

Abstract
BACKGROUND
Extraskeletal myxoid chondrosarcoma (EMC) is a genetically distinct sarcoma with a propensity for local recurrence and metastasis despite an indolent course. To the authors’ knowledge, there are limited data examining chemotherapy outcomes as a guide to therapeutic decisions for unresectable disease.
METHODS
The clinical behavior and treatment responses of 87 patients with EMC who were seen at 2 institutions between 1975 and 2008 were examined.
RESULTS
The median age of the patients at the time of diagnosis was 49.5 years, with a male-to-female ratio of 2:1. For patients presenting without metastases, 37% developed local recurrence (median time of 3.3 years) and 26% developed distal recurrence (median time of 3.2 years). Approximately 13% of patients presented with metastases. The 5-year, 10-year, and 15-year overall survival rates were 82%, 65%, and 58%, respectively. Twenty-one patients received 32 evaluable courses of chemotherapy. No significant radiologic or clinical responses were noted. The median time to disease progression while receiving chemotherapy was 5.2 months. The best physician-assessed response to chemotherapy was stable disease for at least 6 months in 25% of patients, stable disease for <6 months in 41% of patients, and disease progression in 34% of patients. The estimated progression-free survival rates at 3 months, 4 months, 6 months, and 9 months were 69%, 65%, 40%, and 26%, respectively.
CONCLUSIONS
This retrospective review highlights the poor response rate to chemotherapy and emphasizes aggressive control of localized disease as the primary approach to management. Although there are biases inherent in retrospective analyses, these data provide a benchmark for time to disease progression for the study of new agents for the treatment of patients with this diagnosis.
Keywords: soft-tissue sarcoma, extraskeletal myxoid chondrosarcoma, retrospective analysis, chemotherapy, chromosomal translocation
Extraskeletal myxoid chondrosarcoma (EMC) is a rare soft-tissue malignancy that is distinguished from other sarcomas by its unique histology and a characteristic chromosomal translocation, typically t(9;22)(q22;q12.2), fusing EWSR1 to NR4A3 (genes formerly termed EWS and CHN, TEC, or NOR1, respectively).1–3 A small proportion of EMC have a different translocation, t(9;17)(q22;q11.2), which results in a RBP56-NR4A3 fusion gene and neuroendocrine differentiation in some cases.4 The chromosomal translocations result in fusion gene products responsible for alterations in cellular growth and differentiation.5
Histologically, the tumor has a vague resemblance to human cartilage, but with a marked phenotypic plasticity that overlaps with other mesenchymal malignancies,6 and an uncertain histogenesis.7–9 Since its recognition as a distinct clinicopathologic entity, the debate regarding tumor behavior has shifted toward an understanding of its nature as an intermediate-grade rather than a low-grade neoplasm.10 This idea is substantiated by studies with long median follow-up that demonstrate a high rate of local tumor recurrence and distant spread despite a prolonged clinical course.11–13
Currently, the only curative option for EMC is early wide local resection with or without radiation for localized disease.13,14 To our knowledge, few published data exist to date examining responses to systemic therapy for disseminated disease.15 We therefore assembled a retrospective series of patients from 2 large referral centers to examine treatment outcomes and progression-free survival (PFS) with systemic therapy. Understanding that we would find little activity from chemotherapy, our goal was to provide a metric for metastatic EMC patients that can be used as a comparator for studies of new agents.
MATERIALS AND METHODS
Patient Selection
After obtaining Institutional Review Board permission, 86 patients of EMC were retrieved from the databases of the Memorial Sloan-Kettering Cancer Center in New York City and the Royal Marsden Hospital in London. Patients were seen and treated at each institution between 1975 and 2006; approximately two-thirds of patients examined were diagnosed after 1995. Patients with a definite diagnosis of EMC by expert pathologic review of the primary tumor were included in the study. In situations in which a clear diagnosis was not established on pathology, confirmation was made by reverse-transcriptase polymerase chain reaction (RT-PCR) or fluorescence in situ hybridization (FISH) analysis for the EWSR1-NR4A3 translocation t(9;22). Data were censored on March 1, 2008.
Demographics and Statistical Methods
Demographic data examined as part of this retrospective study included patient age at diagnosis, patient sex, tumor location, and primary tumor size. Pathology and surgical reports were used to determine the completeness of resection (R0: complete resection, negative margins; R1, complete macroscopic resection, positive microscopic margins; and R2, incomplete resection). Estimates of survival with the endpoints of local, distant, and any recurrence; metastasis, and overall survival (OS) were determined by Kaplan-Meier analysis using the date of wide local excision (WLE) as Time 0, irrespective of previous procedures. The prognostic significance of sex (male or female), age (<50 years or ≥50 years), tumor site (located in the extremities or in other sites), and tumor size (<5 cm, 5–10 cm, or >10 cm) in terms of local, distant, and any recurrence was determined with a univariate analysis using the log-rank test. A multivariate analysis of local recurrence and metastatic disease in addition to the above factors affecting OS was performed using the Cox proportional hazards model after a similar univariate analysis.
