

Summary
Abnormalities of striatal function have been implicated in several major neurological and psychiatric disorders, including Parkinson's disease, schizophrenia, and depression. Adenosine, by activation of A2A receptors, antagonizes dopamine signaling at D2 receptors and A2A receptor antagonists have been tested as therapeutic agents for Parkinson's disease. We report here a direct physical interaction between the G protein-coupled A2A receptor and the receptor tyrosine kinase FGF receptor. Concomitant activation of these two classes of receptors, but not individual activation of either one alone, causes a robust activation of the MAPK/ERK pathway, differentiation and neurite extension of PC12 cells, spine morphogenesis in primary neuronal cultures, and cortico-striatal plasticity induced by a novel A2AR/FGFR-dependent mechanism. The discovery of a direct physical interaction between the A2A and FGF receptors and the robust physiological consequences of this association shed light on the mechanism underlying FGF functions as a co-transmitter and open new avenues for therapeutic interventions.
Introduction
The striatum, a component of the basal ganglia, receives dopaminergic innervation from the substantia nigra as well as glutamatergic innervation from the cortex. It moderates the control of complex motor activity, as well as cognitive aspects of motor control. Medium spiny neurons, which represent over 90% of all striatal neurons, are the only known efferent neurons of the striatum. Roughly half of these neurons (the striatopallidal projection) contain A2A receptors (A2ARs). A2ARs play an important role in the biology of the striatum through their coupling to Gs/Golf proteins. Their signaling actions are opposed by those of D2 dopamine receptors, which are located on the same cells and are coupled to Gi/o and Gq proteins 1. Striatal A2ARs are activated by adenosine generated by ecto-nucleotidase degradation of ATP released by neurons and astrocytes 2. The stimulatory effects of caffeine on locomotor activity are attributable to blockade of A2ARs 3. Conversely, A2AR agonists inhibit movement 4. Abnormalities in striatal A2AR signaling have been implicated in Parkinson's disease (PD), schizophrenia, Attention Deficit Hyperactivity Disorder (ADHD) and drug abuse (for a review see 5), leading to a concerted effort to develop pharmacological agents acting at these receptors. Thus, clarification of the signal transduction mechanisms associated with A2ARs may help in design of improved therapeutic agents.
FGFs are polypeptides that signal through a family of four major receptors 6. For a long time FGFs were known to only play a trophic role in basic cells such as fibroblasts. Recently, FGFs have been shown to play crucial roles in neural induction, neural plate patterning, neuronal proliferation and survival, as well as in presynaptic organization 7,8,9,10. Several recent studies have provided evidence that the expression of FGF is regulated in the brain by different stimuli including dopamine, cocaine and stress 11,12. Like other growth factors, FGF has also been hypothesized to play an important role in the adult nervous system 6. One of the functions of tyrosine kinase-linked receptors in the adult brain is in the regulation of synaptic plasticity thought to underlie learning and memory. For example, BDNF is a key regulator of synaptic plasticity in the adult hippocampus. Both FGF and FGF receptors are abundantly expressed in the striatum 13. Moreover, there is increasing evidence for a neuroprotective role of FGFs and FGFRs in the striatum, suggesting that this growth factor may affect synaptic plasticity in this brain region.
Here we present direct evidence for a profound role of the FGF system in the modulation of synaptic plasticity, specifically in the cortico-striato-pallidal pathway. Our data demonstrate that A2AR-FGFR co-stimulation causes a dramatic synergistic increase in neurite formation and spine density, and induces a form of cortico-striatal LTP in striatopallidal neurons. The mechanism underlying these phenomena appears to involve a direct physical interaction between the A2AR and the FGFR. This interaction leads to synergistic activation of MEK1/2 and consequently to a robust increase of ERK1/2 phosphorylation, which occurs rapidly and is long lasting. A “proximity model” is proposed to account for the synergistic interaction observed.
Results
Identification of FGFR as an A2AR-interacting protein
In a search for novel molecular mechanisms that might affect the actions of the dopamine modulator adenosine, we used the yeast two-hybrid method to identify new proteins that could selectively bind to the A2AR. The cDNA corresponding to the last 133 aa of the intracellular C-terminal sequence of the A2AR (residues 278-410) was subcloned into a pAS2-GAL4-derived plasmid and used as bait (A2AR-278-410). We screened 25 × 106 diploid clones from a pACT2 cDNA library. Of more than 1000 histidine-positive clones, 71 clones were also positive for the β-galactosidase reporter gene. Unexpectedly, we found that 7 of the 50 sequenced double-positive clones shared sequence similarities with the cytoplasmic tail of the fibroblast growth factor receptor 1 (FGFR1).
To determine the specificity of interaction, the FGFR prey plasmid corresponding to the smallest fragment of the FGFR (666-822) rescued from yeast was amplified in E. coli and transformed into yeast that harbored (i) the original A2AR-278-410 bait plasmid, (ii) the empty bait plasmid (-), or (iii) one of four irrelevant baits (two viral proteins and two yeast proteins). The interaction was specific since only the FGFR (666-822) and A2AR (278-410) interaction was positive (Fig. 1A). To confirm the A2AR-FGFR interaction, a GST pull-down assay was employed. Radiolabeled FGFR domain (666-822) or full length FGFR (FGFR.FL) was incubated with the same amount of either GST or GST-A2AR-278-410. GST-A2AR-278-410 fusion protein but not GST alone pulled down both FGFR-666-822 (data not shown) and FGFR.FL (Fig. 1B, left panel) as demonstrated by SDS-PAGE analysis. As a control, GST-A2AR-278-410 was found not to interact with radiolabeled luciferase (Fig. 1B, right panel).
Figure 1. Characterization of the physical interaction between A2A and FGF receptors.
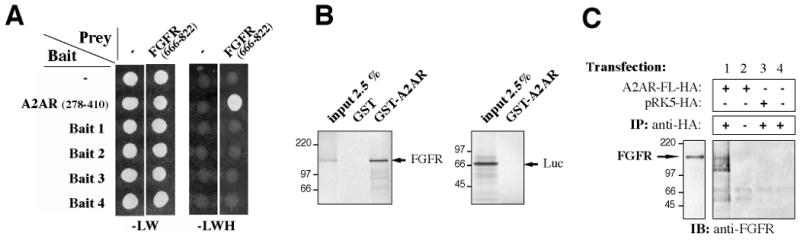
(A) The bait and prey constructs are indicated, on the left and top, respectively; (-) indicates that the corresponding bait or prey plasmid did not contain any insert in fusion with GAL4. Double transformants were spotted and grown for 3 days at 30C on -LW medium or on -LWH to check for histidine auxotrophy. All the clones grew on -LW medium (growth control), but only coexpression of A2AR (aa 278-410) and FGFR (aa 666-822) allowed growth in the absence of histidine (-LWH).
(B) Immobilized GST or GST-A2AR-278-410 (GST-A2AR) fusion protein was incubated with 35S-labeled, in-vitro translated FGFR or luciferase as indicated. Bound material was subjected to SDS-PAGE and visualized by autoradiography. 2.5% of the 35S-labeled input is shown.
(C) Full-length A2AR receptor, HA-tagged (A2AR-FL-HA), or the empty plasmid (pRK5-HA) was expressed in COS-7 cells. Cells were lysed and immunoprecipitation (IP) was carried out using a monoclonal anti-HA antibody. Proteins were analyzed by SDS-PAGE and immunoblotting (IB) with anti-FGFR antibody. An aliquot of the cell lysate was also immunoblotted with anti-FGFR (left panel).
