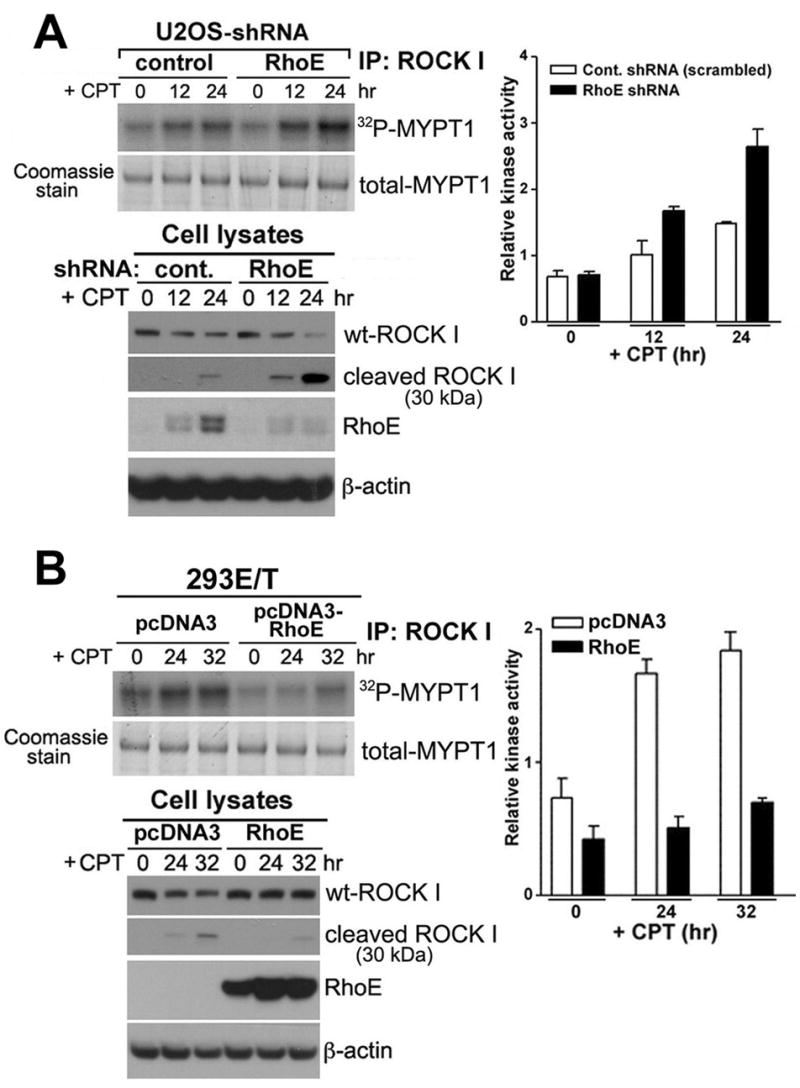
(A) Inhibition of RhoE expression enhances ROCK I kinase activity in wt-p53 expressing cells. U2OS cells stably expressing scrambled-RhoE-shRNA and RhoE-shRNA were treated with CPT for 12 or 24 hr. Cell lysates were subjected to immunoprecipitation (IP) with 2 μg of ROCK I antibody and western blotted to examine the levels of ROCK I and RhoE. The IP pallets were assayed for Rho kinase activity using MYPT1 as the substrate. The assay mixture was then subjected to SDS-PAGE. The gel was dried and autoradiographed. The band corresponding to MYPT1 was excised and quantified by scintillation counting. Error bars represent the standard deviation from three individual experiments.
(B) Effects of RhoE over-expression on ROCK I kinase activity in 293ET cells containing dysfunctional p53. 293ET cells were transfected with control and RhoE expression vector for 24 hr. The cells were then treated with 600 nM CPT for 24 and 32 hr, immunoprecipitated, and analyzed as described in (A).
Given that RhoE expression inhibits ROCK I function and exerts a pro-survival effect in response to p53 and p53-dependent genotoxic stress, we investigated the possibility that ROCK I-mediated apoptosis is one of the apoptotic pathways activated in response to genotoxic stress, and that RhoE modulates ROCK I-induced apoptosis. However, the role of ROCK I in apoptosis remains controversial [15,17,18,20,21], with studies carried out in different cell types using a variety of death inducing agents. Therefore, we first needed to confirm that genotoxic stress induced ROCK I activation and cell death.
