

Abstract
RNA-binding proteins constitute key factors of the post-transcriptional machinery. These regulatory proteins recognize specific elements within target transcripts to promote, for example, maturation, translation, or stability of mRNAs. In Ustilago maydis, evidence is accumulating that post-transcriptional processes are important to determine pathogenicity. Deletion of khd4, encoding a predicted RNA-binding protein with five K homology (KH) domains, causes aberrant cell morphology and reduced virulence. Here, we demonstrate that Khd4 recognizes the sequence AUACCC in vivo via its tandem KH domains 3 and 4. This sequence most likely functions as a regulatory RNA element in U. maydis, since it accumulates in 3′ untranslated regions. Consistently, an independent mRNA expression profiling approach revealed that the binding motif is significantly enriched in transcripts showing altered expression levels in khd4Δ strains. Since the vast majority of potential Khd4 target mRNAs exhibit increased amounts in deletion mutants, Khd4 might promote mRNA instability. Mutants that fail to bind AUACCC resemble deletion mutants, which exhibit altered cell morphology, disturbed filamentous growth, and severely reduced virulence. Hence, RNA binding is essential for function of Khd4, stressing the importance of post-transcriptional control in regulating morphology and pathogenicity.
Keywords: three-hybrid screen, K homology domain, microarray, plant pathogen
INTRODUCTION
Post-transcriptional regulation is an important mechanism to determine gene expression in eukaryotic cells (Moore 2005; Keene 2007). Key factors are RNA-binding proteins that interact with target RNAs to form higher-order ribonucleoprotein complexes (Dreyfuss et al. 2002). These regulators orchestrate various post-transcriptional events such as maturation, localization, stability, and translation of mRNAs. A characteristic feature of these proteins is the presence of at least one RNA-binding domain. A well-studied example is the K homology (KH) domain that was first identified in the heterogenous nuclear ribonucleoprotein K (Siomi et al. 1993). The domain comprises ∼70 amino acids with a structurally conserved KH core motif consisting of two parallel β-sheets and two α-helices connected by a characteristic GXXG loop (Valverde et al. 2008). KH domain proteins are found in archea, bacteria, and eukaryotes. They are involved in various cellular functions such as control of transcription, splicing, and translation (Valverde et al. 2008).
We study the model fungus Ustilago maydis, the etiologic agent of smut disease on corn. Prerequisite for infection is a drastic morphological switch from cylindrical yeast-like cells that proliferate by budding to elongated filaments. An important transcriptional regulator of this program is the homeodomain transcription factor bE/bW, whose activity is regulated at the level of heterodimerization. Since subunits must originate from different mating-types, bE/bW activity is efficiently coupled to cell fusion during mating (Bölker 2001; Feldbrügge et al. 2004). The active heterodimer elicits a defined transcriptional cascade leading to the formation of infectious filaments that expand at the apical pole and insert retraction septa at the basal pole. After penetration, filamentously growing cells form a multicellular mycelium. A characteristic disease symptom is the formation of tumors on stems, leaves, and flowers, in which black teliospores develop (Kahmann and Kämper 2004; Feldbrügge et al. 2006). In recent years, accumulating evidence suggests that, in addition to extensive transcriptional regulation, post-transcriptional processes are important for pathogenicity. A reverse genetic approach, in which 18 genes encoding predicted RNA-binding proteins were deleted, identified Rrm4 and Khd4 as post-transcriptional regulators of pathogenic development (Becht et al. 2005). Rrm4 mediates microtubule-dependent transport of distinct mRNAs encoding, for example, polarity factors involved in the formation of infectious filaments (Becht et al. 2006; König et al. 2009). Analysis of the second candidate revealed that Khd4, which contains five predicted KH domains in its central region, belongs to a new class of fungal multi-KH domain proteins with homologs in important human pathogens such as Cryptococcus neoformans, Aspergillus fumigatus, and Candida albicans (Fig. 1; Becht et al. 2005). khd4Δ mutants are disturbed in cell morphology, exhibit a reduced pheromone response, and are severely affected in virulence. Thus, this predicted RNA-binding protein appears to be important during several critical steps of the life cycle (Becht et al. 2005). Here, we demonstrate that Khd4 specifically recognizes the sequence AUACCC via its third and fourth KH domain in vivo. This sequence is significantly enriched in regulatory 3′ untranslated regions (UTRs) as well as in mRNAs that exhibit increased levels in khd4Δ strains. Thus, AUACCC-containing transcripts are promising candidates for Khd4 target mRNAs. Importantly, the tandem KH domains 3 and 4 are essential for determination of cell shape, cytokinesis, and plant infection. Thus, Khd4-mediated post-transcriptional processes are crucial for the regulation of morphology and pathogenicity in U. maydis.
FIGURE 1.

Khd4 is the founding member of a novel class of fungal multi-KH domain proteins. A schematic representation of the domain structure of six fungal KH domain proteins is depicted in the upper part (U.m., Ustilago maydis; C.n., Cryptococcus neoformans; A.f., Aspergillus fumigatus; C.a., Candida albicans; S.c., Saccharomyces cerevisiae; and S.p., Schizosaccharomyces pombe). KH domains were predicted according to SMART (dark gray rectangle; Letunic et al. 2009). Additional potential KH domains that were only found using the schnipsel database (Letunic et al. 2009) or that were predicted according to our sequence comparison (given below) are indicated by light gray or open rectangles, respectively. A bold line indicates a conserved linker region flanked by KH domains 2a and 3. In the lower part, a sequence comparison of the tandem KH domains 3 and 4 is shown (dashed line flanked by filled arrowheads; flanking numbers correspond to amino acid positions in Khd4; amino acid positions of KH3 and KH4 are 746–815 and 816–891, respectively). GKKN and GVGG below the sequence indicate the amino acids within potential GXXG loops in Khd4 that were mutated to AAAA.
RESULTS
AUACCC is necessary and sufficient for Khd4 interaction
In order to characterize RNA binding of Khd4 in vivo we applied the reverse yeast three-hybrid system (Fig. 2A; SenGupta et al. 1996; Hook et al. 2005) using the following components: Hybrid 1 was a protein consisting of the LexA DNA-binding domain fused to the RNA-binding coat protein of phage MS2. Hybrid 2 was an RNA containing the MS2 binding site and a variable RNA sequence based on genomic DNA of U. maydis (the insert size was 50–250 base-pair [bp] fragments on average; see Materials and Methods). Hybrid 3 was Khd4 with N-terminal Gal4 activation domain and C-terminal Gfp (enhanced version of the green fluorescence protein, Clontech; Khd4AG, Fig. 2A). We omitted the last 135 amino acids of Khd4 containing a glutamine-rich stretch, since such regions are known to activate transcription (Triezenberg 1995). Interaction of Khd4 with a hybrid target RNA resulted in formation of a tripartite complex, which triggered expression of HIS3 and lacZ genes (encoding imidazole-glycerol-phosphate dehydratase and β-galactosidase, respectively) via lexA operator sequences in their regulatory regions (Fig. 2A). Thus, this powerful in vivo technique enables investigation of RNA/protein interactions by simple detection of reporter gene expression (SenGupta et al. 1996; Hook et al. 2005; Stumpf et al. 2008). We screened 4 × 108 colonies for HIS3 reporter gene expression on selection plates (SC –his + 5 mM 3-AT, 3-aminotriazol; see Materials and Methods). Of 98 selected colonies expressing both reporter genes 73 remained after plasmid rescue and retransformation. Sequence analysis revealed that the respective inserts corresponded to 45 independent U. maydis genomic loci (Fig. 2B; Supplemental Table S1; Kämper et al. 2006; http://mips.gsf.de/genre/proj/ustilago/). Twenty sequences mapped to predicted transcripts: 13 to open reading frames (ORFs), two to 5′ UTRs, and five to 3′ UTRs. Twenty-five sequences corresponded to intergenic (four) and antisense (21) regions. Some of the potential target mRNAs encoded promising regulatory proteins related to a histone deacetylase, a serine/threonine-specific kinase, and a splice factor (Supplemental Table S1, um10914, um11195, um00305, respectively). Applying the MEME pattern search algorithm to the identified sequences (Bailey et al. 2006) revealed that the consensus sequence AUACCC is a potential binding motif of Khd4 (Fig. 2C). Importantly, AUACCC was present at least once in all identified sequences. For experimental verification we chose insert S35, which corresponds to the 3′ UTR of um04083 encoding a conserved hypothetical protein (Supplemental Table S1, MUMDB, http://mips.gsf.de/genre/proj/ustilago/). We divided the sequence in two parts that either contained or did not contain the AUACCC sequence (S35b or S35a, respectively). Only S35b containing the potential binding site mediated growth on selection plates (Fig. 2D). Furthermore, the variant S35b* carrying a mutated version (AGAUCU) did not confer growth on selection plates (Fig. 2D), confirming that AUACCC functions as a binding motif of Khd4. Thus, AUACCC is necessary for Khd4 binding. To assay whether this motif is also sufficient for binding, we compared hexameric repeats of the sequences AUACCC and AGAUCU. We observed that [AUACCC]6, but not the mutated version, mediated growth on selection plates (Fig. 2D), demonstrating that the motif AUACCC alone is sufficient to confer binding of Khd4. In essence, the motif AUACCC is necessary and sufficient for Khd4 binding in vivo.