To investigate the interaction between full-length A2AR and native FGFR in mammalian cells, full-length HA-tagged A2AR was expressed in COS-7 cells. A2AR-HA was immunoprecipitated using an anti-HA antibody, and bound FGFR was detected by immunoblotting with an anti-FGFR antibody. Endogenous FGFR was co-immunoprecipitated with A2AR-HA (Fig. 1C, lane 1), whereas no FGFR was detected in immunoprecipitates in the absence of the anti-HA antibody (Fig. 1C, lane 2), from cells transfected with the empty plasmid (Fig. 1C, lane 3), or from non-transfected cells (Fig. 1C, lane 4).
Characterization of the domains of interaction between A2AR and FGFR
Fragments corresponding to different regions of A2AR-278-410 and FGF-666-822 were cloned into the bait and prey plasmids, respectively. The constructs were transformed in yeast, and their expression confirmed by immunoblot using an anti-Gal4 antibody. We found that amino acids 278-315 of A2AR were sufficient for interaction with amino acids 666-822 of FGFR (Fig. 2A). Conversely, amino acids 666-788 of FGFR were necessary for interaction with amino acids 278-410 of A2AR (Fig. 2B). Amino acids 666-788 of FGFR contain two tyrosine residues (Tyr730 and Tyr766), which are phosphorylation sites and are extremely well conserved among growth factor tyrosine kinase receptors. Phosphorylation on Tyr766 leads to activation of phospholipase C-gamma and is involved in the internalization of the FGFR1 14,15, whereas the function of the phosphorylation of Tyr730 is still not clear. To test the possibility that one or both of the tyrosines are implicated in the A2AR/FGFR interaction, we made single alanine and aspartic acid mutants for each position. By GST pull-down, we found that mutant Y766D, but not Y766A, Y730D or Y730A, showed reduced binding to A2AR-278-410, indicating that phosphorylation at Tyr-766 may negatively regulate the A2AR/FGFR interaction (Supplementary Fig. 1).
Figure 2. Characterization of the domains of interaction.
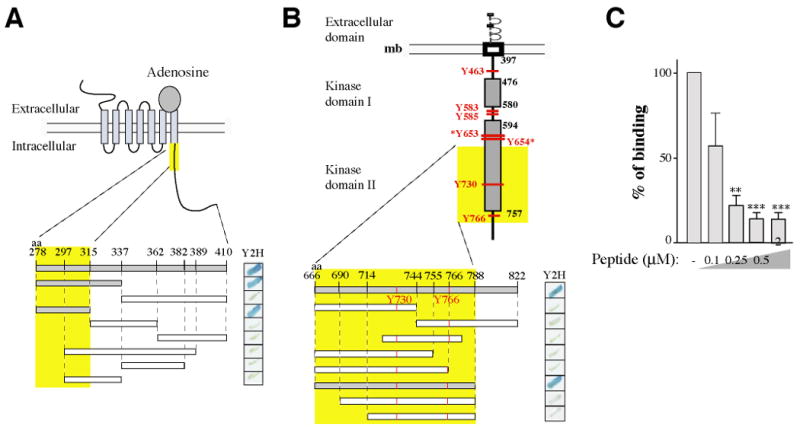
(A) Schematic representation of the A2A receptor. Various truncated GAL4-A2AR fusion proteins were expressed as bait in S. cerevisiae and tested in a yeast two-hybrid -galactosidase assay for their capacity to interact with the GAL4-FGFR prey (aa 666-822). (B) Schematic representation of the FGF receptor. Tyrosine phosphorylation sites are indicated in red. Various truncated GAL4-FGFR fusion proteins were expressed as prey in S. cerevisiae and tested for their capacity to interact with the GAL4-A2AR bait (aa 278-410).
In (A) and (B), positive interactions are indicated by the blue stain (right panels). Interacting fragments are indicated in grey and the minimal domain of interaction found is highlighted by the yellow boxes.
(C) Competition of the A2AR/FGFR interaction analyzed by GST pull-down with a synthetic peptide corresponding to the A2AR minimal domain of interaction (aa 278-315). Immobilized GST-A2AR-278-410 fusion protein was incubated with radio labeled in-vitro translated FGFR. Bound material was subjected to SDS-PAGE and visualized by autoradiography. Signals were quantified and statistical analysis performed as described in Methods.
A synthetic peptide corresponding to the minimal region of interaction found for the A2AR (aa 278-315) was designed. The resulting synthetic peptide effectively disrupted the pre-formed interaction in a GST pull-down assay experiment using a GST-A2AR fusion protein and a radiolabeled in vitro transcribed FGFR (Fig. 2C).
The A2AR agonist CGS21680 and FGF synergistically activate the ERK/MAPK pathway
Functional studies to evaluate the physiological consequences of the interaction of the A2A and FGF receptors initially used PC12 cells, a cell line that was confirmed to express the A2AR and FGFRs (data not shown)16, 17. After 48 hrs of serum starvation (0.5% fetal calf serum), PC12 cells were treated with the A2AR agonist (CGS21680) alone, aFGF alone, or a combination of CGS21680 and aFGF. As a biochemical readout of possible cross-talk between activation of A2ARs and FGFRs, we measured phosphorylation of ERK1/2, a known downstream target of each receptor (Fig. 3A,B). With either CGS21680 alone (250 nM) or aFGF alone (5 ng/ml), a significant increase in ERK1/2 phosphorylation was not seen until 30 min of incubation (Fig. 3A,B). In contrast, at 10 min of incubation, the simultaneous addition of CGS21680 and aFGF resulted in a greater than 15-fold increase in ERK1/2 phosphorylation. Dose response studies indicated that a combination of CGS21680 at 1 μM and aFGF at 20 ng/ml resulted in maximal synergistic phosphorylation of ERK1/2 (Fig. 3C,D). The specificity for CGS21680 acting at A2ARs was confirmed with ZM241385, a selective A2AR antagonist, which induced a dose-dependent inhibition of the CGS21680/aFGF-dependent increase in ERK1/2 phosphorylation (Fig. 3E). We have also found that the FGFR inhibitor PLX052 abolished the synergistic effects observed in a concentration-dependent manner (Fig. 3F). Finally we found that a penetrating peptide corresponding to the minimal A2AR domain of interaction (see Fig. 2), blocked the synergistic activation of ERK (Fig. 3G). Stimulation with CGS21680 or aFGF alone leads in the same conditions to only a weak stimulation of ERK phosphorylation and only at the highest doses (Fig. 3H).
Figure 3. Synergistic activation of the MAPK pathway is induced by co-activation of A2A and FGF receptors.

(A) Serum-starved PC12 cells were treated with CGS21680 (250 nM) and/or aFGF (5 ng/ml) for various periods of time (from 0 to 60 min), lysed and the cell extracts were analyzed by immunoblotting using phospho-p44/42 ERK1/2 (Thr202/Tyr204) antibody.
(B) Quantification of the data. Data were normalized to actin values and compared to untreated cell values. Data are presented as means ± SEM (n=4).
(C-D) Analysis of ERK1/2 phosphorylation in response to various doses of A2AR agonist (CGS21680) with a fixed concentration of aFGF (C) or various concentrations of aFGF with a fixed concentration of CGS21680 (D). Serum-starved PC12 cells were treated with increasing concentrations of CGS21680 or aFGF for 10 min, and cell extracts analyzed by immunoblotting as described above. Cumulative results (n=6) are presented as histograms (means ± SEM).
(E) Competition of A2AR antagonist ZM 241385 and CGS21680. Cells were treated with aFGF and/or CGS21680 in the presence of various concentrations of ZM 241385 and samples analysed as described above.