A response to systemic treatment was judged by either radiographic (World Health Organization [WHO] or Response Evaluation Criteria for Solid Tumors [RECIST]) or clinical evidence of change in tumor size. PFS was examined as the primary endpoint for treatment with data censored for toxicity.
RESULTS
Clinical Features of Local and Metastatic EMC
Of the 86 evaluable patients, 57 were men and 29 were women, for a male-to-female ratio of 2:1. The median age at diagnosis was 49.5 years (range, 15–82 years) (Fig. 1). Approximately 50% of the patients (44 of 86 patients) presented between the ages of 41 and 60 years. Approximately 62% had a primary site in the lower extremities; 17% in the upper extremities; 13% in the abdomen, retroperitoneum, or pelvis; and 8% in other areas (Table 1). The size of the primary tumor ranged from 1 to 30 cm (median, 6.8 cm) in patients with documented measurements (n = 68).
FIGURE 1.
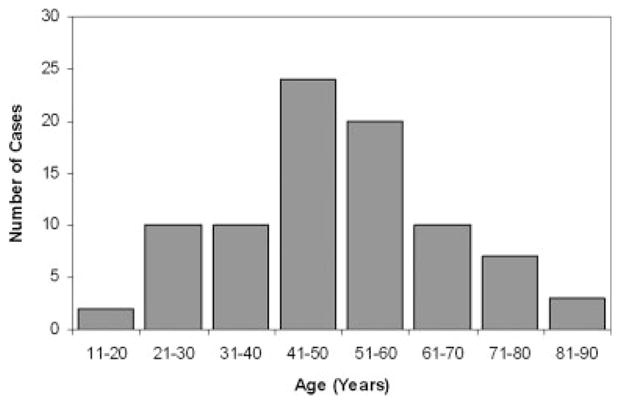
Age distribution at the time of diagnosis of 86 patients of extraskeletal myxoid chondrosarcoma.
TABLE 1.
Anatomic Distribution of the Primary Tumor in 86 Patients of EMC
| No. of Patients | Percentage of Patients | M/F, No. | Median Age, Years | Range | |
|---|---|---|---|---|---|
| Lower extremity | 53 | 62% | 34/19 | 47 | 15–82 |
| Upper extremity | 15 | 17% | 11/4 | 46 | 29–72 |
| Abdomen/retroperitoneum/pelvis | 11 | 13% | 7/4 | 54 | 20–73 |
| Other* | 7 | 8% | 5/2 | 58 | 30–76 |
EMC indicates extraskeletal myxoid chondrosarcoma; M/F, male/female.
“Other” includes 5 patients presenting in the trunk (chest wall and abdominal wall), 1 in the intrathoracic region, and 1 head and neck primary tumor.
Approximately 87% (76 of 86 patients) of patients presented with primary localized disease. The median follow-up from the initial date of diagnosis was 3.6 years. A total of 34% of patients were either diagnosed with or progressed to metastatic disease. The first site of metastasis was the lung in 80% of cases (63% with metastasis confined to the lungs and 17% with metastasis at other sites concurrent with lung disease). Other metastatic sites were observed in 20% of patients. The median survival time from the diagnosis of metastatic disease was 1.7 years.
Treatment Outcomes for Localized Disease
Seventy-six patients presented with primarily localized disease, 73 of whom received subsequent treatment with curative intent. Of these patients, 70% (n = 51) had wide local excision only as primary therapy, whereas 30% (n = 22 patients) underwent both wide excision and radiotherapy in the form of neoadjuvant, intraoperative, or adjuvant therapy. Recurrence rates for surgery with and without radiation are detailed in Table 2.
TABLE 2.
Recurrence Rates After Primary Therapy for 73 Patients of Localized EMC
| Primary Therapy With Curative Intent: Surgery ± RT (n=73) |
||||||
|---|---|---|---|---|---|---|
| Median Time to Disease Recurrence, Years |
Recurrence-Free Survival |
|||||
| Type of Recurrence | Surgery Alone (n=51) | Surgery + RT (n=22) | 5 Years | 10 Years | ||
| Local | 35% (18) | 41% (9) | 37% (27) | 3.3 | 57% | 31% |
| Distant | 20% (10) | 41% (9) | 26% (19) | 3.2 | 71% | 58% |
| Any | 45% (23) | 59% (13) | 49% (36) | 3.2 | 42% | 21% |
EMC indicates extraskeletal myxoid chondrosarcoma; RT, radiotherapy.
