

Abstract
Background
Corin, a transmembrane serine protease expressed in cardiomyocytes, cleaves proANP and proBNP into biologically active peptide hormones. The minor corin I555(P568) allele, defined by the T555I and Q568P mutations, is common in persons of African ancestry and associated with increased risk for hypertension and cardiac concentric hypertrophy. The corin gene product containing the T555I and Q568P mutations has significantly reduced natriuretic peptide processing capacity. We hypothesized that the corin I555(P568) allele would be associated with adverse outcomes and impaired BNP processing in African-Americans with systolic heart failure.
Methods and Results
This is a retrospective study of 354 subjects in the African-America Heart Failure Trial (A-HeFT) Genetic Risk Assessment in Heart Failure (GRAHF) sub-study. In the corin variant group (N=50) compared to corin non-variant group (N=300), BNP-32 (amino acids 77-108) was lower (190pg/ml versus 340 pg/ml, p=0.007), but the ratio of unprocessed BNP 1-108/processed BNP-32 was significantly higher (P= 0.05). Stratified analyses were conducted because of evidence of significant interaction between the corin I555(P568) allele and treatment assignment. In the placebo arm, multivariable analysis demonstrated that the corin I555(P568) allele was associated with increased risk for death or heart failure hospitalization (RR 3.49; 95% CI 1.45-8.39; P=0.005); however, in the treatment arm (fixed-dose combination isosorbide-dinitrate/hydralazine), the corin I555(P568) allele was not associated with adverse outcomes.
Conclusions
We have identified a pharmacogenomic interaction in African-Americans with systolic heart failure. The corin I555(P568) allele is associated with an increased risk for death or heart failure hospitalization in patients receiving standard neurohormonal blockade, but the addition of fixed dose combination isosorbide-dinitrate/hydralazine (FDC I/H) ameliorates this risk. A plausible mechanism for this pharmacogenomic interaction is the impaired processing of BNP in carriers of the corin I555(P568) allele as compared to non-carriers.
Keywords: genetics, epidemiology, heart failure, natriuretic peptides, pharmacology
Introduction
Corin is a transmembrane serine protease that is expressed in cardiomyocytes and cleaves pro-ANP and proBNP into biologically active peptide hormones.1, 2 A minor allele in corin, defined by two coding variants in complete linkage disequilibrium (T555I and Q568P), is common and almost exclusively expressed in persons of African ancestry (allelic prevalence 6.7%). Furthermore, the corin I555(P568) allele is associated with increased systolic blood pressure, increased risk for systemic hypertension, and an enhanced concentric cardiac hypertrophic response to increased blood pressure3, 4. Recently, in vitro experiments have demonstrated that the presence of both the T555I and Q568P amino acid substitutions significantly reduce the natriuretic processing activity of mutant I555(P568) corin5.
Adequate natriuretic peptide processing is essential for the endogenous natriuretic peptide system (NPS) to function in maintaining cardiovascular homeostasis. The human gene for brain natriuretic peptide (BNP) encodes a 134–amino acid preproBNP precursor, which after removal of a 26-amino acid signal peptide gives rise to a 108-amino acid proBNP polypeptide (proBNP1–108). Further processing of proBNP1-108 by the type 2 transmembrane serine protease corin results in the physiologically active 32-amino acid carboxyl-terminal BNP molecule (BNP32), derived from amino acids 77-108, and an inactive N-terminal fragment (NT-BNP), derived from amino acids 1-76. Recently, cell-based studies using recombinant BNP 1-108 have shown that unprocessed BNP 1-108 is unable to activate the type-A natriuretic peptide receptor (NPR-A).6, 7 Therefore, abnormalities in corin-mediated natriuretic peptide processing would be expected to attenuate the in vivo biological activity of the natriuretic peptide system. Although elevated BNP levels are associated with adverse outcomes in heart failure, the multiple biological actions of ANP and BNP within target tissues are “compensatory” in nature and include: vasodilatation of arteries and veins,8 opposing activation of adverse neurohormonal systems, including the renin-angiotensin-aldosterone and sympathetic systems, 9 enhancement of natriuresis by opposing distal tubule sodium reabsorption10-12 and direct inhibition of endothelin release by the renal vascular endothelium.13
Given the recognized importance of the natriuretic peptide system for the maintenance of homeostasis in systolic heart failure, and the almost exclusive presence of the I555 (P568) allele in African-Americans, we undertook the present analysis as a substudy within the African-American Heart Failure (A-HeFT) trial to test the hypothesis that in patients with systolic heart failure the corin I555(P568) allele would be associated with an increased risk of death or hospitalization for heart failure. Furthermore, we sought to determine if there was an increase in the degree of impaired processing of BNP in carriers of the corin I555(P568) allele relative to non-carriers. In order to estimate the degree of impaired processing of BNP we measured unprocessed proBNP 1-108 utilizing a novel immunoassay specific for BNP1-108 (Bio-Rad, Hercules, CA)14 and also measured BNP-32 using the Biosite Triage assay. The Biosite Triage assay does demonstrate some cross-reactivity (∼ 19%) with BNP1-108 15. However, since we had no reason to suspect the degree of cross-reactivity would differ between genotype groups, we reasoned that the ratio of proBNP1-108/BNP-32 would serve as a reasonable assessment of BNP processing efficiency in the corin variant compared to non-variant groups.
Methods
Study design
The A-HeFT study design, patient characteristics, end-point definitions, and complete methodology have been published previously in detail.16-19 In brief, A-HeFT was a randomized, placebo-controlled, double-blind trial with self-identified black patients recruited at 169 centers in the United States. The study protocol was reviewed and approved by appropriate institutional review boards. All patients gave written informed consent. Independent committees adjudicated all primary and secondary end points, reviewed data for safety, and oversaw the 2 pre-specified interim analyses done to assess adequacy of sample size only.