(F) The FGFR inhibitor PLX052 inhibits in a dose dependent manner the synergism observed. Cells were treated with aFGF and CGS21680 in the presence or absence of various concentrations of PLX052 and samples analyzed as described above.
(G) An A2AR inhibitory peptide reduces the synergism observed. Cells were treated with aFGF and CGS21680 in the presence or absence of various concentrations of the A2AR inhibitory peptide and samples analyzed as described above.
(H) Doses of CGS21680 or aFGF used in panels C-F were applied individually for 10 min.
We next investigated the signaling steps that might be subject to synergistic regulation by activation of A2ARs and FGFRs. By using various phospho-specific antibodies directed against c-Raf, MEK1/2 and ERK1/2, MEK1/2 was found to be the first protein of the MAP kinase cascade to become highly phosphorylated in a synergistic manner upon simultaneous stimulation by aFGF and CGS21680 (Supplementary Fig. 2). No activation of the p38 MAPK or SAPK/JNK pathways was observed (data not shown). In addition, aFGF failed to alter CGS21680-induced cAMP formation, and CGS21680 failed to affect FGF-induced phosphorylation of the FGF receptor (Supplementary Fig. 3).
To further investigate the nature of the observed synergism, we tested the possibility that FGFR activation may affect A2AR signaling indirectly by altering its cell surface expression or internalization. If FGF was able to increase the cell surface expression of A2AR, such a phenomenon could explain our results. PC12 cells were incubated for either 10 min or 60 min with CGS21680 (1 μM), FGF (100 ng/ml) or both. Cell surface binding sites were determined by incubation at 4°C with the A2A antagonist [3H]ZM241385 and non-specific binding (determined by the addition of a saturating concentration of ZM241385) was subtracted. This assay did not reveal any effect of FGF on A2AR cell surface expression-internalization (data not shown).
An A2AR dominant negative mutant suppresses the A2AR/FGFR-dependent synergistic activation of ERK1/2
The adenosine binding region of the A2AR is located on the extracellular part of the receptor and includes TM5-TM7 (trans-Membrane domains 5 to 7). We designed A2AR deletion mutants in order to disrupt the ligand binding site without affecting the binding of FGFR to A2AR. Four mutants were tested for their capacity to interact in vitro with FGFR in a GST pull-down assay, for their level of expression by immunoblot analysis and for their capacity to be directed to the membrane by immuno-fluorescence. A schematic representation of one of the mutants satisfying all the criteria, designated A2AR-6-7TM (deletion of TM 6 and 7), is shown in comparison to A2AR.FL (Fig. 4A).
Figure 4. Synergistic activation of the MAPK pathway is reduced by an A2AR dominant negative mutant.
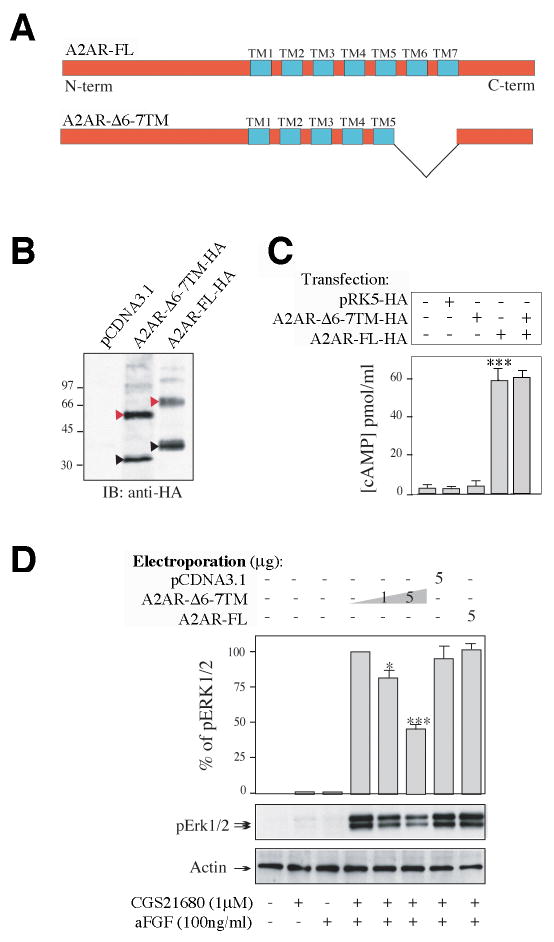
(A) Schematic representation of the full-length (top) and the truncated mutant (bottom) A2AR. Trans-membrane domains (TM) are indicated in blue. Domains 6 and 7 are deleted in the mutant construct (A2AR-6-7TM).
(B) Comparison of the level of expression of the two constructs after expression in COS-7 cells. Cells were lysed and proteins were analyzed by SDS-PAGE and immunoblotting (IB) using a monoclonal anti-HA antibody. Black and red triangles indicate positions of the monomeric and dimeric receptors respectively. Empty plasmid (pCDNA3.1) was used as negative control.
(C) Comparison of the abilities of the full-length and mutant A2AR to generate cAMP in COS-7 cells. Cells were incubated with 1μM CGS21680 for 10 min, lysed, and cAMP content of cell lysates analyzed by ELISA (Sigma). PRK5, empty control vector.
(D) Expression of the mutant A2AR in PC12 cells reduces synergistic regulation of ERK1/2 by A2AR/FGFR. Mutant and full-length A2AR were expressed in PC12 cells using electroporation. Empty plasmid was electroporated as a control. Cells were incubated with CGS21680 and aFGF, and phospho-ERK1/2 was measured as described above. Values were normalized to the amount of total protein, determined by immunoblot analysis using an anti-actin antibody, and to the electroporation efficiency determined by immunofluorescent analysis using an anti-GFP antibody. Data are presented as means ± SEM (n=4).
The levels of expression of the two receptors (A2AR and A2AR-6-7TM) were similar in COS-7 cells (Fig. 4B). We compared their capacity to be activated by CGS21680 by measuring cAMP production (Fig. 4C). Wild type A2AR was active and positively coupled to adenylyl cyclase, as shown by cAMP production upon CGS21680 stimulation. In contrast, the A2AR-6-7TM mutant did not respond to CGS21680 with the production of cAMP, nor did it alter the response of wild type A2AR to CGS21680 (Fig. 4C). However, expression of A2AR-6-7TM resulted in 52% reduction of the ERK1/2 phosphorylation seen in response to CGS21680/aFGF (Fig. 4D). These results provide strong support for the conclusion that a direct physical interaction between the A2AR and the FGFR is required for the synergistic activation of ERK1/2.
Morphological consequences of the interaction between activated A2ARs and FGFRs
PC12 cells manifest certain morphological aspects of neuronal differentiation in the presence of growth factors such as FGF, as well as agents that stimulate cAMP levels. Moreover, activation of the MAP kinase pathway has been implicated in the differentiation of PC12 cells and in the elaboration of neurites 18. PC12 cells were incubated with aFGF and/or CGS21680 for 4 days (Fig. 5A). In the absence of ligand, PC12 cells were spherical. After activation with CGS21680 or aFGF, cells began to differentiate and neurite extensions appeared. However, when both ligands were applied simultaneously, there was a dramatic increase in the number of cells with extensions and in the average number of extensions per cell (Fig. 5B, C). Preincubation with the penetrating A2AR inhibitory peptide (Pep, 50 μM) or the FGFR inhibitor PLX052 (PLX, 10 μM) blocked the synergistic effects of CGS21680/aFGF on neurite extension (Fig. 5A, B, C). PC12 cells co-stimulated with CGS21680/aFGF in presence of the A2AR antagonist ZM241385 were similar to PC12 cells incubated with aFGF alone (not shown).