For the analysis of recurrence-free survival, recurrence from the date of WLE was used, regardless of whether patients received radiotherapy (P =.79, Fisher exact test). Local disease recurrence was noted in 37% of patients (n = 27) who received primary therapy with curative intent (surgery with or without radiation), with a median time to disease recurrence of 3.3 years. The median local PFS was 7.0 years (95% confidence interval [95% CI], 4.4 years–9.6 years). Local recurrence-free survival was 57% at 5 years and 31% at 10 years (Fig. 2A). Patients receiving radiotherapy had a trend toward a higher risk of distant disease recurrence (P =.08, Fisher exact test), suggesting that patients with more aggressive tumors were selected for radiotherapy. In a subset of 43 patients in whom there were data regarding quality of the resection (R0, R1, and R2), only 2 of 24 patients who underwent an R0 resection developed local disease recurrence, whereas 3 of 12 patients with an R1 resection and 5 of 7 with an R2 resection experienced local disease recurrence (P < .01, Fisher exact test).
FIGURE 2.
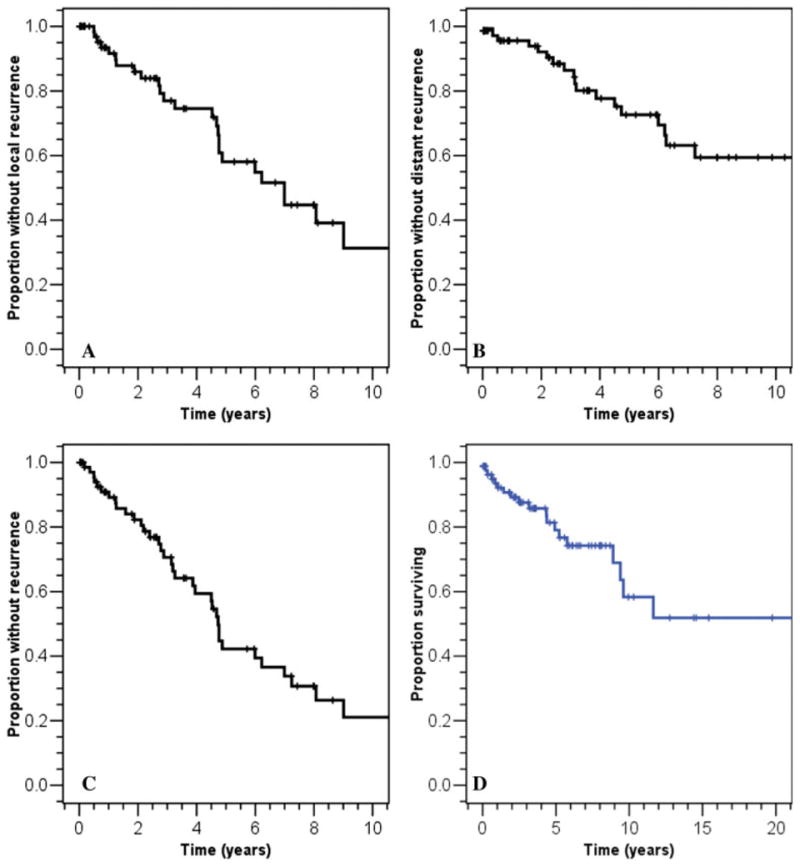
Survival curves for this patient cohort (n = 73 unless otherwise specified). (A) Local recurrence-free survival. (B) Distant recurrence-free survival. (C) Disease-free survival. (D) Overall survival (n = 86). Please note the different abscissa.
Distant disease recurrence was observed in 26% of patients (n = 19), with a median time of 3.2 years. The distant recurrence-free survival rate at 5 years and 10 years was 71% and 58%, respectively (Fig. 2B). The median distant time to disease progression was not reached at the time of last follow-up. Approximately 49% of patients experienced any form of disease recurrence (n = 36 patients). The disease-free survival rate in these patients was 42% at 5 years and 21% at 10 years (Fig. 2C). The median time to any disease recurrence was 4.7 years (95% CI, 4.4 years–5.0 years). By univariate analysis, the local, distant, and overall recurrence-free survival rates were not affected by age or sex. An extremity primary tumor location was found to be a favorable prognostic factor for local recurrence-free survival. Extremity and small primary tumor size were found to be marginally associated with a favorable risk for any recurrence (Table 3).
TABLE 3.
Univariate Analysis of Potential Prognostic Factors for Recurrence-free Survival*
| Local Disease Recurrence |
Distant Disease Recurrence |
Any Disease Recurrence |
|
|---|---|---|---|
| Factor | P | P | P |
| Age ≥50 y | .16 | .77 | .95 |
| Male gender | .95 | .23 | .95 |
| Extremity primary tumor | .005 | .85 | .051 |
| Primary tumor size <5 cm | .38 | .11 | .06 |
The number of patients was 73, except in the case of tumor size, for which documented measurements were available in only 53 patients.