FIGURE 2.

AUACCC is necessary and sufficient for interaction with Khd4 in vivo. (A) Schematic representation of the reverse yeast three-hybrid system. For simplicity only one MS2 protein (MS2-CP) and binding site are indicated (MS2-CP binds as a dimer and the RNA hybrid contains two MS2 binding sites). The label “kis” symbolizes the presence of a Khd4 interacting sequence. Activation domain and operator are abbreviated AD and Op, respectively. (B) Diagram representing the distribution of 45 sequences identified in the yeast three-hybrid screen corresponding to the following regions: 5′ UTR, ORF, 3′ UTR, and other (intergenic and antisense). (C) Graphic representation of the AUACCC consensus sequence. The absolute height of each column and the relative height of each letter depict overall conservation and relative frequency, respectively (Crooks et al. 2004). (D) Yeast three-hybrid analysis demonstrating that AUACCC is necessary and sufficient for Khd4 binding. The AUACCC-containing sequence S35 derived from the 3′ UTR of um04083 was selected as an example (sequence given above). Fragments S35a and S35b (marked with lines) are indicated above. AUACCC is highlighted. The sequence AGAUCU indicates point mutations introduced in S35b*. The presence of protein-encoding and RNA-encoding plasmids was verified by growth on transformation plates (SC medium –ura –leu). Khd4 binding was scored as growth on selection plates (SC medium –his). The filled triangle indicates decreasing amounts of cells (dilution factor 1:5). (E,F) Yeast three-hybrid analysis demonstrating that KH domains 3 and 4 are necessary for binding. (E) Expression of Khd4AG variants (indicated below) was determined in strains expressing three different hybrid RNAs (inlay) by quantifying relative Gfp fluorescence per OD600. (F) Relative β-galactosidase activity was determined using a fluorogenic substrate. To account for the difference in protein expression, enzyme activity is given relative to Gfp fluorescence intensity (ΔRFUGal per minute per RFUGfp). Different Khd4AG variants (bottom) were tested with three different hybrid RNAs (inlay).
The tandem KH domains 3 and 4 of Khd4 are essential for AUACCC binding
Khd4 is predicted to contain five KH domains. In order to map the region of AUACCC binding we inserted four alanine block mutations in the GXXG loop of KH domains 3, 4, and 5, which exhibited highest sequence conservation (Fig. 1; see Materials and Methods). The conserved loop region was important for nucleic acid binding in various examples of KH domains (Valverde et al. 2008), and mutations in this region are known to severely impair binding (Vanrobays et al. 2008). To assay RNA binding we tested the Khd4AG variants (Khd4m3AG, Khd4m4AG, and Khd4m5AG) against two hybrid RNAs containing AUACCC (S35b and [AUACCC]6) or a mutated control ([AGAUCU]6). Analysis of Gfp fluorescence in the various combinations verified that all three mutant variants were expressed, although levels of Khd4m5AG were reduced in the presence of sequences S35b and [AUACCC]6 (Fig. 2E). To compensate for varying Khd4AG levels, we calculated the substrate turnover over time relative to Gfp fluorescence (Fig. 2F, ΔRFUGal/min*RFUGfp; König et al. 2007). lacZ reporter gene expression was scored by measuring β-galactosidase activity using a fluorogenic substrate. As expected, Khd4AG recognized both AUACCC-containing sequences, but not the mutated control RNA. Mutations in KH domains 3 and 4 completely abolished binding, while mutations in KH domain 5 had no influence on this interaction (Fig. 2F). Although we cannot exclude that KH domains 1 and 2 add to the RNA binding specificity of Khd4, we conclude that tandem KH domains 3 and 4 are essential for AUACCC recognition in vivo.
AUACCC is significantly enriched in mRNAs that exhibit increased levels in khd4Δ strains
Since AUACCC is sufficient for Khd4 binding, we evaluated the regulatory potential of Khd4 by searching for the presence of AUACCC in the transcribed regions of 6785 manually annotated U. maydis genes (MUMDB; http://mips.gsf.de/genre/proj/ustilago/; start and end of transcripts were defined as 300 nucleotides [nt] upstream of the start codon and downstream from the stop codon, respectively); 1581 transcripts (23%) contained at least one AUACCC sequence, suggesting that Khd4 has a wide regulatory range (1883 motifs in total; 19% in the 5′ UTR; 55% in the ORF, and 26% in the 3′ UTR). In order to address this experimentally we investigated whether mRNAs were differentially expressed in the absence of Khd4. Using Affymetrix microarrays (Scherer et al. 2006; Zarnack et al. 2008; see Materials and Methods), we compared the expression profiles of FB2khd4Δ and wild-type strain FB2 in two independent transformants and two replicates growing in minimal medium, respectively (Supplemental Table S2). Seventy-two mRNAs were differentially expressed (1.5% of 4786 detected transcripts), whose abundance was significantly changed at least twofold in the deletion strain. These could be divided into 57 mRNAs with increased and 15 mRNAs with decreased levels in FB2khd4Δ (Supplemental Table S2). Regulation of three examples, um00748, um12098, and um03706.2 encoding subunit 5 of splice factor 3B and two proteins with unknown function, respectively, was verified in Northern analysis. Consistent with the microarray data, the