Figure 5. CGS21680 and aFGF synergistically induce neurite formation in PC12 cells and increase spine density in primary neuronal cultures.
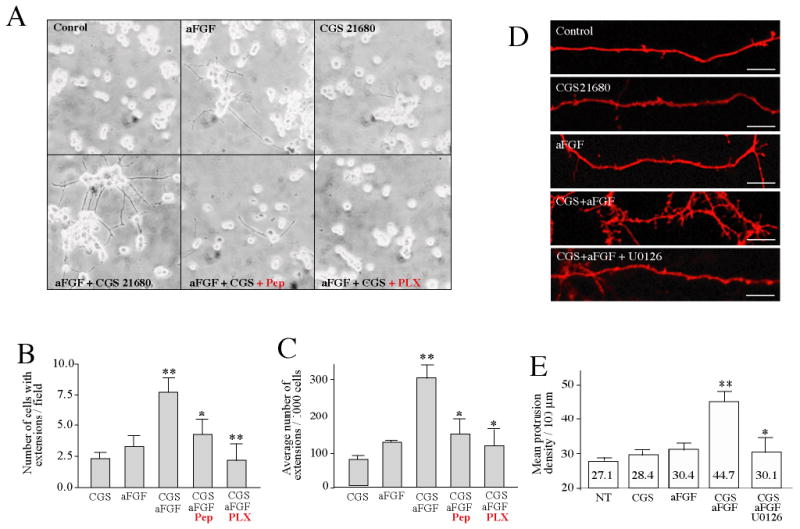
(A-C) PC12 cells, (A) Serum-starved PC12 cells were treated without (Control) or with aFGF (100 ng/ml) and/or CGS21680 (1 μM), or with aFGF and CGS21680 in the presence of U0126 (10 μM), or the A2AR inhibitory peptide (Pep, 50 μM), or PLX052 (PLX, 10 μM) as indicated. After two days, the medium was replaced and fresh ligands were added. After a total incubation time of 4 days, the cells were fixed and photographed. Protrusions were analyzed under light microscopy.
(B) Number of cells with extensions per field.
(C) Average number of extensions per 1,000 cells. Data are presented as means ± SEM.
(D) Primary hippocampal neurons (11 DIV), starved for 3 hrs, were treated for 60 min without (Control) or with aFGF (100 ng/ml) and/or CGS21680 (1 μM) as indicated. The number of protrusions was determined after immunofluorescent labeling.
(E) Data from (D) are presented as means ± SEM.
The MAP kinase pathway has been implicated in various aspects of neuronal development and synaptic plasticity, including regulation of dendritic spine morphogenesis 19,20,21. In view of the strong ERK activation observed in the present study, we investigated the possibility that the synergistic action observed between CGS21680 and aFGF could influence protrusion density in cultured hippocampal neurons. Neuronal cultures were incubated for 60 min in the absence or presence of CGS21680 and/or aFGF. Representative segments of dendrites from neurons expressing CD8 as a volume marker are shown in Fig. 5D. In control neurons, the protrusion density was 27.1 ± 3.2 per 100 μm. Treatment with CGS21680 alone, aFGF alone or CGS21680 and aFGF, caused increases in mean density of protrusions per 100 m of 4.8% ± 0.8, 12.2% ± 1.1, and 65% ± 1.7%, respectively, compared with paired untreated control cultures (Fig. 5 D,E). Treatment with the MEK inhibitor, U0126, but not with an inactive analog, U0124, inhibited the effect of co-application of CGS21680 and aFGF treatment by 83% (Fig. 5D, E).
Synergistic facilitation of corticostriatal LTP
Given the ability of the A2AR/FGFR interaction to synergistically activate ERK1/2 and to synergistically increase spine morphogenesis, we tested the possibility that combined activation of A2ARs and FGFRs might play a role in regulation of corticostriatal synaptic plasticity. Plasticity at glutamatergic, corticostriatal synapses was studied in parasagital brain slices (Fig. 6A) from BAC transgenic mice in which either D1 receptor-expressing striatonigral MSNs or D2 receptor-expressing striatopallidal MSNs were labeled with green fluorescent protein. As previously reported22, activation of A2ARs with the selective agonist CGS 21680 (10 nM) had no effect on basal AMPA receptor mediated corticostriatal synaptic transmission in either type of MSN (data not shown, n=2); FGF (10 ng/ml) did not affect basal synaptic transmission either (data not shown; n=3).
Figure 6. Co-activation of FGF receptors and A2A receptors facilitates the induction of LTP at corticostriatal synapses of striatopallidal MSNs.
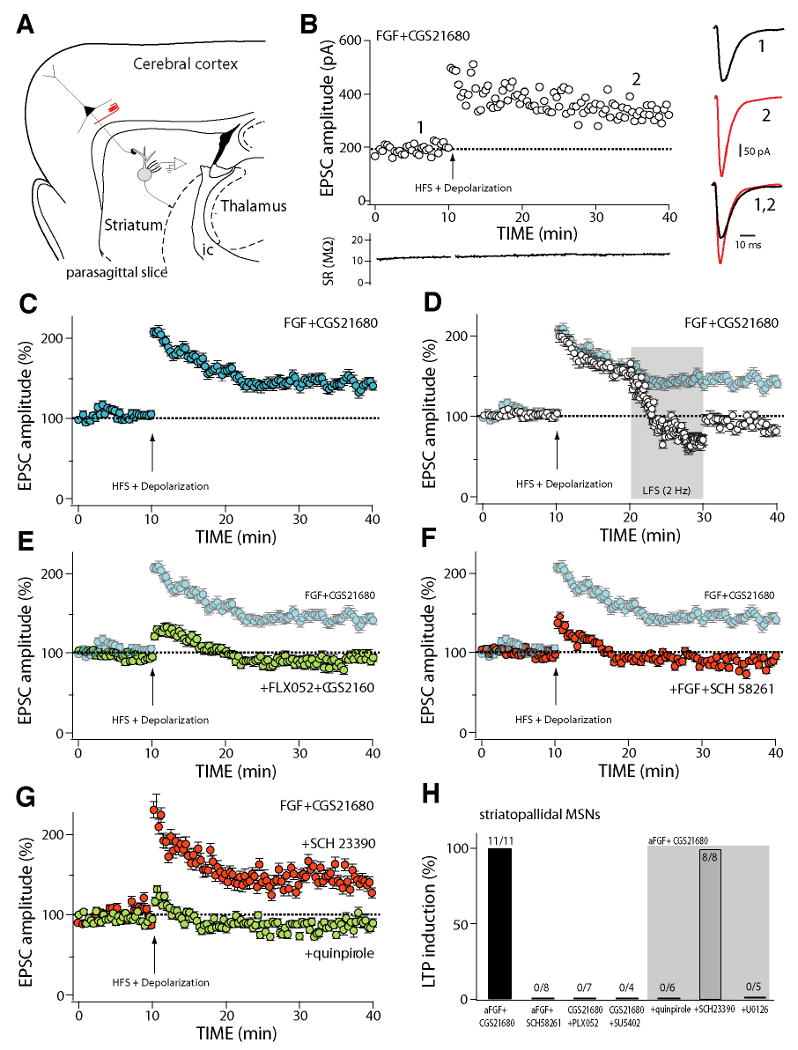
(A) Schematic of a parasagittal slice preparation used to study corticostriatal synaptic plasticity. Axons of corticostriatal neurons were electrically stimulated and medium spiny neurons were recorded with patch clamp techniques.