Follow-up and OS
Follow-up data were available for all 86 patients studied, with a median follow-up time of 3.6 years (range, 0.2 years–24.6 years). At the time of last follow-up, 22 patients were alive with disease, 14 were dead of disease, and 4 were dead of other causes but with disease. Approximately 63% of patients were free of disease after primary therapy with curative intent (46 of 73 patients). Kaplan-Meier estimates of OS at 5, 10, and 15 years were 82%, 65%, and 58%, respectively (Fig. 2D). Adverse prognostic factors affecting OS that were identified on univariate analysis were size >10 cm (P =.005) and the presence of metastases (P =.001). On multivariate analysis, only presentation with metastatic disease was found to affect OS (P =.005). Median survival after the detection of metastases for the 28 patients with follow-up data was 17.8 months.
Chemotherapy Outcomes
Twenty-one of 86 EMC patients received a combined total of 39 courses of chemotherapy: 2 as neoadjuvant therapy, 3 as adjuvant therapy, 2 for local disease recurrence, and 32 for metastatic disease. Response data were available from 32 of these courses in 21 patients. Treatments were highly heterogeneous; 25 different regimens were administered, with doxorubicin-containing regimens comprising the largest subgroup (Table 4). The remaining regimens were comprised of conventional or experimental agents.
TABLE 4.
Best Radiologic or Clinical Response After 32 Courses of Chemotherapy for EMC
| Agent Classification | Agents | PD | SD <6 Months | SD ≥6 Months |
|---|---|---|---|---|
| Doxorubicin-containing (10 courses) | CyVADIC | 1 | ||
| Doxorubicin | 1 | 1 | ||
| Liposomal doxorubicin | 1 | |||
| Doxorubicin + cisplatin | 1 | 1 | ||
| Doxorubicin + ifosfamide | 2 | 1 | ||
| CHOP | 1 | |||
| Doxorubicin-containing | 3 | 5 | 2 | |
| Other agents (22 courses) | Ifosfamide | 1 | ||
| Ifosfamide + topotecan | 1 | |||
| Gemcitabine + docetaxel | 1 | 1 | 1 | |
| Etoposide | 1 | |||
| Etoposide + cisplatin | 1 | |||
| Gemcitabine | 1 | 1 | ||
| Imatinib | 1 | |||
| Interferon | 1 | |||
| Methotrexate | 1 | |||
| Sorafenib | 1 | |||
| Thalidomide | 1 | |||
| Investigational agents | 2 | 3 | 3 | |
| Other agents | 8 | 8 | 6 | |
| Total | All lines of therapy | 11 | 13 | 8 |
EMC indicates extraskeletal myxoid chondrosarcoma; PD, progressive disease; SD, stable disease; CyVADIC, cyclophosphamide, vincristine, doxorubicin, and dacarbazine; CHOP, cyclophosphamide, doxorubicin, vincristine, and prednisone.
No radiologic (according to RECIST or WHO criteria) complete responses (CRs) or partial responses (PRs) were noted. For doxorubicin-containing regimens, best objective response was stable disease (SD) lasting ≥6 months in 2 patients, SD lasting <6 months in 5 patients, and progressive disease (PD) in 3 patients. For the other regimens, best response was SD lasting ≥6 months in 6 patients, SD lasting <6 months in 8 patients, and PD in 8 patients. For all courses of therapy combined, 25% (8 of 32 patients) of patients had a best result of SD lasting ≥6 months, 41% (13 of 32 patients) had a best result of SD lasting <6 months, and 34% (11 of 32 patietns) had a best response of PD. Time to disease progression while receiving therapy was documented or could be determined by radiologic review in 29 of 32 courses of chemotherapy associated with an indication of response. The median PFS was 5.2 months (Kaplan-Meier 95% CI, 3.4 months–7.1 months). Estimated PFS rates (Kaplan-Meier) at 3 months, 4 months, 6 months, and 9 months were 69%, 65%, 40%, and 26%, respectively (Fig. 3).
FIGURE 3.
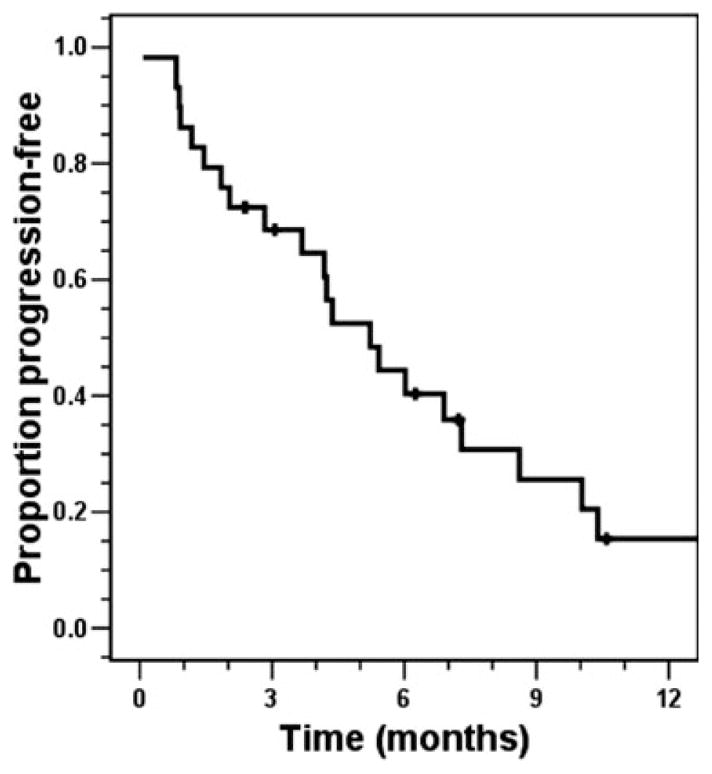
Progression-free survival from the time that chemotherapy was initiated.