Inclusion criteria
Patients 18 years and older, self-identified as African American and with New York Heart Association class III or IV heart failure for at least 3 months, were eligible for screening. Patients were required to be undergoing standard background heart failure therapy, as determined by their physician, which included ACE inhibitors (ACEIs), angiotensin-receptor blockers (ARBs), β-blockers for at least 3 months before randomization, digoxin, spironolactone, and diuretics. Evidence of LV dysfunction within the 6 months preceding randomization was required and consisted of either a resting LV ejection fraction less than 35% or a resting LV ejection fraction less than 45% with an LV internal diastolic diameter >2.9 cm/m2 of body surface area or >6.5 cm by echocardiography.
Endpoints
The primary efficacy end point for the A-HeFT trial was a novel composite score that weighted all-cause mortality, first hospitalization for heart failure throughout the 18-month follow-up period, and change in quality of life at 6 months. For the present analysis we used the composite endpoint of death from any cause or first hospitalization for heart failure.
Genotyping
Subjects were enrolled in the Genetic Risk Assessment in Heart Failure (GRAHF) sub-study at the 6-month visit and DNA was isolated from peripheral blood by leukocyte centrifugation and cell lysis (PureGene, Gentra Systems, Minn). The corin Q568P polymorphism (in complete linkage disequilibrium with the T555I locus) was genotyped in 350 subjects participating in A-HeFT using the TaqMan SNP Genotyping Assay and the Applied Biosystems 7000 (Applied Biosystems, Foster City, CA).
Measurement of proBNP and BNP
Using a novel immunoassay, we measured baseline plasma levels of unprocessed BNP 1-108 (Bio-Rad, Hercules CA)14 in 695 of 1050 subjects in the African-American Heart Failure Trial (239 of the 350 subjects who consented for genetic studies in GRAHF) who consented for measurement at baseline and at 6-month follow-up. This immunoassay has been demonstrated to have no cross-reactivity with either recombinant BNP-32 or NT-BNP and is thereforehighly specific for proBNP 1-108. Brain natriuretic peptide (BNP) was also measured in these patients using the Biosite Triage immunoassay. A recent Expert Consensus Panel analyzed the cross-reactivity patterns of available commercial BNP immunoassays. The Biosite Triage immunoassay has minimal cross-reactivity (19%; 95% CI 18%-20%) with glycosylated recombinant BNP 1-108 expressed in mammalian cells15. We reasoned that the ratio of BNP1-108 (Bio-Rad) to BNP-32 (Biosite) would serve as a reasonable assessment of BNP processing efficiency.
Statistical analysis
Subjects were followed to an endpoint of death or heart failure hospitalization. Left ventricular function was assessed by transthoracic echocardiography at baseline and six months. We compared continuous variables by linear ANOVA when appropriate. ProBNP (BNP1-108) and BNP values were log-transformed to produce a normal distribution of values permitting standard parametric testing (Students t-test) of means. In the case of the ratio of ProBNP to BNP defined as BNP 1-108/BNP 77-108 this variable did not satisfy assumption of normality; therefore, we statistically tested for differences in the ProBNP/BNP ratio between the corin variant (I555/P568) and non-variant groups using the non-parametric Van der Waerden normal quantile test20.
Comparison of event free survival (death or first hospitalization for heart failure) by genotype class was analyzed with Kaplan-Meier survival analysis and log-rank methods. Multivariable survival analysis were conducted using Cox-proportional hazards modeling21 and randomization assignment was adjusted for using the intention-to-treat principle. In these models the risk for persons in the corin variant group, defined as GRAHF participants heterozygous for the corin I555(P568) allele, was compared to the non-variant group. In GRAHF all carriers of the corin I555(P568) allele were heterozygous, a finding which is consistent with our results examining the genotype prevalence in two, large and independent population-based cohorts of self-identified African-Americans4, 22. The multivariable Cox-proportional hazards included the following covariates: age, sex, log-transformed (BNP), ischemic etiology of systolic dysfunction, diabetes mellitus, and baseline use of beta-blocker. We conducted two multivariable Cox-proportional hazards models that differed only in the inclusion or exclusion of Ln (BNP) since we reasoned this may be causally related to the prognostic importance of the corin I555(P568) allele. Interaction between corin genotype status and randomization assignment on risk for the composite endpoint was assessed statistically by the introduction of a multiplicative interaction term in the multivariable model with the a priori decision to explore potential interactions with a P-value < 0.1.
Results
Baseline characteristics
Approximately 14% of the GRAHF participants were heterozygous for the corin I555(P568) allele consistent with previous findings from population-based samples4, 22. We did not identify any subjects homozygous for the corin I555(P568) allele. However, this is not unexpected given the small sample size in A-HeFT and the fact that in our previous population based studies, the homozygous genotype frequency for the corin I555(P568) allele was extremely low (< 0.2%). The corin I555(P568) allele was in Hardy-Weinberg equilibrium (P=0.43). Comparing the corin variant group (heterozygous for I555/P568 allele) to the non-variant group, there were no significant differences noted in baseline characteristics (Table 1) or medication use other than the corin variant group being younger (53.4 years versus 57.8 years, p=0.02). The corin variant and non-variant groups when stratified by treatment assignment (BiDil versus placebo) also did not substantially differ in baseline characteristics except that the corin variant group as compared to the corin non-variant group was younger in the group randomized to treatment with BiDil (Table 2).