(B) Plot of the amplitude of corticostriatal EPSCs evoked in a representative striatopallidal MSN as a function of time before and after LTP induction by pairing high-frequency stimulation (HFS)(see Methods) and postsynaptic depolarization (DP) after incubation with aFGF (10 ng/ml) and CGS 21680 (10 nM). Induction (denoted by the arrow) began 10 minutes after the beginning of recording. At the right, representative current traces from before (black)(1) and after (red)(2) induction. At the bottom, access resistance as a function of time showing that induction did not alter recording condition.
(C) Average (±SEM) EPSC amplitude as a function of time before and after LTP induction in striatopallidal MSNs incubated with FGF and CGS 21680 (n=11).
(D) LTP induced by HFS-DP pairing in the presence of FGF and CGS21680 was reversed (depotentiated) by low frequency stimulation (2 Hz) of the corticostriatal axons; plot shows average EPSC amplitude (±SEM) in a sample of cells (n=6) where LTP induction was followed 10 minutes later by LFS. Background points are taken from panel (C) for comparison.
(E) HFS-DP did not induce LTP when FGFRs were blocked by FLX052 (10 μM), an FGFR tyrosine kinase inhibitor, even though A2A receptors were activated by CGS 21680; plot shows average EPSC amplitude (±SEM) in striatopallidal MSNs (n=7). Background points are taken from (c) for comparison. (f) HFS-DP did not induce LTP when A2A receptors were blocked by SCH 58261 (0.1 μM), even though FGF was present; plot shows average EPSC amplitude (±SEM) in striatopallidal MSNs (n=8). Background points are taken from (C) for comparison.
(G) HFS-DP LTP induced following FGFR/A2AR co-activation in striatopallidal MSNs was not altered by D1 receptor blockade (red circles) with SCH2330 (3 μM), but was disrupted by activation of D2 dopamine receptors with quinpirole (5 μM) (green circles); plot shows average EPSC amplitude (±SEM) in cells studied with the D1 receptor antagonist (n=8) and the D2 receptor agonist (n=6).
(H) Summary showing the percentage of striatopallidal MSNs in which HFS-DP induced a significant (>15%) change in EPSC amplitude at the 40 minute time point in various pharmacological conditions. The ratios at the top of each bar give the number of cells in which LTP was induced and the sample size. SU5402, a tyrosine kinase inhibitor. Gray area marks samples in which slices were preincubated with FGF and CGS21680 and one of three other agents: D2 agonist quinpirole, D1 receptor antagonist SCH 23390 or MEK inhibitor U 0126.
ERK1/2 activation has been implicated in forms of long-term potentiation (LTP) induced by pairing high frequency stimulation (HFS) of glutamatergic synapses with postsynaptic depolarization (DP) 23. In parasagital brain slices that preserved the connectivity between the cerebral cortex and the striatum, pairing of corticostriatal HFS with DP of striatopallidal MSNs led to a modest LTP in about one of every three neurons examined (26 ± 8% in 6 of 20 neurons); LTP was not observed in any of the striatonigral MSNs tested (n=5). Preincubation of slices with both A2AR agonist CGS 21680 (10 nM) and FGF (10 ng/ml) dramatically enhanced the induction of LTP by HFS-DP pairing in striatopallidal MSNs (n=11; Fig. 6B, C, H), whereas it had no affect on LTP induction in striatonigral MSNs (n=5, data not shown). This form of LTP resembled that previously described in MSNs 24,25 as it was abolished by blockade of NMDA receptors with APV (50 μM, n=3) and was reversed (depotentiated) by low frequency (2 Hz) afferent stimulation (Fig. 6D, n=6). Coactivation of A2AR and FGFR was necessary for LTP induction as exposure of slices to either the FGFR inhibitor FLX 052 (10 μM, Fig. 6E, H), the A2AR antagonist SCH 58261 (0.1 μM, Fig. 6F, H), or the tyrosine kinase inhibitor SU5402 (10 μM, Fig. 6H) abolished LTP induction. Although coactivation of A2AR and FGFR alone did not affect AMPA receptor mediated synaptic transmission, it did significantly enhance NMDA receptor mediated current in D2 MSNs (Supplemental Fig. 4). In contrast to previous reports, the induction of LTP by pairing HFS-DP was unaffected by antagonizing D1 dopamine receptors with SCH 23390 (3 μM, n=8, Fig. 6G,H). However, induction was blocked by the D2 receptor agonist quinpirole (5 μM, n=5, Fig. 6G,H), which is known to antagonize A2AR signaling. Consistent with the biochemical analysis of the A2AR/FGFR synergy, inhibition of MEK with U0126 (10 μM), but not with an inactive analog, U0124 (not shown), completely prevented LTP induction (n=5, Fig. 6H).
Discussion
Numerous studies, since the early 1970s, have investigated the biological consequences of receptor oligomerization 26. In the late 1980s, an allosteric receptor oligomerization model was proposed for the tyrosine kinase receptors 27. In the 1990s, models proposing GPCR homodimers, as well as oligomers, appeared 28. Gradually the classical 1:1:1 stoichiometry models (one ligand, one receptor and one G-protein) are being replaced by models involving higher-order complexes 29. However, in spite of the growing number of publications relating to receptor dimerization (for reviews see: 30,31,5) little is known about the structural basis of receptor-receptor interactions or about the functional consequences of oligomer formation.
The results of the present study strongly suggest that a direct physical interaction between the A2AR and the FGFR plays an important role in ERK-dependent regulation of morphological and physiological synaptic plasticity at striatopallidal synapses. Notably, the A2AR is highly expressed in striatopallidal neurons and the FGF receptors are also known to be expressed in medium spiny neurons 32. What is the molecular basis by which the physical interaction of A2ARs and FGFRs produces the pronounced synergistic activation of the ERK/MAP kinase pathway described here. The FGFR is coupled to the ERK/MAPK pathway through a Grb2/Ras/Raf-cascade. The ERK/MAPK pathway can also be activated by the A2AR via cAMP/PKA-EPAC/Raf-dependent stimulation of MEK1/2. Our studies provide evidence against an ability of A2ARs to increase tyrosine phosphorylation of the FGFR, or an ability of activation of FGFRs to increase cAMP levels or A2AR localization on the cell surface. We propose a “physical proximity” model to explain the synergistic action resulting from co-stimulation of A2ARs and FGFRs. This model postulates that the physical interaction between A2ARs and FGFRs, by confining both receptors to the same micro-cellular environment, results in dual phosphorylation of a key downstream signaling molecule. The ERK/MAPK pathway activation is believed to be entirely dependent upon protein kinase phosphorylation events and the observed synergism is consistent with the fact that MEK activation requires phosphorylation on at least two serine residues in the activation loop. However, it is conceivable that protein phosphatases, by dephosphorylating inhibitory sites, could also play a role in activating or keeping a given signaling complex in its optimal configuration. A recent study implicating a protein phosphatase cascade in the regulation of ERK in the striatum supports this possibility 33. In summary, the model suggests that MEK/ERK activation occurring under conditions of relatively low receptor occupancy by adenosine and FGF would serve to drastically amplify the coincident activation of the ERK/MAPK pathway by A2ARs and FGFRs. Based on the high degree of identity (98%) between the tyrosine kinase domains of the various FGFR isoforms, it is possible that not only FGFR1, the one found in this study, but FGFR2 and 3 may also play a role since these isoforms are expressed in MSNs 32.