DISCUSSION
EMC is distinguished by a biology that is distinct from the genetic heterogeneity observed in other forms of chondrosarcoma.13 The majority of patients are characterized by translocations that lead to abnormal gene products. The most common of these is t(9;22)(q22;q11), resulting in the juxtaposition of the genes EWSR1 (formerly called EWS) on chromosome 22 and NR4A3 (also termed TEC, NOR1, or CHN) on chromosome 9.16–18 Variant translocations have been described in recent years, notably t(9;17)(q22;q11) coding for NR4A3-TAF15,19–22 and t(9;15)(q22;q21), encoding NR4A3-TCF12/HTF4.23,24 These fusion products are postulated to modify transcriptional activity and regulate cellular differentiation and growth, leading to tumorigenesis.16–18,25 Ultrastructural studies of EMC have also uncovered evidence of markers of neuroendocrine differentiation7,8 such as class III β-tubulin and microtubule-associated protein-2.9 These findings argue against a chondrocytic or prechondrocytic origin of this malignancy, and further distinguish EMC as a unique entity among sarcomas.
The results of the current study are consistent with the demographics of EMC annotated in previous studies, with a predilection for a primary site in the extremities, and typical presentation during the fifth and sixth decades of life.12,26,27 Our follow-up was relatively short compared with other series, perhaps because of our difficulty in confirming diagnoses and finding complete records in patients treated before 1985, and patients lost to follow-up after successful surgery or death after the development of metastatic disease. A consistently high rate of local recurrence has been described for these tumors, ranging from 14% to 64% in some series,10–13,26–28 with progression to metastatic disease reported in up to 31% of patients.13
The French Federation of Cancer Centers (FNCLCC) staging system lists EMC as a grade 2/3 tumor29; the reported median survival of <18 months from the onset of metastasis is consistent with an intermediate-grade to high-grade sarcoma. The only satisfactory chance of cure of these tumors is surgical intervention in patients with localized disease.14 In a recent multi-institutional study by Kawaguchi et al,13 the role of WLE despite previous procedures or disease recurrence was emphasized as a means of reducing rates of local disease recurrence and improving OS.
To our knowledge to date, no chemotherapeutic agent or combination of agents has demonstrated a durable efficacy against EMC, despite a few reports of chemotherapeutic responses (Table 5). Patel et al15 observed no objective responses in 10 patients receiving a median of 4 cycles of therapy with what were largely doxorubicin-based and dacarbazine-based regimens. Saleh et al11 were similarly unable to document any significant responses in 7 patients treated with chemotherapy (in 1 case, a “slight and temporary response” was observed). McGrory et al10 found that 2 of 6 patients responded to multiagent chemotherapy for systemic disease (1 PR and 1 CR), and these patients were followed for up to 6 years without evidence of PD. Data regarding the agents used were unavailable. A study by Rubinger et al30 documented a PR to interferon-α-2b with stable residual disease reported to last for 12 months after the completion of therapy. Given these data, we believe that EMC has yet to display a consistent response to any known systemic therapy used for other soft-tissue sarcomas.
TABLE 5.
Outcomes With Chemotherapy for EMC
| Study (No. of Courses/No. of Patients) | CR | PR | SD | PD |
|---|---|---|---|---|
| Rubinger 199530 (1/1) | 0 | 1 | 0 | 0 |
| McGrory 200110 (6/6) | 1 | 1 | 0 | 4 |
| Saleh 199211 (7/7) | 0 | 0 | 0 | 7 |
| Patel 199515 (23/10) | 0 | 0 | 14 | 9 |
| Current study (32/21) | 0 | 0 | 21 | 11 |
EMC indicates extraskeletal myxoid chondrosarcoma; CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease.