Table 1. Baseline Characteristics According to Presence or Absence of Corin I555(P568) Allele.
| Corin+/+ | Corin+/- | P | |
|---|---|---|---|
| Number (%) | 300 (86%) | 50 (14%) | |
| Age, mean, (SD) | 57.9 | 53.4 | 0.02 |
| Men, (%) | 62 | 48 | 0.12 |
| EF, (%) | 34.8 | 33.5 | 0.35 |
| LVIDD | 6.37 | 6.65 | 0.19 |
| SBP | 127.2 | 125.2 | 0.44 |
| DBP | 76.6 | 77.3 | 0.67 |
| Heart rate | 73.1 | 74.8 | 0.34 |
| Creatinine | 1.23 | 1.20 | 0.70 |
| QOL score | 49.9 | 53.4 | 0.38 |
| Etiology, (%) | |||
| -Ischemic | 26.7 | 16.0 | 0.12 |
| -Hypertensive | 37.3 | 30.0 | 0.42 |
| -Idiopathic | 24.0 | 32.0 | 0.16 |
| -Valve-related | 2.7 | 0.0 | 0.61 |
| Diabetes | 44.0 | 42.7 | 0.88 |
| Medications | |||
| -B-blocker | 83.0 | 92.0 | 0.14 |
| -ACEI | 77.3 | 70.0 | 0.31 |
| -ARB | 21.3 | 24.0 | 0.71 |
| -Spironolactone | 36.3 | 34.0 | 0.87 |
Corin +/+ = wild-type
Corin +/- = heterozygous for corin I555(P568) allele.
Table 2. Baseline Characteristics According to Presence of Absence of Corin I555 (P568) Allele Stratified by Randomization Assignment to FDC I/H or Placebo.
| BiDil | Placebo | |||||
|---|---|---|---|---|---|---|
| Corin+/+ | Corin+/- | P | Corin+/+ | Corin+/- | P | |
| Number (%) | 139 (85%) | 24 (15%) | 161 (86%) | 26 (14%) | ||
| Age, mean, (SD) | 58.6 | 53.2 | 0.01 | 57.2 | 55.1 | 0.45 |
| Men, (%) | 58.3 | 37.5 | 0.09 | 65.2 | 57.7 | 0.51 |
| EF, (%) | 34.8 | 34.9 | 0.90 | 34.7 | 33.4 | 0.51 |
| LVIDD | 6.40 | 6.33 | 0.74 | 6.34 | 6.73 | 0.19 |
| SBP | 129.8 | 125.1 | 0.169 | 124.9 | 125.2 | 0.92 |
| DBP | 78.8 | 78.1 | 0.75 | 74.8 | 76.6 | 0.43 |
| Heart rate | 73.2 | 72.0 | 0.47 | 72.9 | 72.8 | 0.95 |
| Creatinine | 1.22 | 1.08 | 0.16 | 1.24 | 1.31 | 0.53 |
| QOL score | 50.3 | 54.9 | 0.42 | 49.7 | 51.3 | 0.67 |
| Etiology, (%) | ||||||
| -Ischemic | 31.6 | 12.5 | 0.09 | 22.4 | 19.2 | 0.95 |
| -Hypertensive | 42.4 | 20.8 | 0.07 | 32.9 | 34.6 | 0.19 |
| -Idiopathic | 15.8 | 37.5 | 0.02 | 31.0 | 30.8 | 0.98 |
| -Valve-related | 2.9 | 0.0 | 0.95 | 2.5 | 0.0 | 0.97 |
| Diabetes | 50.3 | 54.0 | 0.95 | 36.6 | 34.6 | 0.98 |
| Medications | ||||||
| -B-blocker | 84.2 | 91.7 | 0.53 | 82.0 | 92.3 | 0.26 |
| -ACEI | 74.8 | 70.8 | 0.80 | 79.5 | 69.2 | 0.31 |
| -ARB | 23.7 | 29.2 | 0.61 | 19.3 | 19.2 | 1.0 |
| -Spironolactone | 36.0 | 45.8 | 0.37 | 36.6 | 23.1 | 0.27 |
Corin +/+ = wild-type
Corin +/- = heterozygous for corin I555(P568) allele.
FDC I/H=fixed-dose, combination isosorbide-dinitrate/hydralazine (BiDil)
Association of the Corin I555(P568) Allele with Outcomes
When we examined the possibility of interaction between the corin I555(P568) allele and randomization assignment with the composite endpoint (P=0.07) and the endpoint of first hospitalization for heart failure (P=0.03), there was a suggestion for interaction meriting further exploration. Therefore, we stratified our analyses by treatment assignment. As demonstrated in Figure 1, Kaplan-Meier survival curves for the composite endpoint (death or first heart failure hospitalization) were not significantly different between the corin variant and non-variant groups in the overall analysis. However, within the group randomized to placebo, survival free from the composite endpoint was significantly worse in carriers of the corin I555(P568) allele as compared to non-carriers (P=0.029). There was no significant difference in survival between carriers and non-carriers of the I555(P568) allele in the group randomized to treatment with FDC I/H.
Figure 1.
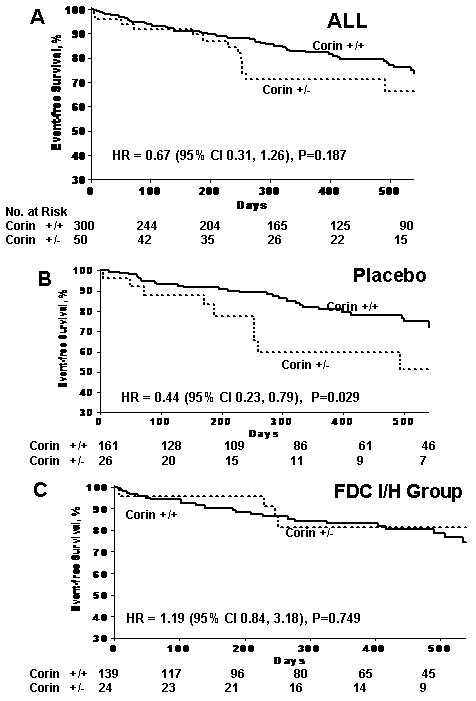
Time to death or first heart failure hospitalization in the overall Genetic Risk Assessment in Heart Failure (GRAHF) study of AHeFT (Fig 1A) and within groups stratified by treatment assignment: Placebo (Figure 1B) versus FDC I/H (Figure 1C). HR denotes hazard ratio.