abundance of all three mRNAs was substantially increased in the khd4Δ strain (Fig. 3). Gene ontology analysis of the mRNAs exhibiting differential expression levels revealed significant enrichment within three functional categories: the overlapping categories “electron transport and membrane-associated energy conservation” and “electron transport” as well as “sodium binding” (Ruepp et al. 2004; http://mips.gsf.de/projects/funcat; P-values < 10−4). It was noteworthy that a significant number of differentially expressed mRNAs contained AUACCC (29 of the 72 mRNAs, 40%; P-value 3.7 × 10−03 according to hypergeometric distribution). The sequence motif was enriched to the same extent among transcripts exhibiting increased and decreased expression levels. These 29 mRNAs were good candidates for direct targets of Khd4 (Table 1). Since most of these transcripts exhibited increased amounts (23 out of 29 mRNAs, 79%), we assume that Khd4 might predominantly promote mRNA instability. The putative target transcripts encoded two regulatory proteins involved in transcription and splicing, 14 proteins with predicted enzymatic function, and 13 novel proteins with unknown function. None of the predicted functions can currently be clearly correlated with the mutant phenotype. To support our hypothesis that AUACCC functions as regulatory RNA element, we performed a positional analysis. We initially noted that differentially expressed transcripts in FB2khd4Δ showed enrichment for those target transcripts that carry AUACCC in the 3′ UTR (19%, 44%, and 38% of AUACCC were located in the 5′ UTR, ORF, and 3′ UTR, respectively) (Table 1). For a more detailed analysis, we investigated the positioning of AUACCC in the regulatory 5′ and 3′ UTR as well as in first and last 300 nt of ORFs. For correlation with our genome-wide expression studies, we plotted the log2 ratio of expression levels (FB2 versus FB2khd4Δ) versus the position of AUACCC in the respective transcript region (see Materials and Methods). The antisense sequence GGGUAU as well as the mutated sequence AGAUCU served as controls (Fig. 4A). This positional analysis revealed that AUACCC was enriched toward the first 150 nt of the predicted 3′ UTRs (Fig. 4A). Analyzing the cumulative distribution verified a strong positional bias of AUACCC within the 3′ UTR (Fig. 4B; P-value 2.2 × 10−16 by Wilcoxon rank sum test). A significant, albeit less pronounced, enrichment could also be seen within the 5′ UTR (P-value 3.7 × 10−03). In contrast, the motif displayed an even distribution within the analyzed coding regions. Analysis of the relative positions of motifs along the entire ORF lengths revealed that AUACCC exhibited a slight enrichment toward the beginning of ORFs (Supplemental Fig. S1, P-value 2 × 10−07). Control motifs showed no significant enrichment in any transcript regions with the exception of AGAUCU in the 3′ UTR (P-value 2.4 × 10−06) (Fig. 4B). In this context, it is important to note that the experimentally determined average length of the 5′ and 3′ UTRs is in both cases 114 nt (median determined from 3664 and 3428 expressed sequence tags; 25th percentiles 62 and 68 nt and 75th percentiles 202 and 196 nt, respectively). Thus, the enrichment of AUACCC correlates with the length of 3′ UTRs. It is noteworthy that all up-regulated mRNAs containing AUACCC in the 3′ UTR harbored the binding motif within the first 100 nt. In essence, the Khd4 binding motif is significantly enriched in transcripts exhibiting increased levels in khd4Δ strains as well as within regulatory 3′ UTRs. These two lines of evidence support our hypothesis that AUACCC functions as a regulatory element in U. maydis.
FIGURE 3.
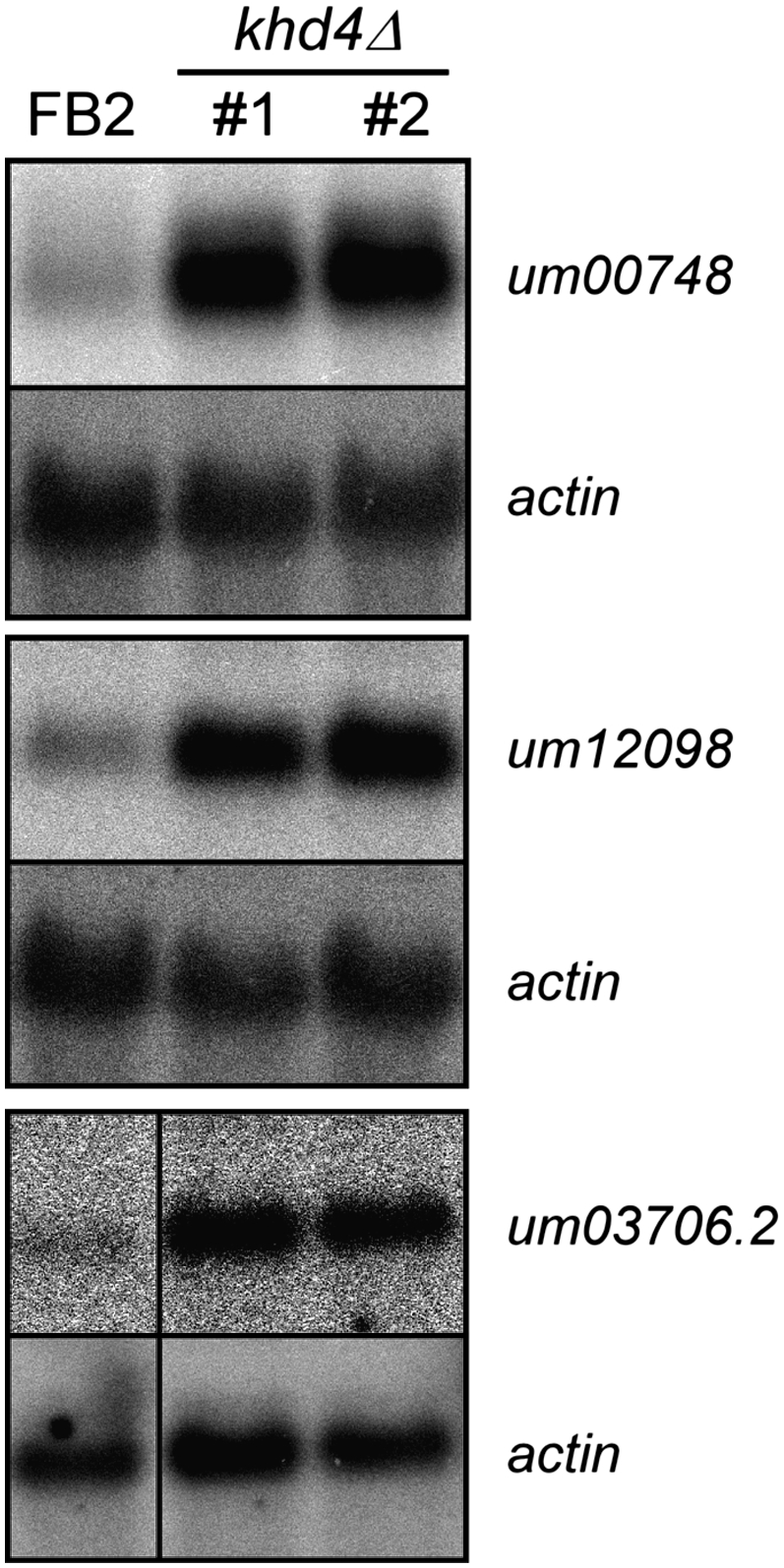
Potential target mRNAs exhibit increased amounts in khd4Δ strains. Northern analysis showing expression of selected transcripts (indicated on the right). um00748, um12098, and um03706.2 encode subunit 5 of splice factor 3B and two proteins of unknown function (MUMDB; http://mips.gsf.de/genre/proj/ustilago/). mRNA levels were increased 5.2 (2.3), 4.7 (3.3), and 5.3 (2.4) fold, respectively (values in brackets indicate microarray results, lower bound of a 90% confidence interval; Supplemental Table S2). Replicates of FB2 and two independent khd4Δ strains were analyzed (indicated above). Detection of actin mRNA served as control.
TABLE 1.
AUACCC-containing Khd4 target mRNAs

FIGURE 4.

Positional analysis of AUACCC. (A) Upper part, schematic representation of an eukaryotic mRNA consisting of 5′ CAP structure (black circle), 5′ UTR, ORF, 3′ UTR, and poly(A) tail (AAAA). Lower part, positions of AUACCC, GGGUAU, and AGAUCU (first, second, and third row, respectively) are plotted against the log2 ratio of the lower bound of fold change of the respective transcripts obtained in microarray analysis comparing FB2khd4Δ versus FB2. The four windows in each row depict 300 nt of 5′ and 3′ UTR as well as the first and last 300 nt of the ORF (indicated by lines). Positions are given relative to the start or stop codon in case of 5′ UTR and the first 300 nt of ORF or the last 300 nt of ORF and 3′ UTR, respectively. (B) Graph depicting the cumulative distribution of AUACCC (black), GGGUAU (light gray), and AGAUCU (medium gray) in the four transcript regions as defined in A. The cumulative fraction of motifs is plotted along the 300 nt of each region.