Despite the lack of sufficient evidence of chemotherapeutic efficacy in EMC, recent investigations into tumor biology have uncovered novel potential approaches to systemic treatment. One strategy involves the modulation of pathways involving the EWSR1-NR4A3 fusion gene.31 Several targets downstream from the translocation protein include SGK1, a serine-threonine kinase that is up-regulated in EMC,32 PGA2, a prostaglandin that transactivates the EWSR1-NR4A3 fusion protein,33 and Six-3, a cofactor that activates the CHN gene, even in the absence of a translocation.34 Client proteins of hsp90 include steroid receptors, which are dependent on hsp90 for maintaining their conformation.35 Because NR4A3 is a member of this family of proteins, it may be a target for hsp90-directed therapy such as derivatives of geldanamycin, which is currently in clinical trials.
Gene profiling studies have recently uncovered an array of diagnostic markers for EMC, the most common of which include the genes DKK1 (involved in Wnt signaling), NMB, DNER, CLCN3, and DEF6.36 Other genes found to be overexpressed in EMC compared with other sarcomas highlighted the Wnt and Myc pathways as possible therapeutic targets, for which several molecules that interfere with signaling are currently being developed. Furthermore, in light of its high levels of expression in the same study, peroxisome proliferator-activated receptors-gamma (PPARG) was identified as another possible therapeutic target for small molecular inhibitors such as thiazolidinediones or O-arylmandelic acid.37
Such hypotheses are ripe for examination in cell lines transfected with the EWSR1-NR4A3 fusion gene, and could be examined in multicenter studies of patients with this rare diagnosis. Two examples of EMC-directed clinical trials are the study of the kinase inhibitor perifosine and the IGF1R antagonist antibody R1507 in patients with EMC and other diagnoses by the Sarcoma Alliance for Research through Collaboration cooperative group (SARC),38,39 the results of which are anticipated within the next 1 to 2 years. In these and future studies, we hope to see an improvement in the time to disease progression and survival data observed in the current study.
Acknowledgments
Supported by National Cancer Institute Program Project Grant P01-CA47179, spin4survival.org.
References
- 1.Sandberg AA. Genetics of chondrosarcoma and related tumors. Curr Opin Oncol. 2004;16:342–354. doi: 10.1097/01.cco.0000129678.72521.e5. [DOI] [PubMed] [Google Scholar]
- 2.Hirabayashi Y, Ishida T, Yoshida MA, et al. Translocation (9;22)(q22;q12). A recurrent chromosome abnormality in extraskeletal myxoid chondrosarcoma. Cancer Genet Cytogenet. 1995;81:33–37. doi: 10.1016/0165-4608(94)00201-0. [DOI] [PubMed] [Google Scholar]
- 3.Stenman G, Andersson H, Mandahl N, Meis-Kindblom JM, Kindblom LG. Translocation t(9;22)(q22;q12) is a primary cytogenetic abnormality in extraskeletal myxoid chondrosarcoma. Int J Cancer. 1995;62:398–402. doi: 10.1002/ijc.2910620407. [DOI] [PubMed] [Google Scholar]
- 4.Panagopoulos I, Mertens F, Isaksson M, et al. Molecular genetic characterization of the EWS/CHN and RBP56/CHN fusion genes in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer. 2002;35:340–352. doi: 10.1002/gcc.10127. [DOI] [PubMed] [Google Scholar]
- 5.Filion C, Labelle Y. The oncogenic fusion protein EWS/NOR-1 induces transformation of CFK2 chondrogenic cells. Exp Cell Res. 2004;297:585–592. doi: 10.1016/j.yexcr.2004.03.040. [DOI] [PubMed] [Google Scholar]
- 6.Oliveira AM, Nascimento AG. Phenotypic plasticity and prognostic factors in extraskeletal myxoid chondrosarcoma. Adv Anat Pathol. 2000;7:73–78. doi: 10.1097/00125480-200007020-00001. [DOI] [PubMed] [Google Scholar]
- 7.Aigner T, Oliveira AM, Nascimento AG. Extraskeletal myxoid chondrosarcomas do not show a chondrocytic phenotype. Mod Pathol. 2004;17:214–221. doi: 10.1038/modpathol.3800036. [DOI] [PubMed] [Google Scholar]
