

Abstract
Inducible epidermal deletion of JunB and c-Jun in adult mice causes a psoriasis-like inflammatory skin disease. Increased levels of the proinflammatory cytokine TNFα play a major role in this phenotype. Here we define the underlying molecular mechanism using genetic mouse models. We show that Jun proteins control TNFα shedding in the epidermis by direct transcriptional activation of tissue inhibitor of metalloproteinase-3 (TIMP-3), an inhibitor of the TNFα-converting enzyme (TACE). TIMP-3 is down-regulated and TACE activity is specifically increased, leading to massive, cell-autonomous TNFα shedding upon loss of both JunB and c-Jun. Consequently, a prominent TNFα-dependent cytokine cascade is initiated in the epidermis, inducing severe skin inflammation and perinatal death of newborns from exhaustion of energy reservoirs such as glycogen and lipids. Importantly, this metabolic “cachectic” phenotype can be genetically rescued in a TNFR1-deficient background or by epidermis-specific re-expression of TIMP-3. These findings reveal that Jun proteins are essential physiological regulators of TNFα shedding by controlling the TIMP-3/TACE pathway. This novel mechanism describing how Jun proteins control skin inflammation offers potential targets for the treatment of skin pathologies associated with increased TNFα levels.
Keywords: Jun, AP-1, epidermis, TNFα, inflamation, TIMP-3
The epidermis provides a protective barrier at the body surface against harmful pathogens and prevents dehydration. This thin layer of stratified squamous epithelium rests on top of a basement membrane of extracellular matrix, separating it and its appendages from the underlying mesenchymally derived dermis. Composed mainly of keratinocytes, the epidermis possesses the capacity to undergo continuous self-renewal, which is sustained by epidermal stem cells (Jones et al. 2007; Fuchs and Horsley 2008). Skin homeostasis is maintained through a complex interplay of cytokines and growth factors, regulating proliferation and differentiation of keratinocytes (Szabowski et al. 2000; Werner and Smola 2001).
The dimeric transcription factor AP-1 controls multiple biological processes and is composed of basic region leucine zipper (bZIP) proteins. Regulation of AP-1 activity can occur at the level of transcription, mRNA turnover, protein stability, post-translational modifications, or interactions with other transcription factors (Eferl and Wagner 2003). In mammals, the most abundant AP-1 proteins are the Jun (c-Jun, JunB, and JunD) and Fos (c-Fos, FosB, Fra1, and Fra2) family members, which regulate gene expression at multiple levels. In the mouse or human epidermis, all Jun and Fos members are expressed in the basal layer, whereas they are differentially expressed in the overlaying epidermal sheets (Angel et al. 2001; Mehic et al. 2005). In the epidermis, AP-1 has been shown to regulate differentiation, carcinogenesis, UV response, photo-aging, and wound repair processes (Zenz and Wagner 2006; Ezhkova et al. 2009).
Using mouse models, we showed genetically that epidermal c-Jun modulates keratinocyte proliferation and differentiation through controlling EGFR and HB-EGF expression (Zenz et al. 2003). Moreover, mice lacking JunB in the epidermis develop a multiorgan disease, likely due to increased G-CSF and IL-6 expression (Meixner et al. 2008). Importantly, inducible epidermal deletion of both JunB and c-Jun in adult mice leads to a psoriasis-like disease and arthritis, in part mediated by increased expression of S100A8 and S100A9 proteins (Zenz et al. 2005; Gebhardt et al. 2006). In this model, increased expression of TNFα and TNFα-dependent cytokines contributes to disease development, since in the absence of TNFR1 the skin phenotype is improved and arthritis is prevented (Zenz et al. 2005).
TNFα is a pleiotropic proinflammatory cytokine produced by many cell types and is a central mediator of diverse cellular events, including cell proliferation, differentiation, apoptosis, and necrosis (Aggarwal 2003). TNFα was first identified as an inducer of cell death and cachexia, which is a TNFα-driven chronic disease (Beutler et al. 1985). The main characteristics are progressive body weight loss, anorexia, anemia, and net losses of protein, glycogen, and lipid, the main energy stores of the body, ultimately leading to death (Spiegelman and Hotamisligil 1993). TNFα can trigger, through the activation of NF-κB and AP-1, local expression of cytokines and chemokines such as IL-1, IL-6, IL-8, and INF-γ, which also contribute to the cachectic phenotype (Gelin et al. 1991; Matthys et al. 1991; Barton 2001).
The biological activity of TNFα is controlled mainly at the post-transcriptional level (Aggarwal 2003; Kollias 2005). TNFα is produced as a type II transmembrane protein (mTNFα/pro-TNFα) that is physiologically shed by the metalloprotease TNFα-converting enzyme (TACE) (Black et al. 1997; Moss et al. 1997; Overall and Blobel 2007; Murphy et al. 2008). TACE, also known as ADAM17 (a disintegrin and metalloprotease), is found in its processed and active form in most tissues, including the epidermis (Black 2002). Interestingly, TACE activity is specifically inhibited by TIMP-3, a member of the tissue inhibitor of metalloproteinase (TIMP) family and a highly potent TACE inhibitor (Amour et al. 1998; Mohammed et al. 2004; Wisniewska et al. 2008). In addition, TACE is also responsible for the cleavage at the cell membrane of many receptors and factors, such as TNFR1, EGFR, and HB-EGF, thereby regulating their activity (Peschon et al. 1998; Sunnarborg et al. 2002).
To molecularly define how JunB and c-Jun control TNFα and TNFα-dependent cytokines in the skin of newborn and adult mice, JunB and c-Jun were deleted in the epidermis. Here we show that epidermal JunB/c-Jun proteins control TNFα shedding by direct transcriptional regulation of TIMP-3, an inhibitor of TACE. As a consequence, a TNFα-dependent cytokine cascade is initiated in the epidermis, leading to skin inflammation and TNFα-dependent cachexia in newborns and a psoriasis-like phenotype in adults. Interestingly, these phenotypes are rescued in the absence of TNFR1 or by epidermal-specific re-expression of TIMP-3.
Results
Perinatal lethality due to cachexia in epidermal JunB/c-Jun-deficient pups
Double-mutant (DKO) pups were born at expected frequencies and were indistinguishable from their littermate controls, with the exception that mice lacking only epidermal c-Jun showed an “eyes open at birth” phenotype (Zenz et al. 2003). All DKO pups were recognized at postnatal day 1 (P1) by their failure to thrive, and died ∼48 h after birth, showing signs of cachexia (Fig. 1A,B). Prior to death, DKO pups were frail and underweight and presented a wasting syndrome, impaired movements, and reddish skin, when compared with littermate controls. However, all DKO newborns had a normal suckling behavior, and milk was always found in their stomachs. Detailed analyses of DKO pups demonstrated efficient deletion of JunB and c-Jun by quantitative PCR (qRT–PCR) and Western blot in epidermis and forestomach, but not in other keratin 5-expressing tissues such as the tongue, esophagus, or thymus (Fig. 1C,D; data not shown). Histological analyses of DKO skin at P2 revealed normal epidermal stratification and differentiation as revealed by keratin 14, keratin 6, and keratin 10 immunofluorescence (Fig. 1E; Supplemental Fig. 1A). However, subcorneal pustules and increased numbers of Ki67-positive cells, probably linked to the presence of inflammatory cells, were observed (Fig. 1E, arrowhead/arrows; Supplemental Fig. 2A). Flow cytometric analysis of both epidermis and dermis-derived cell suspensions showed a massive increase in granulocytes in DKO skin (Supplemental Fig. 2B). Furthermore, the numbers of dendritic cells (DCs) were comparable, but displayed higher levels of CD80+, CD86+, and MHC class II+, suggestive of an activated state (Supplemental Fig. 2C). This is indicative of increased proinflammatory cytokine levels in the epidermal/dermal compartments of DKO pups.
Figure 1.