Our studies revealed that coactivation of A2ARs and FGFRs, resulting in stimulation of ERK1/2, was necessary for the induction of LTP by HFS-DP pairing at corticostriatal synapses of striatopallidal MSNs, but not neighboring striatonigral MSNs. The induction of plasticity is likely to be due to direct postsynaptic effects of FGF and A2A receptors in striatopallidal MSNs for several reasons. Our reliance upon parasagital brain slices that preserve the connectivity between MSNs and cortical pyramidal neurons34 allowed the selective activation of corticostriatal axons during the induction protocol and not fibers passing through the striatum or the heterogenous population of neurons intrinsic to the striatum. Although corticostriatal axons potently activate fast spiking GABAergic interneurons 35, blocking GABAergic receptors had no effect on the induction of LTP, arguing that they were not a factor in the induction of plasticity 36. Cholinergic interneurons, which can promote the induction of LTP at MSN glutamatergic synapses 36 are only weakly activated by cortical inputs; moreover, adenosine reduces the opening of voltage-dependent Ca2+ channels controlling the release of acetylcholine 37, arguing against any role for these interneurons in our results. The restriction of the plasticity to corticostriatal synapses formed on striatopallidal MSNs is consistent with previous studies showing that A2A receptor expression in the striatum is almost entirely limited to neurons that co-express D2 dopamine receptors and the releasable peptide enkephalin 37,38,39. The cellular specificity also is consistent with the observation that corticostriatal LTP, measured with field potential recording, is reduced but not eliminated by genetic deletion of A2A receptors, as this approach will average changes in both striatonigral and striatopallidal MSNs.
However, in contrast to previous reports 40,41, the NMDA receptor dependent LTP in striatopallidal MSNs did not require activation of D1 dopamine receptors. Because the D1 receptors are localized almost exclusively in striatonigral MSNs 42,43 the present results are understandable. Our results suggest that in striatopallidal MSNs, dopamine is a negative modulator of LTP induction. This observation is in agreement with a variety of studies showing that D2 and A2A receptors are capable of inhibiting one another 38,39, and provide strong evidence that an interaction between the A2A/FGF receptors plays an important role in the regulation of synaptic plasticity in striatopallidal MSNs. The ability of D2 receptors to disrupt LTP induction in striatopallidal MSNs, and to promote the induction of LTD 44,25, has fundamental implications for striatum-based models of reinforcement learning. We have focused our analysis on the role of the interaction of A2AR and FGFR in synaptic plasticity of striatopallidal neurons, but our study raises the possibility that this interaction might also be relevant to signaling in other neurons in the brain that co-express the two proteins.
The demonstration of synergy between the A2AR and FGFR pathways has major clinical implications. A2ARs, through their localization in striatopallidal neurons, play an important role in modulating the actions of dopamine acting at D2 receptors. Thus, signaling through the A2AR pathway has become an increasingly attractive target for the development of therapeutic agents for Parkinson's disease and schizophrenia. In addition, a variety of growth factors and neurotrophic factors have been tested for clinical efficacy in several neurological and psychiatric disorders. For instance, enhanced neuroprotective effects of injected basic FGF in regional brain ischemia were recently observed after conjugation to a blood-brain barrier delivery vector 45. The discovery of cross-talk between the A2AR and the FGFR provides a new range of possibilities for therapeutic interventions affecting both pathways.
Methods
Yeast two-hybrid analysis
Yeast two-hybrid experiments were carried out as described 46. Briefly, the yeast strain Y187 was co-transformed with bait and prey plasmids using the LiAc/TE protocol, protein expression of baits and preys was analysed by immunoblotting using an anti-GAL4-BD antibody, and -galactosidase expression was measured by an overlay assay.
Production of Recombinant Proteins
cDNAs corresponding to the different proteins of interest were amplified by PCR using standard methods. PCR fragments were purified and digested by the appropriate restriction enzymes and subcloned into the pGEX4T1 vector, which expresses the recombinant protein with an N-terminal GST moiety. The resulting expression vectors were transformed into the bacterial host strain BL21 (DE3), and expression of protein was induced at mid-log phase by adding 1 mM IPTG for 4 hrs at 37°C. Recombinant proteins were purified by affinity chromatography using standard methods 47.
Glutathione-S-transferase pull down assays and affinity chromatography
The C-termini of the A2AR as well as the different deletion mutants were PCR amplified from the full-length A2AR cDNA construct and ligated in-frame to the coding sequence of glutathione-S-transferase (GST) using the EcoRI and BamHI restriction sites of pGEX-4T1 (Amersham Pharmacia Biotech). GST-fusion proteins expressed in E. coli were purified by affinity chromatography using glutathione-sepharose beads and used for radioactively labeled pull-down assays as described 46. Bound proteins were eluted by boiling in SDS sample buffer, separated by SDS polyacrylamide gel electrophoresis (SDS-PAGE), and detected by immunoblotting with specific antibodies.
Co-immunoprecipitation and antibodies
Co-immunoprecipitation experiments were done following standard protocols 47. FGFR antibody was purchased from Oncogene. Phospho-Tyr 4G10 antibody was purchased from Upstate. CD8 antibody was purchased from Caltag. Antibodies corresponding to the MAPK pathways (p-c-raf, pERK1/2, phospho-p38, p-SAPK/JNK) were purchased from Cell Signaling Technology™ (Beverly, MA). HA-Tag antibody was purchased from Clontech.
Signal Transduction Analysis in PC12 cells
Rat pheochromocytoma PC12 cells were maintained in RPMI 1640 medium with L-glutamine supplemented with 10% v/v heat inactivated horse serum and 5% v/v fetal bovine serum. For experiments, 1.5 × 105 PC12 cells/well (12 well plate coated with poly-D-lysine) were grown in 2 ml/well of supplemented medium for 48 hrs. The cells were washed once and serum starved in RPMI 1640/0.25% horse serum/0.25% fetal bovine serum for 48 hrs to prevent serum activated phosphorylation. Cells were treated with ligands as described in the figure legends, and lysed by removing the medium and adding 50 μl/well of 1× sample buffer. The extracts were sonicated for 10 sec to reduce sample viscosity. Samples (5 μl) were analyzed by SDS/PAGE and imunoblotting. Immunoblots were quantified using NIH Image 1.52. The data (at least 4 repeats for each experiment) are presented as means ± SEM, and statistical differences were determined by Student's t test.
PC12 Cell Differentiation
Sterile poly-D-lysine coated cover slips were used to grow PC12 cells at a density of 2 × 105/10 cm2. Plated cells were grown in 3 ml of non-supplemented medium for 24 hrs. Ligands were added to the cells and incubated at 37°C for 48 hours, new ligands were added to fresh non-supplemented medium and incubation was continued for 48 hrs at 37°C. Cells were then fixed with 4% cold paraformaldehyde, the cover slips mounted on slides, and the cells analyzed by light/phase-contrast microscopy.
Hippocampal neuronal cultures
Primary hippocampal cultures from embryonic day 18-19 Sprague-Dawley rats were transfected with CD8 as a cell-morphology marker 48. Neurons were transfected at DIV6. At DIV11, neurons were starved for 3 hrs prior to performing the different treatments. All treatments were for 3 hrs and the neurons fixed for immunocytochemistry.
Mammalian cellular expression
Transfection of COS-7 cells was performed using standard methods. Cells were grown to 60% confluence and then transfected with the indicated constructs with FuGENE according to the manufacturer's protocol (Roche Applied Science). After transfection, cell extracts were prepared according to the measurement protocol.
cAMP enzyme immunoassay
cAMP production from stimulated cells was quantified using a non-radioactive cAMP enzyme immunoassay kit following the manufacturer's protocol (Sigma).