Tandem KH domains 3 and 4 are crucial for Khd4 function during regulation of cell morphology and filamentous growth
Using the yeast three-hybrid system we demonstrated that Khd4 recognizes the binding motif AUACCC via its KH domains 3 and 4. To address whether these domains are functionally important in U. maydis, we expressed Khd4 variants carrying the same mutations in KH domains 3–5 (see above). Khd4 variants were fused at the C terminus to eGfp and tandem affinity purification tag (Becht et al. 2006). In order to investigate morphology and filamentous growth we replaced the wild-type khd4 with the tagged variants in strain AB33 by homologous recombination using a SfiI-mediated gene replacement system (Brachmann et al. 2004; see Materials and Methods). AB33 is a derivative of wild-type strain FB2 that contains an active bE/bW heterodimeric transcription factor under control of the nitrogen source-regulatable promoter Pnar1. Thereby, b-dependent filamentation can be elicited by changing the nitrogen source in the medium (Brachmann et al. 2001; Becht et al. 2006). Microscopic analyses revealed that haploid cells of strain AB33khd4GT resembled wild type, indicating that the tagged protein was fully functional (Fig. 5A). Khd4GT predominantly localized to the cytoplasm consistent with a role in regulating the cytoplasmic fate of mRNAs on the level of translation or stability, for instance. The morphology of AB33khd4m5GT cells resembled wild-type cells, indicating that KH domain 5 is not important during regulation of cell morphology. However, strains expressing Khd4m3GT and Khd4m4GT formed cells with aberrant shape and a strong tendency to form aggregates indicative for defects in cytokinesis (Fig. 5A,B). Since this mutant phenotype was reminiscent of the phenotype of khd4Δ cells (Fig. 5A,B; Becht et al. 2005), we concluded that the mutations in KH domains 3 and 4 led to a loss of function of Khd4. Analyzing the subcellular localization of Khd4m3GT, Khd4m4GT, and Khd4m5GT revealed that the mutations impaired neither expression nor the predominantly cytoplasmic localization. Thus, mutant phenotypes were most likely not caused by reduced protein amounts (Fig. 5A).
FIGURE 5.

KH domains 3 and 4 are functionally important for regulating morphology. (A) Microscopic analysis of AB33 and derivatives expressing Khd4GT variants. Inverted micrographs of Gfp fluorescence are given next to DIC images (arrowheads indicate nuclei; size bar, 10 μm). (B) Bar diagrams depicting the percentage of cells with aberrant shape (at least 80 cells of each strain were analyzed [left]) as well as forming cell aggregates (more than two cells; at least 80 cells of each strain were analyzed [right]). Adjacent columns represent independent experiments.
In order to test filament formation, the same set of strains was grown for six hours under filament-inducing conditions. Khd4GT and Khd4m5GT-expressing strains formed long evenly shaped filaments like AB33 (Fig. 6A). In contrast, AB33khd4Δ was disturbed in filament formation. Mutant filaments were irregularly shaped as well as shorter and thicker than wild-type filaments (Fig. 6), indicating that Khd4 influences the growth and morphology of filaments. Strains expressing Khd4m3GT and Khd4m4GT were affected in filament formation to the same extent as AB33khd4Δ (Fig. 6), indicating that AUACCC-binding KH domains 3 and 4 are crucial for regulation of morphology as well as filamentation.
FIGURE 6.

KH domains 3 and 4 are functionally important for regulation of filamentous growth. (A) Microscopic analysis of AB33 and derivatives 6 h after induction of filamentous growth (size bar, 10 μm). (B) Bar diagram showing percentage of filaments within different size categories given at the top (at least 29 cells of each strain were analyzed). (C) Bar diagram indicating the diameter of filaments (at least 10 cells of each strain were measured at three different positions within the filament).
Mutations in KH domain 3 drastically affect pathogenic development
In order to address whether AUACCC binding of Khd4 has an impact on virulence, we introduced alleles khd4m3GT and khd4m5GT in the homologous locus of solopathogenic strain SG200R (SG200 variant expressing cytoplasmic mRfp, monomeric version of the red fluorescent protein) (Bölker et al. 1995; Campbell et al. 2002; Fuchs et al. 2006). This strain enables plant infection experiments without prior mating. SG200khd4Δ served as control (Becht et al. 2005). Filamentous growth was scored on medium plates containing activated charcoal. SG200R formed b-dependent filaments, resulting in the formation of fuzzy colonies (Fig. 7A). Consistent with our results obtained in the AB33 background (see above), filament formation was impaired in SG200khd4Δ and SG200khd4m3GT, but not in SG200khd4m5GT (Fig. 7A). Plant infection experiments revealed that SG200R and SG200khd4m5GT caused comparable disease symptoms on corn seedlings. Two weeks after infection, the majority of infected plants showed typical disease symptoms including ligula swelling, small and large tumors, as well as plant death (Fig. 7B,C). In contrast, infections with strains SG200khd4Δ or SG200khd4m3GT caused only minor disease symptoms such as chlorosis (Fig. 7B,C). Thus, the same mutations that abolished AUACCC binding in vivo also caused a drastic reduction in plant infection. Taken together, these results indicate that mutations in KH domains 3 and 4, which interact with AUACCC in vivo, cause defects in regulation of morphology, filamentous growth, and pathogenicity.
FIGURE 7.

Mutation of KH domain 3 causes a drastic reduction in virulence. (A) Filamentous growth of SG200R and derivatives expressing different Khd4 variants on plates containing charcoal. Inverse images of colony edges are shown (arrowheads point to protruding filaments). (B) Leaves of corn seedlings infected with SG200R and SG200 derivatives expressing different Khd4 variants. Examples of tumors are indicated by arrowheads. (C) Results of plant infection experiments with SG200R and SG200 derivatives expressing different Khd4 variants (two independent cultures per strain were tested). The percentage of plants with typical disease symptoms is given (two experiments, at least 85 plants infected with each strain).
DISCUSSION
Analysis of post-transcriptional gene regulation in pathogenic fungi is still in its infancy (Feldbrügge et al. 2008). Currently, only a few critical RNA-binding proteins have been identified, such as the RRM (RNA recognition motif) proteins Col1 and Rrm4 from the elm pathogen Ophiostoma novo-ulmi and U. maydis, respectively. Deletion of the corresponding genes leads to reduced virulence in both fungi (Pereira et al. 2000; Becht et al. 2005). A key step toward understanding the function of these regulatory factors is a detailed knowledge of their RNA binding capacity. Here, we report the functional characterization of Khd4, a multi-KH domain protein that is the founding member of a novel class of multi-KH domain proteins present in a number of fungal pathogens such as C. neoformans and C. albicans.