During apoptosis, the full-length ROCK I, at 160 kDa (wt-ROCK I), is cleaved by caspase-3 into two fragments, an active short form of ROCK I (s-ROCK I), at 130 kDa, and a 30 kDa cleaved fragment [14,15,22]. Cell lysates from CPT or DMSO treated U2OS cells were immunoblotted with a C-terminal specific antibody that detects the full-length wt-ROCK I and the 30 kDa fragment (154C, Becton Dickinson) or an N-terminal specific antibody (H-85, Santa Cruz) that recognizes wt-ROCK I and active s-ROCK I. ROCK I cleavage was detected in the CPT treated cells, but not in the DMSO treated cells (Figure 4A). Transient transfection of ROCK I shRNA (RK1 siRNA sequence) resulted in a decrease of both forms of ROCK I in CPT treated cells, while control shRNA had no effect on ROCK I levels (Figure 4A). To exclude potential off-target effects, we compared our designed ROCK I shRNA with that of the commercially available siRNA (Santa Cruz), both of which reduced ROCK I expression (data not shown). In all subsequent experiments, our designed RK1 siRNA sequence was used. While ROCK I shRNA had no effect on the induction of RhoE following DNA damage, RhoE shRNA treatment resulted in increased ROCK I activation (Figure 4A). Cleavage of PARP, an indicator of caspase-3 activation, was also reduced in ROCK I knock-down cells upon CPT treatment as compared to the control cells (Figure 4A), suggesting that ROCK I could affect caspase-3 activation through a feed-forward loop. A recent study also indicated that constitutively active ROCK I resulted in an increase in caspase 3 activity and inhibiting ROCK I by shRNA suppressed further activation of caspase 3 upon ceramide treatment [23].
Figure 4. Effect of ROCK I inhibition on apoptosis.

(A) ROCK I activation/cleavage upon genotoxic stress. U2OS cells stably transfected with control (scrambled), ROCK I, or RhoE shRNA plasmids were treated with 600 nM CPT for 24 hr. The cleavage of ROCK I was assessed by immunoblotting with antibodies to both the C-terminus (top panel) and N-terminus of ROCK I (bottom panel). The cell lysates were further blotted for cleaved PARP, RhoE, p53, and β-actin.
(B&C) shRNA ablation of ROCK I expression inhibits the rate of apoptosis. U2OS (B) and HCT116 p53+/+ cells (C) expressing the shRNA plasmids were treated with 600 nM CPT for the indicated times. Apoptotic cell death was quantified by TUNEL assay and enumerated by flow cytometry. The left panel shows a representative experiment (B) and the percentage of TUNEL positive cells in the graphs is shown as the mean ± SEM (n=3).
(D) Effect of ROCK I inhibitor (Y-27632) on CPT-mediated apoptosis.
U2OS cells were treated with either DMSO or Y-27632 (3 μM) for 24 hr, and exposed to 600 nM CPT for the indicated time periods (24 and 48 hr). Cells were then assessed for apoptosis by TUNEL assay by both fluorescence microscopy and flow cytometry as described for (B&C).
To directly determine whether inhibition of ROCK I could influence genotoxic stress-induced apoptosis, we used the TUNEL assay to analyze the level of apoptosis in CPT treated U2OS cells in the presence of ROCK I or controlshRNA. Following the reduction of ROCK I expression, the percentage of cells undergoing apoptosis after CPT treatment was significantly decreased, compared to control cells, while shRNA against RhoE resulted in an increase in cell death in response to CPT (Figure 4B). This data was confirmed using PI staining and a DNA fragmentation assay (Figure S4). Similar results were obtained in HCT116 cells (Figure 4C) and mouse embryonic fibroblasts (MEF) (Figure S4), confirming this result is not cell type dependent. We also examined whether the ROCK I inhibitor Y-27632 [24] could affect the DNA damage-induced apoptotic response. Y-27632 prevented CPT-induced cell death to a level similar to that achieved by ROCK I shRNA transfection, as measured by TUNEL assay (Figure 4D).
Since the 130 kDa s-ROCK I is known to be the active form of ROCK I generated in response to DNA damage [14,15], we investigated whether over-expression of s-ROCK I itself could result in apoptosis. U2OS cells were infected with adenoviruses (Ad) expressing s-ROCK I or wt-ROCK I. Overexpression of s-ROCK I caused a significant induction of cell death as measured by the number of TUNEL positive cells (Figure S5), but expression of wt-ROCK I failed to have any apoptotic effect. The ability of s-ROCK I to induce apoptosis was markedly abrogated in cells co-infected with Ad-s-ROCK I and Ad-RhoE (Figure S5). Consistent with previous studies [23], this data demonstrates a direct correlation between the apoptotic response and ROCK I activation. Together, this data indicates that ROCK I may be an important mediator in apoptosis and that RhoE is a critical negative regulator of ROCK I activity, including ROCK I mediated membrane blebbing and cell death.