- 8.Goh YW, Spagnolo DV, Platten M, et al. Extraskeletal myxoid chondrosarcoma: a light microscopic, immunohistochemical, ultrastructural and immuno-ultrastructural study indicating neuroendocrine differentiation. Histopathology. 2001;39:514–524. doi: 10.1046/j.1365-2559.2001.01277.x. [DOI] [PubMed] [Google Scholar]
- 9.Hisaoka M, Okamoto S, Koyama S, et al. Microtubule-associated protein-2 and class III beta-tubulin are expressed in extraskeletal myxoid chondrosarcoma. Mod Pathol. 2003;16:453–459. doi: 10.1097/01.MP.0000067422.61241.64. [DOI] [PubMed] [Google Scholar]
- 10.McGrory JE, Rock MG, Nascimento AG, Oliveira AM. Extraskeletal myxoid chondrosarcoma. Clin Orthop Relat Res. 2001;(382):185–190. doi: 10.1097/00003086-200101000-00025. [DOI] [PubMed] [Google Scholar]
- 11.Saleh G, Evans HL, Ro JY, Ayala AG. Extraskeletal myxoid chondrosarcoma. A clinicopathologic study of ten patients with long-term follow-up. Cancer. 1992;70:2827–2830. doi: 10.1002/1097-0142(19921215)70:12<2827::aid-cncr2820701217>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 12.Meis-Kindblom JM, Bergh P, Gunterberg B, Kindblom LG. Extraskeletal myxoid chondrosarcoma: a reappraisal of its morphologic spectrum and prognostic factors based on 117 cases. Am J Surg Pathol. 1999;23:636–650. doi: 10.1097/00000478-199906000-00002. [DOI] [PubMed] [Google Scholar]
- 13.Kawaguchi S, Wada T, Nagoya S, et al. Extraskeletal myxoid chondrosarcoma: a multi-institutional study of 42 cases in Japan. Cancer. 2003;97:1285–1292. doi: 10.1002/cncr.11162. [DOI] [PubMed] [Google Scholar]
- 14.Enneking WF, Spanier SS, Goodman MA. A system for the surgical staging of musculoskeletal sarcoma. Clin Orthop Relat Res. 2003;415:4–18. doi: 10.1097/01.blo.0000093891.12372.0f. 1980. [DOI] [PubMed] [Google Scholar]
- 15.Patel SR, Burgess MA, Papadopoulos NE, Linke KA, Benjamin RS. Extraskeletal myxoid chondrosarcoma. Long-term experience with chemotherapy. Am J Clin Oncol. 1995;18:161–163. doi: 10.1097/00000421-199504000-00014. [DOI] [PubMed] [Google Scholar]
- 16.Hisaoka M, Ishida T, Imamura T, Hashimoto H. TFG is a novel fusion partner of NOR1 in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer. 2004;40:325–328. doi: 10.1002/gcc.20044. [DOI] [PubMed] [Google Scholar]
- 17.Maltais A, Filion C, Labelle Y. The AF2 domain of the orphan nuclear receptor TEC is essential for the transcriptional activity of the oncogenic fusion protein EWS/TEC. Cancer Lett. 2002;183:87–94. doi: 10.1016/s0304-3835(02)00104-0. [DOI] [PubMed] [Google Scholar]
- 18.Labelle Y, Bussieres J, Courjal F, Goldring MB. The EWS/TEC fusion protein encoded by the t(9;22) chromosomal translocation in human chondrosarcomas is a highly potent transcriptional activator. Oncogene. 1999;18:3303–3308. doi: 10.1038/sj.onc.1202675. [DOI] [PubMed] [Google Scholar]
- 19.Attwooll C, Oddi S, Cartwright P, et al. A novel repressive E2F6 complex containing the polycomb group protein, EPC1, that interacts with EZH2 in a proliferation-specific manner. J Biol Chem. 2005;280:1199–1208. doi: 10.1074/jbc.M412509200. [DOI] [PubMed] [Google Scholar]
- 20.Kim S, Lee J, Kim J. Regulation of oncogenic transcription factor hTAF(II)68-TEC activity by human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) Biochem J. 2007;404:197–206. doi: 10.1042/BJ20061297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Attwooll C, Tariq M, Harris M, Coyne JD, Telford N, Varley JM. Identification of a novel fusion gene involving hTA-FII68 and CHN from a t(9;17)(q22;q11.2) translocation in an extraskeletal myxoid chondrosarcoma. Oncogene. 1999;18:7599–7601. doi: 10.1038/sj.onc.1203156. [DOI] [PubMed] [Google Scholar]
- 22.Bjerkehagen B, Dietrich C, Reed W, et al. Extraskeletal myxoid chondrosarcoma: multimodal diagnosis and identification of a new cytogenetic subgroup characterized by t(9;17)(q22;q11) Virchows Arch. 1999;435:524–530. doi: 10.1007/s004280050437. [DOI] [PubMed] [Google Scholar]
- 23.Sjogren H, Wedell B, Meis-Kindblom JM, Kindblom LG, Stenman G. Fusion of the NH2-terminal domain of the basic helix-loop-helix protein TCF12 to TEC in extraskeletal myxoid chondrosarcoma with translocation t(9;15)(q22;q21) Cancer Res. 2000;60:6832–6835. [PubMed] [Google Scholar]