In multivariable analysis (Table 3), we conducted two models, differing only in the exclusion or inclusion of log-transformed BNP. The other covariates that we included in our multivariable models included: sex, ejection fraction, systolic blood pressure, age, ischemic etiology, diabetes mellitus, and beta-blocker use. In the placebo arm, with multivariable analysis adjusting for all covariates except Ln(BNP), the corin I555(P568) minor allele remained significantly associated with an increased risk for the composite endpoint of death or heart failure hospitalization (RR 2.33; 95% CI 1.07-5.05; P=0.03). Multivariable analysis including log-transformed BNP demonstrated that participants heterozygous for the corin I555(P568) minor allele were at significantly increased risk for the composite endpoint (RR 3.49; 95 CI 1.45-8.39; P=0.005). However, in the group of participants randomized to treatment with FDC H/I (BiDil) the corin I555(P568) allele was not associated with adverse outcomes in multivariable analysis regardless of the inclusion or exclusion of BNP levels.
Table 3. Impact of Corin Variant on Event-free Survival (Death or First Hospitalization for Heart Failure) in GRAHF (sub-study of A-HeFT).
| Group | Corin+/+ N(%) |
Corin+/- N (%) |
Hazard ratio | 95% CI | P |
|---|---|---|---|---|---|
| All | 300 (86%) | 50 (14%) | |||
| Unadjusted | 1.499 | 0.816-2.752 | 0.192 | ||
| Adjusted | 1.644 | 0.851-3.177 | 0.139 | ||
| Adjusted w/o Ln(BNP) | 1.475 | 0.791-2.751 | 0.222 | ||
| Placebo | 161 (86%) | 26 (14%) | |||
| Unadjusted | 2.266 | 1.068-4.806 | 0.033 | ||
| Adjusted | 3.485 | 1.447-8.394 | 0.0054 | ||
| Adjusted w/o Ln(BNP) | 2.326 | 1.072-5.045 | 0.033 | ||
| BiDil | 139 (85%) | 24 (15%) | |||
| Unadjusted | 0.842 | 0.293-2.420 | 0.749 | ||
| Adjusted | 0.698 | 0.226-2.159 | 0.532 | ||
| Adjusted w/o Ln(BNP) | 0.639 | 0.211-1.931 | 0.427 | ||
Adjusted analysis included the following covariates: sex, ejection fraction, systolic blood pressure, age, Ln(BNP), ischemic etiology (yes or no), diabetes mellitus, beta-blocker use.
Corin +/+ = wild-type
Corin +/- = heterozygous for corin I555(P568) allele.
Association of Corin I555(P568) Allele with Baseline BNP Values
The absolute and log-transformed baseline plasma measures of both BNP and proBNP were lower in the corin variant compared to non-variant groups (Table 4). When we calculated the Pearson correlation coefficient, the corin allele was significantly associated with lower BNP values in univariate (P=0.007) and multivariable (P=0.03) analysis that adjusted for age, gender, body mass index, ischemic etiology and use of beta-blockers. Likewise, when calculating the Spearman correlation coefficient, the corin I555(P568) allele was associated with lower BNP values in both unadjusted (P=0.01) and adjusted (P=0.04) analyses.
Table 4. Baseline Ln(BNP) and Ln(ProBNP) Values in Corin Variant (PI555/P568) Compared to Non-Variant Groups.
| Corin | BNP (pg/mL) | ProBNP | Ln(BNP) (pg/mL) | Ln(proBNP) |
|---|---|---|---|---|
| Corin +/+ | 339.8±429.6 | 505.1±773.7 | 5.04±1.44 | 5.22±1.63 |
| Corin +/- | 190.4±249.7 | 236.1±403.0 | 4.37±1.50 | 4.29±1.59 |
BNP and proBNP measures were taken using Biosite and BioRad immunoassays, respectively.
Corin +/+ = wild-type
Corin +/- = heterozygous for corin I555(P568) allele.
Ratio of ProBNP/BNP is increased in Carriers of the Corin I555/P568 Allele
As an index of impaired natriuretic peptide processing, we compared the ratio of proBNP 1-108/BNP-32 using the values measured from plasma samples obtained at the baseline visit in carriers versus non-carriers of the corin I555(P568) allele. We reasoned that a higher ratio would be indicative of greater impairment in BNP 1-108 processing. The mean proBNP 1-108/BNP-32 ratio in corin variant group was 1.92 compared to 1.41 in the non-variant group (37% higher). The median ProBNP/BNP ratio in the corin variant group was 1.64 compared to 1.21 in the non-variant group (36% increase) (Figure 2). The proBNP 1-108/BNP-32 ratios were not normally distributed so we utilized non-parametric testing to compare the ratio between corin genotype groups. The proBNP 1-108/BNP-32 ratio was significantly higher in the corin I555(P568) group (Van der Waerden test P=0.045) as compared to corin wild-type group.
Figure 2.