Electrophysiology procedures
Individual slices were transferred to a submersion-style recording chamber and continuously superfused with ACSF bubbled with 95% O2 and 5% CO2 at a rate of 2-3 ml/min at 31-33°C. Visualized, whole-cell voltage recordings were performed on striatal medium spiny neurons using infrared-differential interference contrast (IR-DIC) video microscopy with an Olympus OLY-150 camera/controller system (Olympus, Japan). Recordings were obtained using a computer controlled MultiClamp 700A amplifier (Molecular Devices, USA). In all experiments, 10 μM (-)bicuculline methiodide was added to the superfusion medium to block GABAA receptor-mediated synaptic responses. Patch electrodes were made by pulling BF150-86-10 glass on a P-97 Flaming/Brown micropipette puller (Sutter Instrument Co.) and fire polished before use. Pipette resistance was typically 2-5 MΩ after filling with internal solution. The internal pipette solution contained the following (in mM): 120 CsMeSO3, 5 NaCl, 10 TEA-Cl, 10 HEPES, 5 QX-314, 0.2 EGTA, 4 ATP-Mg2, 0.3 GTP-Na2, 10 phosphocreatine, pH 7.2 adjusted with CsOH, 280-290 mOsm/L. For evoked EPSC (eEPSC) recordings, medium spiny neurons were voltage-clamped at −70 mV. Test stimuli were delivered with an S48 stimulator (W. Warwick, USA) and DS3 isolator (Digitimer Ltd., UK) at a frequency of 0.05 Hz or 0.1 Hz through a concentric electrode (Frederick Haer & Co, ME) placed in the cerebral cortex near the region of interest within the striatum. After a stable eEPSC baseline had been established (>10 min), the cortex was stimulated at high-frequency (HFS). The HFS protocol consisted of four one second episodes during which the cortex was stimulated at 100 Hz; episodes were separated by 10 second intervals. During the HFS protocol, neurons were depolarized to 0 mV from a holding potential of −70 mV. Access resistance was monitored on-line; if it changed by >20% during the course of recording, the cell was discarded. Synaptic responses were analyzed using pClamp. Traces shown in figure 6 were averages of 10-15 individual sweeps in a given recording. In pre-incubation experiments, FGF and/or CGS 21680 was added to the solution bathing the slices for at least 30 min before recording.
Chemicals and reagents
QX-314, (-)-quinpirole hydrochloride, CGS 21680 hydrochloride, NBQX, ifenprodil, SCH 58261, and SCH 23390 hydrochloride were obtained from Tocris. U0124 (1,4-Diamino-2,3-dicyano-1,4-bis(methylthio)butadiene), U0126 (1,4-Diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene) and SU5402 (3-[3-(2-Carboxyethyl)-4-methylpyrrol-2-methylidenyl]-2-indolinone) were purchased from Calbiochem. FLX052 was a gift from Dr. J. Schlessinger. All other chemicals were from Sigma/RBI. The poly-Arg-A2AR inhibitory sequence is the following: MRRRRRRRRRRRGVNPFIYAYRIREFRGTFRKIIRTHVLRRQEPFQAGG
Statistical analysis
Statistical differences were determined by one-way ANOVA. *: p<0.05; **: p<0.01; ***: p<0.001
Supplementary Material
Acknowledgments
We are grateful to Dr. J. Rogers, and Dr. H. Rebholz for critical reading of the manuscript, and to Dr. G. Fisone for helpful discussions. This work was supported in part by grants from the National Institutes of Health (P.G., A.C.N. and D.J.S.) MH074866, (D.J.S) NS34696, The Simons Foundation, The Picower Foundation, and by award W81XWH-04-2-0009 from The USA Medical Research and Materiel Command NETRP program to Intra-Cellular Therapies, Inc. Marc Flajolet's project was supported in part by a 2006 NARSAD Young Investigator Award.
Footnotes
Supplementary Information is linked to the online version of the paper.
References
- 1.Fredholm BB. Purinoceptors in the nervous system. Pharmacol Toxicol. 1995;76:228–239. doi: 10.1111/j.1600-0773.1995.tb00135.x. [DOI] [PubMed] [Google Scholar]
- 2.Zimmermann H. Biochemistry, localization and functional roles of ecto-nucleotidases in the nervous system. Progress in neurobiology. 1996;49:589–618. doi: 10.1016/0301-0082(96)00026-3. [DOI] [PubMed] [Google Scholar]
- 3.Lindskog M, et al. Involvement of DARPP-32 phosphorylation in the stimulant action of caffeine. Nature. 2002;418:774–778. doi: 10.1038/nature00817. [DOI] [PubMed] [Google Scholar]
- 4.Durcan MJ, Morgan PF. Evidence for adenosine A2 receptor involvement in the hypomobility effects of adenosine analogues in mice. Eur J Pharmacol. 1989;168:285–290. doi: 10.1016/0014-2999(89)90789-9. [DOI] [PubMed] [Google Scholar]
- 5.Fuxe K, et al. Intramembrane receptor-receptor interactions: a novel principle in molecular medicine. J Neural Transm. 2007;114:49–75. doi: 10.1007/s00702-006-0589-0. [DOI] [PubMed] [Google Scholar]
- 6.Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-3-reviews3005. REVIEWS3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dono R. Fibroblast growth factors as regulators of central nervous system development and function. Am J Physiol Regul Integr Comp Physiol. 2003;284:R867–881. doi: 10.1152/ajpregu.00533.2002. [DOI] [PubMed] [Google Scholar]
- 8.Reuss B, von Bohlen und Halbach O. Fibroblast growth factors and their receptors in the central nervous system. Cell Tissue Res. 2003;313:139–157. doi: 10.1007/s00441-003-0756-7. [DOI] [PubMed] [Google Scholar]
- 9.Umemori H, Linhoff MW, Ornitz DM, Sanes JR. FGF22 and its close relatives are presynaptic organizing molecules in the mammalian brain. Cell. 2004;118:257–270. doi: 10.1016/j.cell.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 10.Forget C, Stewart J, Trudeau LE. Impact of basic FGF expression in astrocytes on dopamine neuron synaptic function and development. Eur J Neurosci. 2006;23:608–616. doi: 10.1111/j.1460-9568.2006.04570.x. [DOI] [PubMed] [Google Scholar]
- 11.Fumagalli F, Di Pasquale L, Caffino L, Racagni G, Riva MA. Stress and cocaine interact to modulate basic fibroblast growth factor (FGF-2) expression in rat brain. Psychopharmacology (Berl) 2007 doi: 10.1007/s00213-007-0966-x. [DOI] [PubMed] [Google Scholar]
- 12.Grothe C, Timmer M. The physiological and pharmacological role of basic fibroblast growth factor in the dopaminergic nigrostriatal system. Brain research reviews. 2007;54:80–91. doi: 10.1016/j.brainresrev.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 13.Claus P, Werner S, Timmer M, Grothe C. Expression of the fibroblast growth factor-2 isoforms and the FGF receptor 1-4 transcripts in the rat model system of Parkinson's disease. Neuroscience letters. 2004;360:117–120. doi: 10.1016/j.neulet.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 14.Mohammadi M, et al. A tyrosine-phosphorylated carboxy-terminal peptide of the fibroblast growth factor receptor (Flg) is a binding site for the SH2 domain of phospholipase C-gamma 1. Mol Cell Biol. 1991;11:5068–5078. doi: 10.1128/mcb.11.10.5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sorokin A, Mohammadi M, Huang J, Schlessinger J. Internalization of fibroblast growth factor receptor is inhibited by a point mutation at tyrosine 766. J Biol Chem. 1994;269:17056–17061. [PubMed] [Google Scholar]