Our results demonstrate that RhoE is a novel transcriptional target gene of p53 that confers pro-survival effects in the p53-dependent response to genotoxic stress. Studies on RhoE indicate that it has two distinct functions, one in regulating the actin cytoskeleton and another in cell proliferation by inhibiting cell cycle progression [13]. Recently it was shown that RhoE expression is significantly reduced in prostate cancer cells compared to normal prostate cells [25], suggesting that RhoE has anti-proliferative effects [10]. However, RhoE expression is elevated in pancreatic tumors, colon cancer cells and melanomas [26-28], suggesting various roles for RhoE in different types of cancer. Our data points to RhoE acting as a novel pro-survival factor whereupon induction of RhoE leads to the survival and propagation of cells that have sustained DNA damage. This is supported by the fact that cells with silenced RhoE show an increased propensity to undergo apoptosis in response to DNA damage. p53 pro-survival effectors such as RhoE, which are induced in damaged wt-p53 containing cells, play an important role in tissue damage and repair in response to DNA damaging agents. Therefore, identifying the components of the novel p53 → RhoE/ROCK I signaling pathway will provide an opportunity for therapeutic intervention by revealing novel targets for drug design that will potentially maximize the p53 dependent response and sensitive tumor cells to therapeutic drugs.
Supplemental Data
Supplemental Experimental Procedures and figures can be found with this article online.
Acknowledgments
We thank the members of the CBRC for helpful discussion and are grateful to J. Settleman for providing RhoE antibody, and S. Narumiya (Kyoto University, Kyoto Japan) for the ROCK I cDNA. This work was supported by NIH grants (CA80058, CA127247, CA097216, TG-AR007098-31), American Diabetes Association (7-05-PPG-02) and Shiseido Research Core funding.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 2.Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Rev Cancer. 2002;2:133–142. doi: 10.1038/nrc725. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt A, Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 2002;16:1587–1609. doi: 10.1101/gad.1003302. [DOI] [PubMed] [Google Scholar]
- 4.Moon SY, Zheng Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003;13:13–22. doi: 10.1016/s0962-8924(02)00004-1. [DOI] [PubMed] [Google Scholar]
- 5.Coleman ML, Marshall CJ, Olson MF. RAS and RHO GTPases in G1-phase cell-cycle regulation. Nat Rev Mol Cell Biol. 2004;5:355–366. doi: 10.1038/nrm1365. [DOI] [PubMed] [Google Scholar]
- 6.Chardin P. GTPase regulation: getting aRnd Rock and Rho inhibition. Curr Biol. 2003;13:R702–704. doi: 10.1016/j.cub.2003.08.042. [DOI] [PubMed] [Google Scholar]
- 7.Foster R, Hu KQ, Lu Y, Nolan KM, Thissen J, Settleman J. Identification of a novel human Rho protein with unusual properties: GTPase deficiency and in vivo farnesylation. Mol Cell Biol. 1996;16:2689–2699. doi: 10.1128/mcb.16.6.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guasch RM, Scambler P, Jones GE, Ridley AJ. RhoE regulates actin cytoskeleton organization and cell migration. Mol Cell Biol. 1998;18:4761–4771. doi: 10.1128/mcb.18.8.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nobes CD, Lauritzen I, Mattei MG, Paris S, Hall A, Chardin P. A new member of the Rho family, Rnd1, promotes disassembly of actin filament structures and loss of cell adhesion. J Cell Biol. 1998;141:187–197. doi: 10.1083/jcb.141.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chardin P. Function and regulation of Rnd proteins. Nat Rev Mol Cell Biol. 2006;7:54–62. doi: 10.1038/nrm1788. [DOI] [PubMed] [Google Scholar]
- 11.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 12.Riento K, Guasch RM, Garg R, Jin B, Ridley AJ. RhoE binds to ROCK I and inhibits downstream signaling. Mol Cell Biol. 2003;23:4219–4229. doi: 10.1128/MCB.23.12.4219-4229.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villalonga P, Guasch RM, Riento K, Ridley AJ. RhoE inhibits cell cycle progression and Ras-induced transformation. Mol Cell Biol. 2004;24:7829–7840. doi: 10.1128/MCB.24.18.7829-7840.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sebbagh M, Renvoize C, Hamelin J, Riche N, Bertoglio J, Breard J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol. 2001;3:346–352. doi: 10.1038/35070019. [DOI] [PubMed] [Google Scholar]