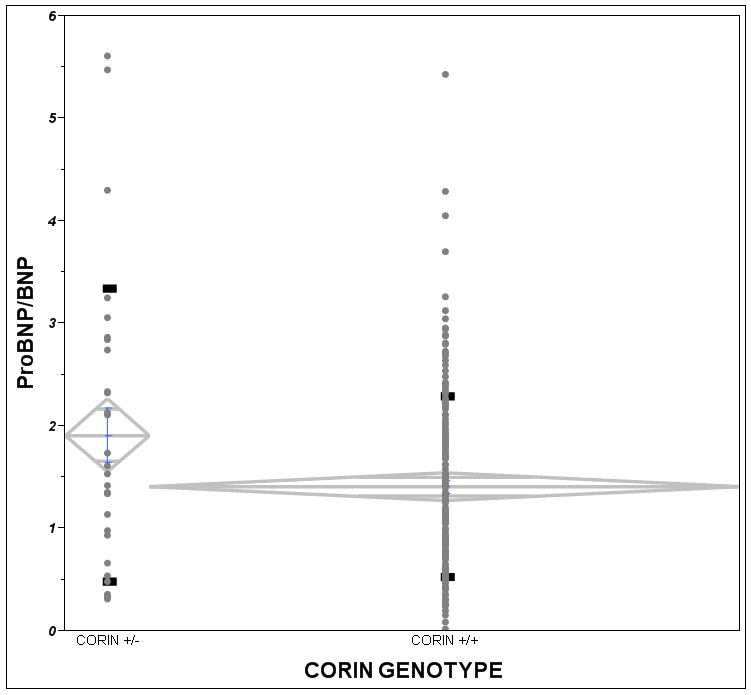
Ratio of proBNP/BNP, an index of unprocessed brain natriuretic peptide, by corin minor allele carrier status. A significant increase in the ratio of proBNP to BNP was identified in corin allele carriers (N=35) versus non-carriers (N=204). The median ProBNP/BNP ratio in the corin variant group was 1.64 compared to 1.21 in the non-variant group. The graph plots individual data points with mean diamonds representing mean and standard error of the mean intervals and black solid rectangles representing the standard deviation. (p=0.045 by non-parametric Van der Waerden normal quantile test).
Discussion
We examined the association of the corin I555(P568) allele with clinically relevant outcomes in African-Americans with moderate to severe systolic heart failure and identified a pharmacogenomic interaction between the presence of the corin I555(P568) allele and the administration of fixed-dose combination isosorbide-dinitrate/hydralzine (FDCI/H) on the risk for death or heart failure hospitalization. We report that African-Americans with systolic heart failure heterozygous for the corin I555(P568) allele who are not on FDCI/H, but are receiving standard neurohormonal blockade of the renin-angiotensin-aldosterone and sympathetic nervous systems with ACE-I/ARB, aldosterone and β-adrenergic receptor blockers, are at significantly increased risk of heart failure progression as evidenced by a significant increased risk for the composite endpoint of death or heart failure hospitalization. This survival difference for the composite endpoint was largely driven by an increased risk for heart failure hospitalization. In contrast, in the arm randomized to treatment with FDC I/H (BiDil), no increase in death or hospitalization for heart failure was observed in carriers of the corin I555(P568) allele, as compared to non-carriers.
Moreover, using an immunoassay for processed BNP 77-108 with a known 20% cross-reactivity to proBNP and a highly specific novel immunoassay for unprocessed BNP 1-108, we report in the African-American carriers of the corin I555(P568) allele a higher proBNP/BNP ratio consistent with greater impairment in BNP processing. There is no reason to suspect that the magnitude of cross-reactivity (∼ 19%) between the Biosite Triage immunoassay and BNP 1-108 would differ between the corin variant as compared to the corin non-variant groups. Therefore, a comparison of the BNP 1-108/BNP 77-108 ratio represented a reasonable method of estimating differences in BNP processing efficiency between corin genotypes. However, the proBNP 1-108/BNP-32 ratio serves as an approximate estimate of impaired processing and future studies are needed to confirm our finding of an increased ratio of unprocessed to processed BNP associated with the corin I555 (P568) allele. Interestingly, despite the increased risk for death or first hospitalization in the corin I555(P568) variant group, the BNP-32 (Biosite) levels were lower in this group, a paradoxical finding given the prognostic importance of an increased BNP in heart failure. In fact, adjusting for the BNP levels increased the risk ratio for the composite endpoint in the corin variant group receiving placebo which is consistent with our hypothesis that impaired processing of brain (and potentially other natriuretic peptides) may be the mechanistic link between the presence of the corin I555(P568) allele and disease progression in congestive heart failure.
The T555I and Q568P amino acid changes in corin are located in conserved amino acids within corin's second cysteine-rich, frizzled domain4. This domain is involved in protein-protein interactions, and previous work had demonstrated this domain to be required for corin catalytic activity23. Recently, in vitro studies using both human embryonic kidney (HEK) 293 cells and murine HL-1 cardiomyocytes, have shown that the corin variant gene product containing both the I555/P568 amino acid substitutions had a reduced activity for processing proANP (38+/-7%, P<0.01) and proBNP (44+/-15%, P<0.05) compared to that of wild type.5 This study also demonstrated that the mechanism explaining the reduced natriuretic processing capacity of the mutant I555(P568) corin to be impaired mutant corin zymogen activation, not a reduction in corin catalytic capacity per se. Interestingly, the presence of each mutation individually did not significantly reduce the biological activity of corin. This is intriguing because within the African-American populations we have studied, the single nucleotide polymorphisms (SNPS) causing the T555I and Q568P amino acid changes are in complete linkage disequilibrium so that both amino acid changes are indeed transcribed into the same corin molecule in subjects that are heterozygous for the corin I555(P568) allele.
It is also important to recognize that although the magnitude of impaired BNP processing was greater in the corin variant group, impaired BNP processing is evident in non-carrier participants with moderate to severe congestive heart failure. Several recent reports have demonstrated that the presence of immature natriuretic peptide precursors is common in patients with congestive heart failure.14, 24, 25 However, the molecular mechanisms underlying this phenomenon remain undetermined. The initial report describing the highly specific immunoassay for proBNP used in our study identified increasing levels of BNP1-108 in patients with congestive heart failure and a correlation with NYHA functional class 14. Furthermore, this study and an early investigation of circulating BNP forms in humans report substantial inter-individual variability in the relative proportions of the various forms of natriuretic peptides. 14, 26 Whether processing efficiency of natriuretic peptides or some other physiologic mechanism can account for differences in the relative proportions of natriuretic peptides remains to be determined. We suspect that genetic determinants, such as the corin I555(P568) allele as well as others yet to be discovered, may explain some of the variability in the processing of natriuretic peptides.