- 16.Hayashi H, Ishisaki A, Suzuki M, Imamura T. BMP-2 augments FGF-induced differentiation of PC12 cells through upregulation of FGF receptor-1 expression. J Cell Sci. 2001;114:1387–1395. doi: 10.1242/jcs.114.7.1387. [DOI] [PubMed] [Google Scholar]
- 17.Kobayashi S, Millhorn DE. Stimulation of expression for the adenosine A2A receptor gene by hypoxia in PC12 cells. A potential role in cell protection. J Biol Chem. 1999;274:20358–20365. doi: 10.1074/jbc.274.29.20358. [DOI] [PubMed] [Google Scholar]
- 18.Vaudry D, Stork PJ, Lazarovici P, Eiden LE. Signaling pathways for PC12 cell differentiation: making the right connections. Science. 2002;296:1648–1649. doi: 10.1126/science.1071552. [DOI] [PubMed] [Google Scholar]
- 19.Wu GY, Deisseroth K, Tsien RW. Spaced stimuli stabilize MAPK pathway activation and its effects on dendritic morphology. Nat Neurosci. 2001;4:151–158. doi: 10.1038/83976. [DOI] [PubMed] [Google Scholar]
- 20.Goldin M, Segal M. Protein kinase C and ERK involvement in dendritic spine plasticity in cultured rodent hippocampal neurons. Eur J Neurosci. 2003;17:2529–2539. doi: 10.1046/j.1460-9568.2003.02694.x. [DOI] [PubMed] [Google Scholar]
- 21.Alonso M, Medina JH, Pozzo-Miller L. ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Learn Mem. 2004;11:172–178. doi: 10.1101/lm.67804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.d'Alcantara P, Ledent C, Swillens S, Schiffmann SN. Inactivation of adenosine A2A receptor impairs long term potentiation in the accumbens nucleus without altering basal synaptic transmission. Neuroscience. 2001;107:455–464. doi: 10.1016/s0306-4522(01)00372-4. [DOI] [PubMed] [Google Scholar]
- 23.Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- 24.Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30:228–235. doi: 10.1016/j.tins.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 25.Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 2007;30:211–219. doi: 10.1016/j.tins.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 26.Lee SP, O'Dowd BF, George SR. Homo- and hetero-oligomerization of G protein-coupled receptors. Life Sci. 2003;74:173–180. doi: 10.1016/j.lfs.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 27.Schlessinger J. Signal transduction by allosteric receptor oligomerization. Trends Biochem Sci. 1988;13:443–447. doi: 10.1016/0968-0004(88)90219-8. [DOI] [PubMed] [Google Scholar]
- 28.Jahangeer S, Rodbell M. The disaggregation theory of signal transduction revisited: further evidence that G proteins are multimeric and disaggregate to monomers when activated. Proc Natl Acad Sci U S A. 1993;90:8782–8786. doi: 10.1073/pnas.90.19.8782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomason PA, Wolanin PM, Stock JB. Signal transduction: receptor clusters as information processing arrays. Curr Biol. 2002;12:R399–401. doi: 10.1016/s0960-9822(02)00885-0. [DOI] [PubMed] [Google Scholar]
- 30.Ferguson SS. Receptor tyrosine kinase transactivation: fine-tuning synaptic transmission. Trends Neurosci. 2003;26:119–122. doi: 10.1016/S0166-2236(03)00022-5. [DOI] [PubMed] [Google Scholar]
- 31.Shah BH, Catt KJ. GPCR-mediated transactivation of RTKs in the CNS: mechanisms and consequences. Trends Neurosci. 2004;27:48–53. doi: 10.1016/j.tins.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 32.Claus P, Werner S, Timmer M, Grothe C. Expression of the fibroblast growth factor-2 isoforms the FGF receptor 1-4 transcripts in the rat model system of Parkinson's disease. Neurosci Lett. 2004;360(3):117–20. doi: 10.1016/j.neulet.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 33.Valjent E, et al. Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci U S A. 2005;102:491–496. doi: 10.1073/pnas.0408305102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ding J, Peterson JD, Surmeier DJ. Corticostriatal and thalamostriatal synapses have distinctive properties. J Neurosci. 2008;28(25):6483. doi: 10.1523/JNEUROSCI.0435-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tepper JM, Wilson CJ, Koos T. Feedforward and feedback inhibition in neostriatal GABAergic spiny neurons. Brain Res Rev. 2007 doi: 10.1016/j.brainresrev.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 2007;30(5):211. doi: 10.1016/j.tins.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 37.Song WJ, Tkatch T, Surmeier DJ. Adenosine receptor expression and modulation of Ca(2+) channels in rat striatal cholinergic interneurons. J Neurophysiol. 2000;83(1):322. doi: 10.1152/jn.2000.83.1.322. [DOI] [PubMed] [Google Scholar]
- 38.Ferre S, Fredholm BB, Morelli M, Popoli P, Fuxe K. Adenosine-dopamine receptor-receptor interactions as an integrative mechanism in the basal ganglia. Trends Neurosci. 1997;20:482–487. doi: 10.1016/s0166-2236(97)01096-5. [DOI] [PubMed] [Google Scholar]
- 39.Ferre S. Adenosine-dopamine interactions in the ventral striatum. Implications for the treatment of schizophrenia. Psychopharmacology (Berl) 1997;133:107–120. doi: 10.1007/s002130050380. [DOI] [PubMed] [Google Scholar]
- 40.Centonze D, Picconi B, Gubellini P, Bernardi G, Calabresi P. Dopaminergic control of synaptic plasticity in the dorsal striatum. Eur J Neurosci. 2001;13:1071–1077. doi: 10.1046/j.0953-816x.2001.01485.x. [DOI] [PubMed] [Google Scholar]
- 41.Kerr JN, Wickens JR. Dopamine D-1/D-5 receptor activation is required for long-term potentiation in the rat neostriatum in vitro. Journal of neurophysiology. 2001;85:117–124. doi: 10.1152/jn.2001.85.1.117. [DOI] [PubMed] [Google Scholar]
- 42.Surmeier DJ, Song WJ, Yan Z. Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J Neurosci. 1996;16:6579–6591. doi: 10.1523/JNEUROSCI.16-20-06579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gerfen CR, et al. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- 44.Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song BW, Vinters HV, Wu D, Pardridge WM. Enhanced neuroprotective effects of basic fibroblast growth factor in regional brain ischemia after conjugation to a blood-brain barrier delivery vector. J Pharmacol Exp Ther. 2002;301:605–610. doi: 10.1124/jpet.301.2.605. [DOI] [PubMed] [Google Scholar]
- 46.Flajolet M, et al. A genomic approach of the hepatitis C virus generates a protein interaction map. Gene. 2000;242:369–379. doi: 10.1016/s0378-1119(99)00511-9. [DOI] [PubMed] [Google Scholar]
- 47.Flajolet M, et al. Protein phosphatase 2C binds selectively to and dephosphorylates metabotropic glutamate receptor 3. Proc Natl Acad Sci U S A. 2003;100:16006–16011. doi: 10.1073/pnas.2136600100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Futter M, et al. Phosphorylation of spinophilin by ERK and cyclin-dependent PK 5 (Cdk5) Proc Natl Acad Sci U S A. 2005;102:3489–3494. doi: 10.1073/pnas.0409802102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.