Khd4 recognizes AUACCC in vivo via tandem KH domains 3 and 4
Based on the presence of multiple KH domains, Khd4 was predicted to be an RNA-binding protein. Deletion mutants were affected in several steps of the life cycle including filamentous growth and tumor formation (Becht et al. 2005). Thus, for functional analysis of Khd4 it was mandatory to verify RNA binding, uncover its specificity, and determine how RNA binding contributes to cellular function. As a first step to answer these questions, we applied the reverse yeast three-hybrid system. This powerful technique enables investigation of interactions between RNA and proteins in vivo (SenGupta et al. 1996, 1999; Stumpf et al. 2008) as well as mapping of the binding domain at ease (König et al. 2007). We discovered that the sequence AUACCC is necessary and sufficient for binding by Khd4. Moreover, tandem KH domains 3 and 4 are crucial for this interaction. These observations are consistent with recent results from Saccharomyces cerevisiae. Bioinformatic analysis of mRNAs, which were co-purified with KH domain-containing protein Yll032C, predicted the consensus binding sequence aAUACCc (Hogan et al. 2008). Interestingly, Yll032C and Khd4 share substantial similarity in the region spanning tandem KH domains 3 and 4 of Khd4 (Fig. 1). However, comparison of the target mRNAs of Yll032C and Khd4 did not reveal any sets of overlapping functions, indicating that these proteins regulate different biological processes. Most KH domain proteins carry multiple copies of the RNA-binding domain that can either bind RNA independently or in a cooperative manner. For example, the two KH domains of bacterial NusA have extensive interdomain contacts and concertedly bind an extended segment of RNA (Valverde et al. 2008), whereas the four KH domains of KSRP have been shown to differ in their binding specificity (Garcia-Mayoral et al. 2008). These examples indicate that it is important to investigate the individual functions of the KH domains in a multi-KH domain protein. For Khd4, we observed that the central KH domains 3 and 4, but not 5, are crucial for binding of AUACCC as well as for regulation of morphology, filamentous growth, and pathogenicity. These observations indicate that the tandem KH domains might cooperate in binding. Consistently, tandem KH domains of the human poly(C)-binding protein, which is involved in translational regulation of folate receptor mRNA, cooperatively recognize a CCC-rich sequence motif (Du et al. 2008). Moreover, KH domains 3 and 4 of the mammalian zipcode-binding protein ZBP1, which contains two N-terminal RRMs and four C-terminal KH domains, bind the sequence ACACCC in vitro (Farina et al. 2003). The same tandem domains are functionally important for RNA localization, granule formation, and cytoskeletal association (Farina et al. 2003). Thus, interaction of A(U/C)ACCC binding sites with tandem KH domains might be a common recognition code in fungi and animals.
The interaction of Khd4 and AUACCC is functionally important in U. maydis
Based on two lines of evidence we propose that the interaction of Khd4 with AUACCC is of functional importance in U. maydis. First, Khd4 binding sites exhibit a strong positional bias toward the first 150 nt of 3′ UTRs. In accordance, 3′ UTRs function as regulatory regions in determining levels of gene expression in various organisms (Wilusz et al. 2001) and can even constitute the primary regulators of expression during germline development (Merritt et al. 2008). Most commonly, sequences in the 3′ UTR affect mRNA stability via interaction of the corresponding RNA-binding proteins with the degradation machinery (Houseley and Tollervey 2009). A significant enrichment for binding sites in the 3′ UTR has previously been reported for PUF family RNA-binding proteins (pumilio-Fem-3-binding factor) (Gerber et al. 2004, 2006; Galgano et al. 2008; Morris et al. 2008). Second, our genome-wide expression studies revealed that AUACCC-containing mRNAs were significantly enriched among differentially expressed transcripts in a khd4Δ strain. This is consistent with our observation that Khd4 interacts with AUACCC and suggests that this interaction influences the amount of target mRNAs. Expression of most differentially expressed transcripts was increased in the absence of Khd4. Hence, one of its functions might be the regulation of mRNA stability (see below). Such regulatory processes are intensively studied during mRNA turnover in higher eukaryotes (Shyu et al. 2008). A prime example is the interaction of RNA-binding proteins with the AU-rich element found in the 3′ UTR of short-lived cytokine and proto-oncogene mRNAs (Chen and Shyu 1995; Bakheet et al. 2001). A plethora of regulators have been described to function via this element. Some of these proteins (for example, TPP and KSRP) promote mRNA degradation, whereas others (like HuR) act antagonistically (Brennan and Steitz 2001; Blackshear 2002; Gherzi et al. 2004). Thus, the amount of target mRNAs at any given point in time is determined by the interplay of different protein effectors. Overall, the motif AUACCC is present in 1581 predicted transcripts of U. maydis. Why is only a small subset of AUACCC-containing transcripts affected in the khd4Δ strain? A parsimonious explanation is that the presence of AUACCC on its own is not sufficient to influence mRNA stability. The simultaneous binding of several factors to a single mRNA might be needed to build a network which determines the complete regulatory response (Hogan et al. 2008). In this context, it is interesting to note that, in S. cerevisiae, only 54 out of 1607 transcripts that carry the predicted binding site of Yll032C were found to be associated with the protein (Hogan et al. 2008). Thus, additional factors might determine the binding capacity and specificity of these RNA-binding proteins. However, we cannot exclude that Khd4 carries out additional regulatory functions at the level of translation or localization (see below).
What is the cellular role of Khd4?
In the present study, we describe the following key aspects of Khd4 function: The tandem KH domains that recognize AUACCC are essential for function, the protein predominantly localizes to the cytoplasm, and the vast majority of differentially expressed transcripts in khd4Δ exhibit increased mRNA levels. Furthermore, loss of Khd4 causes defects in cell shape, cytokinesis, filamentous growth, and pathogenicity. Based on these results we hypothesize that Khd4 regulates expression of morphology determinants or pathogenicity factors at the post-transcriptional level. Potential Khd4 target transcripts encode transcription and splice factors as well as 14 proteins with predicted enzymatic functions. Thus, a distinct transcription or splice program or tight regulation of certain enzymes might be essential for the underlying cellular processes. At present, it is still possible that the role of Khd4 during regulation of mRNA instability is not the prime cause for the mutant phenotype. As mentioned above, Khd4 might function at the level of translational regulation. Consequently, some target genes may not have been identified during mRNA expression profiling. This hypothesis is consistent with recent findings obtained for Khd1 from S. cerevisiae. This KH domain protein recognizes its target transcripts preferentially via binding sites within the ORF and affects mRNA stability of some targets as well as mediates translational control of others (Hasegawa et al. 2008). Promising candidate mRNAs for Khd4 that contain AUACCC, but were not detected as differentially expressed in our microarray analysis, are cdc42, yup1, and spa2, encoding a small G protein, a putative t-SNARE, and a polarisome component, respectively (Wedlich-Söldner et al. 2000; Mahlert et al. 2006; Carbo and Perez-Martin 2008). Deletion of cdc42 leads to defects in cytokinesis and virulence (Mahlert et al. 2006), loss-of-function mutants in yup1 are affected in cell morphology and virulence (Wedlich-Söldner et al. 2000; Fuchs et al. 2006), and spa2Δ strains exhibit increased cell diameters and form shorter filaments, albeit deletion strains are fully pathogenic (Carbo and Perez-Martin 2008). Another candidate for Khd4 regulation is AUACCC-containing mRNA clp1 encoding a key factor of clamp formation, which is essential for pathogenicity. Consistently, Clp1 expression is regulated most likely at the translational level during plant penetration (Scherer et al. 2006). A more detailed analysis of Khd4 targets will be critical to address these questions. In the future, it will be crucial to further elucidate the function of RNA-binding proteins during pathogenicity for a deeper understanding of fungal infection.
MATERIALS AND METHODS
Strains and growth conditions
Escherichia coli K-12 derivates DH5α (Bethesda Research Laboratories) and Top10 (Invitrogen) were used for cloning purposes. The yeast strain L40-coat with the following genotype was used: MATa, ura3–52, leu2–3112, his3Δ200, trp1Δ1, ade2, LexA–MS2coat (TRP1), LYS2∷lexA-op–HIS3, and ura3∷lexA-op–lacZ (SenGupta et al. 1996). Growth conditions for U. maydis strains and source of antibiotics were described previously (Brachmann et al. 2004). U. maydis strains were constructed by transformation of progenitor strains with linear DNA fragments (Supplemental Table S3). Homologous integration events were verified by Southern blot analysis (Brachmann et al. 2004). Filamentous growth of AB33 derivates was induced by shifting cells of an exponential growing culture (OD600 = 0.4–0.5) from liquid complete medium (CM) to nitrate minimal medium (NM). Cells were incubated at 28°C with shaking at 200 rpm. Growth on charcoal-containing CM plates was performed as described in Becht et al. (2005).