There is a plausible biological hypothesis to account for the observed pharmacogenomic interaction in the present study. The natriuretic peptides stimulate cGMP production by activating the particulate guanylate cyclase (pGC) domain, which is a domain contained within the intracellular region of the NPR-A receptor. Fixed-dose combination isosorbide-dinitrate and hydralazine (BiDil) increases intracellular cGMP by activation of soluble guanylate cyclase (sGC). Some investigators report that these two cGMP pools are compartmentalized and activate distinct signaling cascades27. On the other hand, there is also evidence for significant reciprocal regulation of the NO-sGC and the ANP/BNP-pGC cGMP pools in both NPR-A 28 and endothelial nitric oxide synthase (eNOS-/-)29 knockout models implicating cross-talk between the NO-sGC and the NP-pGC pathways in the regulation of cGMP-dependent vasodilatation and pressure-overload induced cardiomyocyte hypertrophy. Assuming that the corin I555(P568) allele reduces BNP processing, especially under conditions of decompensation when BNP transcription substantially increases, this would phenotypically be expected to have the same effect as any of the genetic models that perturbed NPR-A function; namely, an enhanced response to exogenously administered NO-donors. Indeed, although the sample size was small our data suggests an enhanced treatment effect the group of I555(P568) carriers--the corin variant group randomized to FDC I/H (BiDil) demonstrated 64% improvement in event free survival (HR=0.36, p=0.078), as compared to the reported 37% increase in event free survival (HR=0.67, p<0.0001) in the overall cohort of 1050 A-HeFT patients who were randomized to FDC I/H as compared to placebo18.
We recognize the limitations of the present analysis. The sample size carrying the corin I555(P568) allele is small and we have not replicated these results. We recognize that replication is essential to avoid type I error in genetic association studies. Unfortunately, there are limited clinical trials in heart failure with an adequate African-American sample to permit replication. Moreover, the design of A-HeFT was unique and, therefore, it may not be possible to replicate the pharmacogenomic interaction observed. Another concern is the potential for confounding from population stratification. We did not genotype random ancestry-informative markers to allow us to adjust for confounding from population stratification. However, the previous work related to the corin I555(P568) allele is reassuring in this regard. Firstly, in our previous studies of the corin I555(P568) allele with risk for hypertension and concentric cardiac remodeling, adjustment for population stratification using > 1,000 ancestry-informative markers did not reduce the association of the corin I555(P568) allele with our phenotypes of interest.4 Secondly, recent experiments have convincingly demonstrated the functional effects of the two amino acid substitutions on corin; specifically, an ∼ 70% reduction in the ability of mutant corin (I555/P568) to process either proANP or proBNP. Thirdly, we are reassured by the biomarker data that supports the hypothesis of impaired BNP processing in the patients heterozygous for the corin I555(P568) allele. Considered together, these points argue against the results of this study being explained by confounding from population stratification.
There are intriguing clinical implications from our study if it can be confirmed in future analyses. The pharmacogenomic interaction suggest that treatment with FDC I/H may attenuate the adverse prognosis associated with the corin I555(P568) allele in the setting of moderate to severe heart failure. However, many would argue that based on the A-HeFT results from the main trial, there is sufficient rationale to treat all African-Americans fitting the enrollment criteria with BiDil. However, if this pharmacogenomic interaction can be confirmed and further elucidated, it suggests the possibility of tailored treatment of African-Americans heterozygous for the corin I555(P568) allele with AHA/ACC Stage A or B heart failure. In our previous work, untreated and hypertensive African-Americans heterozygous for the corin I555(P568) allele were at increased risk for concentric cardiac remodeling22. Perhaps restoration of cGMP pools with phosphodiesterase inhibitors or FDC I/H would be of particular benefit in ameliorating adverse hypertensive cardiac concentric remodeling in these patients. Another potential clinical implication relates to the observation of the decoupling of plasma BNP levels and prognosis in African-Americans heterozygous for the corin I555(P568) allele, found in approximately 13% of African-Americans at the population level. In the overall AHeFT cohort, plasma BNP remained a powerful prognostic indicator (data not shown), but in the placebo arm, the corin I555(P568) allele carriers demonstrated an increased risk of heart failure hospitalization despite having a lower baseline BNP as compared to non-carriers of the corin allele. If our hypothesis is correct, a low Biosite BNP value in these patients may reflect impaired natriuretic peptide processing and increased risk for heart failure progression, rather than clinical stability and decreased risk for adverse outcomes. Our study also suggests the possibility that simultaneous measurement of BNP using an assay relatively specific for BNP-32 coupled with simultaneous ascertainment of BNP 1-108 in African-Americans with systolic heart failure may improve risk-stratification and the capacity of natriuretic peptides to prognosticate in patients with chronic ambulatory congestive heart failure.
In conclusion, we report that in African-Americans with moderate to severe systolic heart failure, the functional corin I555(P568) allele is associated with an increased risk for heart failure progression in the absence of treatment with FDC I/H. Furthermore, we have identified increased impairment of BNP processing with lower plasma BNP in the corin I555(P568) carriers and hypothesize that this pharmacogenomic interaction might be explained by an amelioration of the adverse prognostic impact of the corin variant allele with FDC I/H by virtue of cross-talk between the soluble and particulate guanylate cyclase cGMP signaling systems. These results are hypothesis generating and require additional confirmation in independent cohorts and animal models. Future studies should focus on further determination of the mechanism and clinical significance of this pharmacogenomic interaction in the management of moderate to severe heart failure and an understanding of the physiologic basis of the relative proportions of the various forms of natriuretic peptides in human heart failure.
Acknowledgments
Sources of Funding. Dr. Dries received funding form the National Institutes of Health RO-1 HL91633-01A2. Eduardo Rame is a recipient of a American Heart Association National Scientist Development Grant (0730375N).