- 15.Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001;3:339–345. doi: 10.1038/35070009. [DOI] [PubMed] [Google Scholar]
- 16.Coleman ML, Olson MF. Rho GTPase signalling pathways in the morphological changes associated with apoptosis. Cell Death Differ. 2002;9:493–504. doi: 10.1038/sj.cdd.4400987. [DOI] [PubMed] [Google Scholar]
- 17.Lane JD, Allan VJ, Woodman PG. Active relocation of chromatin and endoplasmic reticulum into blebs in late apoptotic cells. J Cell Sci. 2005;118:4059–4071. doi: 10.1242/jcs.02529. [DOI] [PubMed] [Google Scholar]
- 18.Croft DR, Coleman ML, Li S, Robertson D, Sullivan T, Stewart CL, Olson MF. Actin-myosin-based contraction is responsible for apoptotic nuclear disintegration. J Cell Biol. 2005;168:245–255. doi: 10.1083/jcb.200409049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riento K, Totty N, Villalonga P, Garg R, Guasch R, Ridley AJ. RhoE function is regulated by ROCK I-mediated phosphorylation. EMBO J. 2005;24:1170–1180. doi: 10.1038/sj.emboj.7600612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lai JM, Hsieh CL, Chang ZF. Caspase activation during phorbol ester-induced apoptosis requires ROCK-dependent myosin-mediated contraction. J Cell Sci. 2003;116:3491–3501. doi: 10.1242/jcs.00660. [DOI] [PubMed] [Google Scholar]
- 21.Minambres R, Guasch RM, Perez-Arago A, Guerri C. The RhoA/ROCK-I/MLC pathway is involved in the ethanol-induced apoptosis by anoikis in astrocytes. J Cell Sci. 2006;119:271–282. doi: 10.1242/jcs.02723. [DOI] [PubMed] [Google Scholar]
- 22.Zihni C, Mitsopoulos C, Tavares IA, Ridley AJ, Morris JD. Prostate-derived sterile 20-like kinase 2 (PSK2) regulates apoptotic morphology via C-Jun N-terminal kinase and RHO kinase-1. J Biol Chem. 2006;281:7317–7323. doi: 10.1074/jbc.M513769200. [DOI] [PubMed] [Google Scholar]
- 23.Chang J, Xie M, Shah VR, Schneider MD, Entman ML, Wei L, Schwartz RJ. Activation of Rho-associated coiled-coil protein kinase 1 (ROCK-1) by caspase-3 cleavage plays an essential role in cardiac myocyte apoptosis. Proc Natl Acad Sci U S A. 2006;103:14495–14500. doi: 10.1073/pnas.0601911103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takahara A, Sugiyama A, Satoh Y, Yoneyama M, Hashimoto K. Cardiovascular effects of Y-27632, a selective Rho-associated kinase inhibitor, assessed in the halothane-anesthetized canine model. Eur J Pharmacol. 2003;460:51–57. doi: 10.1016/s0014-2999(02)02929-1. [DOI] [PubMed] [Google Scholar]
- 25.Bektic J, Pfeil K, Berger AP, Ramoner R, Pelzer A, Schafer G, Kofler K, Bartsch G, Klocker H. Small G-protein RhoE is underexpressed in prostate cancer and induces cell cycle arrest and apoptosis. Prostate. 2005;64:332–340. doi: 10.1002/pros.20243. [DOI] [PubMed] [Google Scholar]
- 26.Gress TM, Muller-Pillasch F, Geng M, Zimmerhackl F, Zehetner G, Friess H, Buchler M, Adler G, Lehrach H. A pancreatic cancer-specific expression profile. Oncogene. 1996;13:1819–1830. [PubMed] [Google Scholar]
- 27.van Groningen JJ, Cornelissen IM, van Muijen GN, Bloemers HP, Swart GW. Simultaneous suppression of progression marker genes in the highly malignant human melanoma cell line BLM after transfection with the adenovirus-5 E1A gene. Biochem Biophys Res Commun. 1996;225:808–816. doi: 10.1006/bbrc.1996.1255. [DOI] [PubMed] [Google Scholar]
- 28.Akashi H, Han HJ, Iizaka M, Nakamura Y. Growth-suppressive effect of non-steroidal anti-inflammatory drugs on 11 colon-cancer cell lines and fluorescence differential display of genes whose expression is influenced by sulindac. Int J Cancer. 2000;88:873–880. doi: 10.1002/1097-0215(20001215)88:6<873::aid-ijc6>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Experimental Procedures and figures can be found with this article online.