Plasmids and plasmid constructions
Plasmids pCR2.1- and pCRII-Topo for cloning (Invitrogen), and pACTIIA as well as pIIIMS2-1 for the yeast three-hybrid analysis were used (SenGupta et al. 1996; König et al. 2007). Plasmid constructions are described in the Supplemental Materials and Methods. All constructions were confirmed by sequencing. All plasmid sequences are available upon request.
Reverse yeast three-hybrid system
Genomic DNA of U. maydis strain 521 was enzymatically cleaved using AciI, BsaHI, HinP1I, HpaII, and TaqI individually as well as in pairwise combinations generating DNA fragments with cohesive 3′ ends (GC). These were size-selected for 50–250 bp using agarose gel electrophoresis followed by electroelution and ligation with vector pIIIMS2-4 (König et al. 2007), which was linearized with ClaI and dephosphorylated. Upon transformation in E. coli, plasmid DNA was extracted from a total of 1.5 × 106 transformants to obtain the library. PCR analysis of 96 single transformants confirmed that >99% of the plasmids contained an insert fragment (data not shown). Using the estimate P = 1 − (1 − f/g)n, according to Clarke and Carbon (1976), suggested that a certain sequence should be included in the library with the probability P = 99.6% (f, fragment size, 150 bp; g, 2× genome size, 4 × 107 bp; n, number of obtained transformants, 1.5 × 106). Reverse three-hybrid analysis was carried out using yeast strain L40-coat harboring genomic integrations of both reporter genes and the gene encoding hybrid protein 1 (SenGupta et al. 1996) as well as plasmid pkhd41–1218G-ACTIIA. Markers for selection during transformation and for detection of plasmid loss were applied as previously described (SenGupta et al. 1999; König et al. 2007). Transformants were incubated on selection plates containing synthetic complete medium lacking amino acids uracil and leucin (SC –ura –leu, 2 d, 28°C) to select for the presence of both plasmids. 4 × 108 colonies were pooled and incubated on SC plates lacking histidine and containing 5 mM 3-aminotriazole (SC –his + 5 mM 3-AT; an inhibitor of His3p). Ninety-eight clones were further analyzed upon plasmid rescue and retransformation for expression of both plasmids, grown on SC –his + 5 mM 3-AT, and β-galactosidase activity, which was determined using standard filter assays with X-gal as chromogenic substrate (Bernstein et al. 2002). Inserts of 73 verified candidates were PCR-amplified using oligonucleotides oMF713 and oMF725 and sequenced. In order to analyze the binding of Khd4 to different sequences (S35, S35a, S35b, S35*), respective plasmids were co-transformed with pkhd41–1218G-ACTIIA in L40-coat and selected as described before. For dilution drop assays, respective clones grown in liquid SC –ura –leu medium (OD600 ∼1) were diluted in four steps in water (1:5) starting at OD600 = 0.5. Drops of 4 μL were spotted on SC –ura –leu as well as SC –his plates and incubated for 2 d at 28°C.
Fluorimetric measurements of β-galactosidase activity
Fluorimetric measurements were performed as described previously (König et al. 2007). Two independent experiments were performed and representative results are shown.
DNA microarray analysis
Strains were grown to an OD600 of 0.3 at 28°C in liquid minimal array medium containing glucose and glutamine (Scherer et al. 2006; Zarnack et al. 2008). RNA isolation, sample preparation, and hybridization were performed as previously described (Scherer et al. 2006; Zarnack et al. 2008). Custom-designed Affymetrix chips based on the U. maydis genome sequence were used (Kämper et al. 2006; Scherer et al. 2006). Initial data analysis was performed using the Affymetrix Micro Array Suite 5.0 software. Quality of replicates was assessed calculating Pearson correlation coefficients using Excel (Microsoft Office). Coefficients were 0.989 and 0.984 (values >0.985 indicating high reproducibility; Zakharkin et al. 2005) for replicate cultures of strain FB2 and two independent transformants of strain FB2khd4Δ, respectively. mRNA expression profiles and respective comparisons performed in this study are listed in Supplemental Table S2 (GEO accession number GSE17956; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE17956). For statistical analysis software dChip1.3 (Li and Hung Wong 2001) was used to calculate mean expression values and fold changes based on a 90% confidence interval (“lower bound of fold change”) allowing a conservative evaluation of fold change. Induced and repressed genes were filtered according to a fold change of at least twofold and minus twofold, respectively, and a minimal mean expression difference of 50 between the compared conditions. As second criterion, differential expression was considered significant, if adjusted P-values were below a threshold of 0.05.
Nucleic acid procedures
Transformation and DNA as well as RNA isolation from U. maydis were performed as described previously (Brachmann et al. 2004). For Northern analysis, total RNA (15 μg/lane) as well as purified mRNA (∼1.5 μg; see below) were separated on MOPS-buffered 1% agarose gels and transferred to Amersham Hybond-XL membranes (GE Healthcare). For detection of um03706.2, poly(A)-mRNA was purified using Oligotex mRNA purification kit (Qiagen) according to the manufacturer's instructions. For Northern analysis, double-stranded probes were amplified with the following oligonucleotide combinations: oSL640 and oSL641 for um00748 (552 bp), oSL642 and oSL643 for um12098 (457 bp), oSL541 and oSL542 for um03706.2 (561 bp), and oSL547 and oSL548 for actin (um05715; 1136 bp).
Microscopy, image processing, and quantitative analysis
Cell suspensions were dropped on glass slides covered by a thin layer of agarose (2% w/v) and analyzed according to König et al. (2009). For quantitative analysis of cell shape and cell aggregates, two independent experiments were performed analyzing at least 80 cells each. In case of filament length and diameter (measured at three different locations), at least 29 filaments and 10 filaments were analyzed, respectively.
Plant infections
Plant infections of corn variety Early Golden Bantam (Olds Seeds) were performed as described previously (Brachmann et al. 2003). Tumor formation was scored after 12 to 14 d. The disease rating was performed using the following categories: no symptoms, chlorosis, ligula swelling, small tumors (<5 mm), large tumors (>5 mm), as well as wilted or dead plants.
Bioinformatics
In order to identify homologous proteins of Khd4 in other fungi (C. neoformans, A. fumigatus, C. albicans, S. cerevisiae, S. pombe), the amino acid sequence obtained from MUMDB (http://mips.gsf.de/genre/proj/ustilago/) was used in NCBI Blast (http://blast.ncbi.nlm.nih.gov/Blast.cgi; Altschul et al. 1990). CNBA5960, XP755682, XP723216, Yll032C, and NP595755 were aligned against Khd4 using CLUSTAL_X (Thompson et al. 1997; amino acids 746 to 891 of Khd4 are shown in Fig. 1). For identifying KH domains, the amino acid sequences of all identified proteins were analyzed using SMART database (http://smart.embl-heidelberg.de/; Schultz et al. 1998; Letunic et al. 2004). In order to identify positions of AUACCC, AGAUCU, and GGGUAU in transcripts, 5′ and 3′ UTRs were defined as 300 nt upstream of the start codon and downstream from the stop codon, respectively. For analysis of positional effects, the positions of motifs within transcripts were correlated to log2 of fold changes (corresponding to a lower bound of a 90% confidence interval). Transcripts were included that showed a minimal mean expression value of >50 in either the wild-type and/or khd4 deletion strain (at least 580 data points for each sequences tested). In some cases of small ORFs (<600 bp), motif positions overlapped in the 300-nt window downstream from the start codon with the one upstream of the stop codon. In these incidences, the positions were only plotted for the first window.