Footnotes
Disclosures. Drs. Sabolinski, Tam and Worcel were employees and stockholders of NitroMed, Inc.
CLINICAL SUMMARY DRIES MANUSCRIPT 866822
The endogenous natriuretic peptide system (NPS), characterized by the ability of the heart to secrete atrial and brain natriureticpeptide)ANP and BNP) into the circulation upon increased cardiac filling pressures, exerts essential compensatory actions in heart failure. Activation of the NPS helps restore volume status and antagonizes adverse neurohormonal systems. In order to function properly, proANP and proBNP must first be cleaved into biologically active peptide hormones by corin, a transmembrane protease expressed by cardiomyocytes. The corin I555(P568) allele results from two amino-acid changing single nucleotide polymorphisms(T555I and Q568P) that are in complete linkage disequilibrium in African-Americans. These two mutations in vitro impair the ability of corin to efficiently process proANP/BNP into biologically active peptides. African-Americans carrying this minor corin allele are at increased risk for heart failure hospitalization in the absence of treatment with BiDil. They also demonstrate evidence of less efficient BNP processing. Inefficient processing of natriuretic peptides would reduce activation of the natriureticpeptide receptor and thereby reduce generation of cyclic GMP by the particulate guanylate cyclase moiety of the natriuretic peptide receptor. By augmenting intracellular cGMP pools independently of the natriuretic peptide receptor, by virtue of activationof soluble guanylate cuyclase, BiDil may ameliorate the adverse prognosis caused by impaired BNP processing in the presence of the corin I555(P568) allelein African-Americans with moderate to severe systolic heart failure.
References
- 1.Yan W, Sheng N, Seto M, Morser J, Wu Q. Corin, a mosaic transmembrane serine protease encoded by a novel cDNA from human heart. J Biol Chem. 1999;274:14926–14935. doi: 10.1074/jbc.274.21.14926. [DOI] [PubMed] [Google Scholar]
- 2.Yan W, Wu F, Morser J, Wu Q. Corin, a transmembrane cardiac serine protease, acts as a pro-atrial natriuretic peptide-converting enzyme. Proc Natl Acad Sci U S A. 2000;97:8525–8529. doi: 10.1073/pnas.150149097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rame JE, Drazner MH, Post W, Peshock R, Lima J, Cooper RS, Dries DL. Corin I555(P568) allele is associated with enhanced cardiac hypertrophic response to increased systemic afterload. Hypertension. 2007;49:857–864. doi: 10.1161/01.HYP.0000258566.95867.9e. [DOI] [PubMed] [Google Scholar]
- 4.Dries DL, Victor RG, Rame JE, Cooper RS, Wu X, Zhu X, Leonard D, Ho SI, Wu Q, Post W, Drazner MH. Corin gene minor allele defined by 2 missense mutations is common in blacks and associated with high blood pressure and hypertension. Circulation. 2005;112:2403–2410. doi: 10.1161/CIRCULATIONAHA.105.568881. [DOI] [PubMed] [Google Scholar]
- 5.Wang W, Liao X, Fukuda K, Knappe S, Wu F, Dries DL, Qin J, Wu Q. Corin variant associated with hypertension and cardiac hypertrophy exhibits impaired zymogen activation and natriuretic peptide processing activity. Circ Res. 2008;103:502–508. doi: 10.1161/CIRCRESAHA.108.177352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heublein DM. Immunoreactivity and cyclic GMP activating actions of various molecular forms of human B-type natriuretic peptide. Hypertension. 2007;49:1114–1119. doi: 10.1161/HYPERTENSIONAHA.106.081083. [DOI] [PubMed] [Google Scholar]
- 7.Liang F, O'Rear J, Schellenberger U, Tai L, Lasecki M, Schreiner GF, Apple FS, Maisel AS, Pollitt NS, Protter AA. Evidence for functional heterogeneity of circulating B-type natriuretic peptide. J Am Coll Cardiol. 2007;49:1071–1078. doi: 10.1016/j.jacc.2006.10.063. [DOI] [PubMed] [Google Scholar]
- 8.Moritoki H, Yoshikawa T, Hisayama T, Takeuchi S. Possible mechanisms of age-associated reduction of vascular relaxation caused by atrial natriuretic peptide. Eur J Pharmacol. 1992;210:61–68. doi: 10.1016/0014-2999(92)90652-k. [DOI] [PubMed] [Google Scholar]
- 9.Kurtz A, Della Bruna R, Pfeilschifter J, Taugner R, Bauer C. Atrial natriuretic peptide inhibits renin release from juxtaglomerular cells by a cGMP-mediated process. Proc Natl Acad Sci U S A. 1986;83:4769–4773. doi: 10.1073/pnas.83.13.4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gunning ME, Brady HR, Otuechere G, Brenner BM, Zeidel ML. Atrial natriuretic peptide(31-67) inhibits Na+ transport in rabbit inner medullary collecting duct cells. Role of prostaglandin E2. J Clin Invest. 1992;89:1411–1417. doi: 10.1172/JCI115730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeidel ML. Renal actions of atrial natriuretic peptide: regulation of collecting duct sodium and water transport. Annu Rev Physiol. 1990;52:747–759. doi: 10.1146/annurev.ph.52.030190.003531. [DOI] [PubMed] [Google Scholar]