- 24.Gan TI, Rowen L, Nesbitt R, et al. Genomic organization of human TCF12 gene and spliced mRNA variants producing isoforms of transcription factor HTF4. Cytogenet Genome Res. 2002;98:245–248. doi: 10.1159/000071042. [DOI] [PubMed] [Google Scholar]
- 25.Ohkura N, Yaguchi H, Tsukada T, Yamaguchi K. The EWS/NOR1 fusion gene product gains a novel activity affecting pre-mRNA splicing. J Biol Chem. 2002;277:535–543. doi: 10.1074/jbc.M109018200. [DOI] [PubMed] [Google Scholar]
- 26.Okamoto S, Hisaoka M, Ishida T, et al. Extraskeletal myxoid chondrosarcoma: a clinicopathologic, immunohistochemical, and molecular analysis of 18 cases. Hum Pathol. 2001;32:1116–1124. doi: 10.1053/hupa.2001.28226. [DOI] [PubMed] [Google Scholar]
- 27.Oliveira AM, Sebo TJ, McGrory JE, Gaffey TA, Rock MG, Nascimento AG. Extraskeletal myxoid chondrosarcoma: a clinicopathologic, immunohistochemical, and ploidy analysis of 23 cases. Mod Pathol. 2000;13:900–908. doi: 10.1038/modpathol.3880161. [DOI] [PubMed] [Google Scholar]
- 28.Tsuneyoshi M, Enjoji M, Iwasaki H, Shinohara N. Extraskeletal myxoid chondrosarcoma—a clinicopathologic and electron microscopic study. Acta Pathol Jpn. 1981;31:439–447. doi: 10.1111/j.1440-1827.1981.tb01387.x. [DOI] [PubMed] [Google Scholar]
- 29.Guillou L, Coindre JM, Bonichon F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol. 1997;15:350–362. doi: 10.1200/JCO.1997.15.1.350. [DOI] [PubMed] [Google Scholar]
- 30.Rubinger M, Plenderleith IH, Lertzman M, Worth AJ. Metastatic extraskeletal myxoid chondrosarcoma. Successful therapy with interferon alfa-2b. Chest. 1995;108:281–282. doi: 10.1378/chest.108.1.281. [DOI] [PubMed] [Google Scholar]
- 31.Hisaoka M, Hashimoto H. Extraskeletal myxoid chondrosarcoma: updated clinicopathological and molecular genetic characteristics. Pathol Int. 2005;55:453–463. doi: 10.1111/j.1440-1827.2005.01853.x. [DOI] [PubMed] [Google Scholar]
- 32.Poulin H, Filion C, Ladanyi M, Labelle Y. Serum- and glucocorticoid-regulated kinase 1 (SGK1) induction by the EWS/NOR1(NR4A3) fusion protein. Biochem Biophys Res Commun. 2006;346:306–313. doi: 10.1016/j.bbrc.2006.05.134. [DOI] [PubMed] [Google Scholar]
- 33.Kagaya S, Ohkura N, Tsukada T, et al. Prostaglandin A2 acts as a transactivator for NOR1 (NR4A3) within the nuclear receptor superfamily. Biol Pharm Bull. 2005;28:1603–1607. doi: 10.1248/bpb.28.1603. [DOI] [PubMed] [Google Scholar]
- 34.Hisaoka M, Okamoto S, Yokoyama K, Hashimoto H. Coexpression of NOR1 and SIX3 proteins in extraskeletal myxoid chondrosarcomas without detectable NR4A3 fusion genes. Cancer Genet Cytogenet. 2004;152:101–107. doi: 10.1016/j.cancergencyto.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 35.Bagatell R, Khan O, Paine-Murrieta G, Taylor CW, Akinaga S, Whitesell L. Destabilization of steroid receptors by heat shock protein 90-binding drugs: a ligand-independent approach to hormonal therapy of breast cancer. Clin Cancer Res. 2001;7:2076–2084. [PubMed] [Google Scholar]
- 36.Subramanian S, West RB, Marinelli RJ, et al. The gene expression profile of extraskeletal myxoid chondrosarcoma. J Pathol. 2005;206:433–444. doi: 10.1002/path.1792. [DOI] [PubMed] [Google Scholar]
- 37.Koyama H, Boueres JK, Han W, et al. 5-Aryl thiazolidine-2,4-diones as selective PPARgamma agonists. Bioorg Med Chem Lett. 2003;13:1801–1804. doi: 10.1016/s0960-894x(03)00257-9. [DOI] [PubMed] [Google Scholar]
- 38.Sponsors and Collaborators: SARC (Sarcoma Alliance for Research through Collaboration) and AOI Pharma, Inc. [Accessed on October 6, 2008];Protocol title: A Trial of Perifosine in Patients With Chemo-Insensitive Sarcomas. 2008 2008 Available at: http://www.Clinicaltrials.gov. Clinical Trial Identifier NCT00401388.
- 39.Sponsors and Collaborators: SARC (Sarcoma Alliance for Research through Collaboration) and Hoffmann-La Roche, Inc. [Accessed on October 6, 2008];Protocol title: A Study of R1507 in Patients With Recurrent or Refractory Sarcoma. 2008 2008 Available at: http://www.Clinicaltrials.gov. Clinical Trial Identifier NCT00642941.