Empirical cumulative distribution plots and Wilcoxon rank sum test calculations were performed using R 2.7.1 (Hornik 2009; http://www.r-project.org/; ecdfplot{latticeExtra}). P-values were calculated against even distribution accepting a significance threshold of <0.01.
SUPPLEMENTAL MATERIAL
Supplemental material can be found at http://www.rnajournal.org.
ACKNOWLEDGMENTS
We thank the laboratory members for valuable discussions and Dr. P. Higgs for a critical review of the manuscript. Special thanks are given to T. Pohlmann, for his initial work on the yeast three-hybrid system, and P. Happel, for excellent technical assistance. We are grateful to Dr. M. Wickens for the generous gift of the yeast three-hybrid system. This work was supported by funds from Bayer CropScience, the MPG, and BMBF 0312738, for genome sequence and DNA microarrays, respectively.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.1817609.
REFERENCES
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Williams N, Misleh C, Li WW. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006;34:W369–W373. doi: 10.1093/nar/gkl198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakheet T, Frevel M, Williams BR, Greer W, Khabar KS. ARED: Human AU-rich element-containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res. 2001;29:246–254. doi: 10.1093/nar/29.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht P, Vollmeister E, Feldbrügge M. Role for RNA-binding proteins implicated in pathogenic development of Ustilago maydis. Euk Cell. 2005;4:121–133. doi: 10.1128/EC.4.1.121-133.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht P, König J, Feldbrügge M. The RNA-binding protein Rrm4 is essential for polarity in Ustilago maydis and shuttles along microtubules. J Cell Sci. 2006;119:4964–4973. doi: 10.1242/jcs.03287. [DOI] [PubMed] [Google Scholar]
- Bernstein DS, Buter N, Stumpf C, Wickens M. Analyzing mRNA–protein complexes using a yeast three-hybrid system. Methods. 2002;26:123–141. doi: 10.1016/S1046-2023(02)00015-4. [DOI] [PubMed] [Google Scholar]
- Blackshear PJ. Tristetraprolin and other CCCH tandem zinc-finger proteins in the regulation of mRNA turnover. Biochem Soc Trans. 2002;30:945–952. doi: 10.1042/bst0300945. [DOI] [PubMed] [Google Scholar]
- Bölker M. Ustilago maydis—a valuable model system for the study of fungal dimorphism and virulence. Microbiology. 2001;147:1395–1401. doi: 10.1099/00221287-147-6-1395. [DOI] [PubMed] [Google Scholar]
- Bölker M, Genin S, Lehmler C, Kahmann R. Genetic regulation of mating, and dimorphism in Ustilago maydis. Can J Bot. 1995;73:320–325. [Google Scholar]
- Brachmann A, Weinzierl G, Kämper J, Kahmann R. Identification of genes in the bW/bE regulatory cascade in Ustilago maydis. Mol Microbiol. 2001;42:1047–1063. doi: 10.1046/j.1365-2958.2001.02699.x. [DOI] [PubMed] [Google Scholar]
- Brachmann A, Schirawski J, Müller P, Kahmann R. An unusual MAP kinase is required for efficient penetration of the plant surface by Ustilago maydis. EMBO J. 2003;22:2199–2210. doi: 10.1093/emboj/cdg198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann A, König J, Julius C, Feldbrügge M. A reverse genetic approach for generating gene replacement mutants in Ustilago maydis. Mol Gen Genom. 2004;272:216–226. doi: 10.1007/s00438-004-1047-z. [DOI] [PubMed] [Google Scholar]
- Brennan CM, Steitz JA. HuR and mRNA stability. Cell Mol Life Sci. 2001;58:266–277. doi: 10.1007/PL00000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbo N, Perez-Martin J. Spa2 is required for morphogenesis but it is dispensable for pathogenicity in the phytopathogenic fungus Ustilago maydis. Fungal Genet Biol. 2008;45:1315–1327. doi: 10.1016/j.fgb.2008.06.010. [DOI] [PubMed] [Google Scholar]
- Chen CY, Shyu AB. AU-rich elements: Characterization and importance in mRNA degradation. Trends Biochem Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- Clarke L, Carbon J. A colony bank containing synthetic Col El hybrid plasmids representative of the entire E. coli genome. Cell. 1976;9:91–99. doi: 10.1016/0092-8674(76)90055-6. [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: A sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol. 2002;3:195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- Du Z, Fenn S, Tjhen R, James TL. Structure of a construct of a human poly(C)-binding protein containing the first and second KH domains reveals insights into its regulatory mechanisms. J Biol Chem. 2008;283:28757–28766. doi: 10.1074/jbc.M803046200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina KL, Hüttelmaier S, Musunuru K, Darnell R, Singer RH. Two ZBP1 KH domains facilitate β-actin mRNA localization, granule formation, and cytoskeletal attachment. J Cell Biol. 2003;160:77–87. doi: 10.1083/jcb.200206003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldbrügge M, Kämper J, Steinberg G, Kahmann R. Regulation of mating and pathogenic development in Ustilago maydis. Curr Op Microbiol. 2004;7:666–672. doi: 10.1016/j.mib.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Feldbrügge M, Bölker M, Steinberg G, Kämper J, Kahmann R. Regulatory and structural netwoks orchestrating mating, dimorphism, cell shape, and pathogenesis in Ustilago maydis. In: Kües U, Fischer R, editors. The Mycota I: Growth, differentiation, and sexuality. Springer; Berlin: 2006. pp. 375–391. [Google Scholar]
- Feldbrügge M, Zarnack K, Vollmeister E, Baumann S, Koepke J, König J, Münsterkötter M, Mannhaupt G. The posttranscriptional machinery of Ustilago maydis. Fungal Genet Biol. 2008;45:S40–S46. doi: 10.1016/j.fgb.2008.03.013. [DOI] [PubMed] [Google Scholar]
- Fuchs U, Hause G, Schuchardt I, Steinberg G. Endocytosis is essential for pathogenic development in the corn smut fungus Ustilago maydis. Plant Cell. 2006;18:2066–2081. doi: 10.1105/tpc.105.039388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galgano A, Forrer M, Jaskiewicz L, Kanitz A, Zavolan M, Gerber AP. Comparative analysis of mRNA targets for human PUF-family proteins suggests extensive interaction with the miRNA regulatory system. PLoS One. 2008;3:e3164. doi: 10.1371/journal.pone.0003164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Mayoral MF, Diaz-Moreno I, Hollingworth D, Ramos A. The sequence selectivity of KSRP explains its flexibility in the recognition of the RNA targets. Nucleic Acids Res. 2008;36:5290–5296. doi: 10.1093/nar/gkn509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber AP, Herschlag D, Brown PO. Extensive association of functionally and cytotopically related mRNAs with Puf family RNA-binding proteins in yeast. PLoS Biol. 2004;2:e79. doi: 10.1371/journal.pbio.0020079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber AP, Luschnig S, Krasnow MA, Brown PO, Herschlag D. Genome-wide identification of mRNAs associated with the translational regulator PUMILIO in Drosophila melanogaster. Proc Natl Acad Sci. 2006;103:4487–4492. doi: 10.1073/pnas.0509260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherzi R, Lee KY, Briata P, Wegmüller D, Moroni C, Karin M, Chen CY. A KH domain RNA binding protein, KSRP, promotes ARE-directed mRNA turnover by recruiting the degradation machinery. Mol Cell. 2004;14:571–583. doi: 10.1016/j.molcel.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Hasegawa Y, Irie K, Gerber AP. Distinct roles for Khd1p in the localization and expression of bud-localized mRNAs in yeast. RNA. 2008;14:2333–2347. doi: 10.1261/rna.1016508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan DJ, Riordan DP, Gerber AP, Herschlag D, Brown PO. Diverse RNA-binding proteins interact with functionally related sets of RNAs, suggesting an extensive regulatory system. PLoS Biol. 2008;6:e255. doi: 10.1371/journal.pbio.0060255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook B, Bernstein D, Zhang B, Wickens M. RNA–protein interactions in the yeast three-hybrid system: Affinity, sensitivity, and enhanced library screening. RNA. 2005;11:227–233. doi: 10.1261/rna.7202705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornik K. The R FAQ. 2009. http://CRAN.R-project.org/doc/FAQ/R-FAQ.html.