- 12.Zeidel ML. Regulation of collecting duct Na+ reabsorption by ANP 31-67. Clin Exp Pharmacol Physiol. 1995;22:121–124. doi: 10.1111/j.1440-1681.1995.tb01967.x. [DOI] [PubMed] [Google Scholar]
- 13.Isono M, Haneda M, Maeda S, Omatsu-Kanbe M, Kikkawa R. Atrial natriuretic peptide inhibits endothelin-1-induced activation of JNK in glomerular mesangial cells. Kidney Int. 1998;53:1133–1142. doi: 10.1046/j.1523-1755.1998.00869.x. [DOI] [PubMed] [Google Scholar]
- 14.Giuliani I, Rieunier F, Larue C, Delagneau JF, Granier C, Pau B, Ferriere M, Saussine M, Cristol JP, Dupuy AM, Merigeon E, Merle D, Villard S. Assay for measurement of intact B-type natriuretic peptide prohormone in blood. Clin Chem. 2006;52:1054–1061. doi: 10.1373/clinchem.2005.061770. [DOI] [PubMed] [Google Scholar]
- 15.Luckenbill KN, Christenson RH, Jaffe AS, Mair J, Ordonez-Llanos J, Pagani F, Tate J, Wu AH, Ler R, Apple FS. Cross-reactivity of BNP, NT-proBNP, and proBNP in commercial BNP and NT-proBNP assays: preliminary observations from the IFCC Committee for Standardization of Markers of Cardiac Damage. Clin Chem. 2008;54:619–621. doi: 10.1373/clinchem.2007.097998. [DOI] [PubMed] [Google Scholar]
- 16.Taylor AL, Cohn JN, Worcel M, Franciosa JA. The African-American Heart Failure Trial: background, rationale and significance. J Natl Med Assoc. 2002;94:762–769. [PMC free article] [PubMed] [Google Scholar]
- 17.Franciosa JA, Taylor AL, Cohn JN, Yancy CW, Ziesche S, Olukotun A, Ofili E, Ferdinand K, Loscalzo J, Worcel M. African-American Heart Failure Trial (A-HeFT): rationale, design, and methodology. J Card Fail. 2002;8:128–135. doi: 10.1054/jcaf.2002.124730. [DOI] [PubMed] [Google Scholar]
- 18.Taylor AL, Ziesche S, Yancy CW, Carson P, Ferdinand K, Taylor M, Adams K, Olukotun AY, Ofili E, Tam SW, Sabolinski ML, Worcel M, Cohn JN. Early and sustained benefit on event-free survival and heart failure hospitalization from fixed-dose combination of isosorbide dinitrate/hydralazine: consistency across subgroups in the African-American Heart Failure Trial. Circulation. 2007;115:1747–1753. doi: 10.1161/CIRCULATIONAHA.106.644013. [DOI] [PubMed] [Google Scholar]
- 19.Anand IS, Tam SW, Rector TS, Taylor AL, Sabolinski ML, Archambault WT, Adams KF, Olukotun AY, Worcel M, Cohn JN. Influence of blood pressure on the effectiveness of a fixed-dose combination of isosorbide dinitrate and hydralazine in the African-American Heart Failure Trial. J Am Coll Cardiol. 2007;49:32–39. doi: 10.1016/j.jacc.2006.04.109. [DOI] [PubMed] [Google Scholar]
- 20.Conover WJ. Practical Nonparametric Statistics. 3rd. 1999. [Google Scholar]
- 21.Cox DR. Regression Models and Life Tables. Journal of the Royal Statistical Society Series. 1972 [Google Scholar]
- 22.Rame JE, Drazner MH, Post W, Peshock R, Lima J, Cooper RS, Dries DL. Corin I555(P568) Allele Is Associated With Enhanced Cardiac Hypertrophic Response to Increased Systemic Afterload. Hypertension. 2007;49:857–864. doi: 10.1161/01.HYP.0000258566.95867.9e. [DOI] [PubMed] [Google Scholar]
- 23.Knappe S, Wu F, Madlansacay MR, Wu Q. Identification of domain structures in the propeptide of corin essential for the processing of proatrial natriuretic peptide. J Biol Chem. 2004;279:34464–34471. doi: 10.1074/jbc.M405041200. [DOI] [PubMed] [Google Scholar]
- 24.Hawkridge AM, Heublein DM, Bergen HR, 3rd, Cataliotti A, Burnett JC, Jr, Muddiman DC. Quantitative mass spectral evidence for the absence of circulating brain natriuretic peptide (BNP-32) in severe human heart failure. Proc Natl Acad Sci U S A. 2005;102:17442–17447. doi: 10.1073/pnas.0508782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waldo SW, Beede J, Isakson S, Villard-Saussine S, Fareh J, Clopton P, Fitzgerald RL, Maisel AS. Pro-B-type natriuretic peptide levels in acute decompensated heart failure. J Am Coll Cardiol. 2008;51:1874–1882. doi: 10.1016/j.jacc.2007.12.051. [DOI] [PubMed] [Google Scholar]
- 26.Tateyama H, Hino J, Minamino N, Kangawa K, Minamino T, Sakai K, Ogihara T, Matsuo H. Concentrations and molecular forms of human brain natriuretic peptide in plasma. Biochemical and biophysical research communications. 1992;185:760–767. doi: 10.1016/0006-291x(92)91691-i. [DOI] [PubMed] [Google Scholar]
- 27.Castro LR, Verde I, Cooper DM, Fischmeister R. Cyclic guanosine monophosphate compartmentation in rat cardiac myocytes. Circulation. 2006;113:2221–2228. doi: 10.1161/CIRCULATIONAHA.105.599241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Madhani M, Okorie M, Hobbs AJ, MacAllister RJ. Reciprocal regulation of human soluble and particulate guanylate cyclases in vivo. Br J Pharmacol. 2006;149:797–801. doi: 10.1038/sj.bjp.0706920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bubikat A, De Windt LJ, Zetsche B, Fabritz L, Sickler H, Eckardt D, Godecke A, Baba HA, Kuhn M. Local atrial natriuretic peptide signaling prevents hypertensive cardiac hypertrophy in endothelial nitric-oxide synthase-deficient mice. J Biol Chem. 2005;280:21594–21599. doi: 10.1074/jbc.M501103200. [DOI] [PubMed] [Google Scholar]