- Houseley J, Tollervey D. The many pathways of RNA degradation. Cell. 2009;136:763–776. doi: 10.1016/j.cell.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Kahmann R, Kämper J. Ustilago maydis: How its biology relates to pathogenic development. New Phytol. 2004;164:31–42. doi: 10.1111/j.1469-8137.2004.01156.x. [DOI] [PubMed] [Google Scholar]
- Kämper J, Kahmann R, Bölker M, Ma LJ, Brefort T, Saville BJ, Banuett F, Kronstad JW, Gold SE, Müller O, et al. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature. 2006;444:97–101. doi: 10.1038/nature05248. [DOI] [PubMed] [Google Scholar]
- Keene JD. RNA regulons: Coordination of post-transcriptional events. Nat Rev Genet. 2007;8:533–543. doi: 10.1038/nrg2111. [DOI] [PubMed] [Google Scholar]
- König J, Julius C, Baumann S, Homann M, Göringer HU, Feldbrügge M. Combining SELEX and yeast three-hybrid system for in vivo selection and classification of RNA aptamers. RNA. 2007;13:614–622. doi: 10.1261/rna.334307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König J, Baumann S, Koepke J, Pohlmann T, Zarnack K, Feldbrügge M. The fungal RNA-binding protein Rrm4 mediates long-distance transport of ubi1 and rho3 mRNAs. EMBO J. 2009;28:1855–1866. doi: 10.1038/emboj.2009.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Copley RR, Schmidt S, Ciccarelli FD, Doerks T, Schultz J, Ponting CP, Bork P. SMART 4.0: Toward genomic data integration. Nucleic Acids Res. 2004;32:D142–D144. doi: 10.1093/nar/gkh088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Doerks T, Bork P. SMART 6: Recent updates and new developments. Nucleic Acids Res. 2009;37:D229–D232. doi: 10.1093/nar/gkn808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Hung Wong W. Model-based analysis of oligonucleotide arrays: Model validation, design issues and standard error application. Genome Biol. 2001;2:research0032.1-0032.11. doi: 10.1186/gb-2001-2-8-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlert M, Leveleki L, Hlubek A, Sandrock B, Bölker M. Rac1 and Cdc42 regulate hyphal growth and cytokinesis in the dimorphic fungus Ustilago maydis. Mol Microbiol. 2006;59:567–578. doi: 10.1111/j.1365-2958.2005.04952.x. [DOI] [PubMed] [Google Scholar]
- Merritt C, Rasoloson D, Ko D, Seydoux G. 3′ UTRs are the primary regulators of gene expression in the C. elegans germline. Curr Biol. 2008;18:1476–1482. doi: 10.1016/j.cub.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ. From birth to death: The complex lives of eukaryotic mRNAs. Science. 2005;309:1514–1518. doi: 10.1126/science.1111443. [DOI] [PubMed] [Google Scholar]
- Morris AR, Mukherjee N, Keene JD. Ribonomic analysis of human Pum1 reveals cis–trans conservation across species despite evolution of diverse mRNA target sets. Mol Cell Biol. 2008;28:4093–4103. doi: 10.1128/MCB.00155-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira V, Royer JC, Hintz WE, Field D, Bowden C, Kokurewicz K, Hubbes M, Horgen PA. A gene associated with filamentous growth in Ophiostoma novo-ulmi has RNA-binding motifs and is similar to a yeast gene involved in mRNA splicing. Curr Genet. 2000;37:94–103. doi: 10.1007/s002940050015. [DOI] [PubMed] [Google Scholar]
- Ruepp A, Zollner A, Maier D, Albermann K, Hani J, Mokrejs M, Tetko I, Guldener U, Mannhaupt G, Münsterkötter M, et al. The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res. 2004;32:5539–5545. doi: 10.1093/nar/gkh894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer M, Heimel K, Starke V, Kämper J. The Clp1 protein is required for clamp formation and pathogenic development of Ustilago maydis. Plant Cell. 2006;18:2388–2401. doi: 10.1105/tpc.106.043521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc Natl Acad Sci. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SenGupta DJ, Zhang B, Kraemer B, Pochart P, Fields S, Wickens M. A three-hybrid system to detect RNA–protein interactions in vivo. Proc Natl Acad Sci. 1996;93:8496–8501. doi: 10.1073/pnas.93.16.8496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SenGupta DJ, Wickens M, Fields S. Identification of RNAs that bind to a specific protein using the yeast three-hybrid system. RNA. 1999;5:596–601. doi: 10.1017/s1355838299002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyu AB, Wilkinson MF, van Hoof A. Messenger RNA regulation: To translate or to degrade. EMBO J. 2008;27:471–481. doi: 10.1038/sj.emboj.7601977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi H, Matunis MJ, Michael WM, Dreyfuss G. The pre-mRNA binding K protein contains a novel evolutionarily conserved motif. Nucleic Acids Res. 1993;21:1193–1198. doi: 10.1093/nar/21.5.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf CR, Opperman L, Wickens M. Analysis of RNA–protein interactions using a yeast three-hybrid system. Methods Enzymol. 2008;449:295–315. doi: 10.1016/S0076-6879(08)02414-2. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triezenberg SJ. Structure and function of transcriptional activation domains. Curr Opin Genet Dev. 1995;5:190–196. doi: 10.1016/0959-437x(95)80007-7. [DOI] [PubMed] [Google Scholar]
- Valverde R, Edwards L, Regan L. Structure and function of KH domains. FEBS J. 2008;275:2712–2726. doi: 10.1111/j.1742-4658.2008.06411.x. [DOI] [PubMed] [Google Scholar]
- Vanrobays E, Leplus A, Osheim YN, Beyer AL, Wacheul L, Lafontaine DL. TOR regulates the subcellular distribution of DIM2, a KH domain protein required for cotranscriptional ribosome assembly and pre-40S ribosome export. RNA. 2008;14:2061–2073. doi: 10.1261/rna.1176708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedlich-Söldner R, Bölker M, Kahmann R, Steinberg G. A putative endosomal t-SNARE links exo- and endocytosis in the phytopathogenic fungus Ustilago maydis. EMBO J. 2000;19:1974–1986. doi: 10.1093/emboj/19.9.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz CJ, Wormington M, Peltz SW. The cap-to-tail guide to mRNA turnover. Nat Rev Mol Cell Biol. 2001;2:237–246. doi: 10.1038/35067025. [DOI] [PubMed] [Google Scholar]
- Zakharkin SO, Kim K, Mehta T, Chen L, Barnes S, Scheirer KE, Parrish RS, Allison DB, Page GP. Sources of variation in Affymetrix microarray experiments. BMC Bioinformatics. 2005;6:214–225. doi: 10.1186/1471-2105-6-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarnack K, Eichhorn H, Kahmann R, Feldbrügge M. Pheromone-regulated target genes respond differentially to MAPK phosphorylation of transcription factor Prf1. Mol Microbiol. 2008;69:1041–1053. doi: 10.1111/j.1365-2958.2008.06345.x. [DOI] [PubMed] [Google Scholar]