Phenotypic and molecular characterization of newborns constitutively deficient for JunB and c-Jun (DKO) in skin epidermis. (A) Pups at P2. (B) Kaplan-Meier survival curves for Co and DKO mice. (Co, n = 23; DKO, n = 27). (C) qRT–PCR for epidermal JunB and c-Jun. Data are shown as mean ± SD relative to Actin. (*) P < 0.05 (Student's t-test). (D) Western blot for epidermal JunB and c-Jun. Actin indicates equivalent loading. (E) Histological analysis of skin sections at P2. Arrows indicate areas with increased cellularity, and arrowheads indicate inflammatory cells in cornified layer (subcorneal pustules). Epidermis and dermis are separated by a dashed line. (Top panel) 20× magnification. (Bottom panel) 40× magnification of the marked area. Epidermis and dermis are separated by a dashed line.
To investigate the cause of death of DKO pups, skin barrier functionality, epidermal tight junction integrity, lipid deposition in the stratum corneum, and trans-epidermal water loss (TEWL) were analyzed. All of these parameters were unaffected (Supplemental Fig. 3A–D), indicating that a skin barrier defect is not likely the cause of lethality of DKO pups. In contrast, Oil-Red-O staining demonstrated lipid deficiency in DKO skin and liver, suggesting a defect in energy metabolism (Fig. 2A). Furthermore, PAS staining and hepatocyte microvesicular vacuolization revealed a reduction of glycogen reservoirs in DKO liver and skeletal muscle (Fig. 2B, arrowheads). In addition, a striking decrease in serum glucose levels of DKO pups was measured (Fig. 2C). Taken together, this phenotype is reminiscent of a lethal cachectic phenotype in which a substantial consumption of the main energy reservoirs leads to perinatal death.
Figure 2.

Phenotypic analyses of newborns constitutively deficient for JunB and c-Jun (DKO) in skin epidermis. (A) Absence of lipids as detected by Oil Red O staining in DKO skin (top panels) and liver (bottom panels) at P2 (20× magnification). (B) Absence of glycogen as detected by PAS staining in DKO liver (top panels) and skeletal muscle (bottom panels) at P2 (20× magnification). (C) Serum glucose levels in Co and DKO pups at P2 (n = 6). Data are shown as mean ± SD. () P < 0.05 (Student's t-test).
Increased levels of proinflammatory cytokines in DKO pups
The cachectic phenotype could be triggered by a massive secretion of proinflammatory cytokines originating in the epidermis that, via the bloodstream, can act systemically and induce cachexia. Indeed, the concentration of many proinflammatory cytokines, implicated in cachexia induction (Tisdale 2002), was found to be dramatically increased in the epidermis of DKO pups—in particular, soluble TNFα (sTNFα), a central proinflammatory cytokine (Fig. 3A). A cytokine ELISA array in the serum of DKO pups revealed prominently increased concentrations of proinflammatory cytokines such as sTNFα, IL-1α, IL-1β, IL-6, and KC/IL-8 that were more abundant at P2 than at P1 (Fig. 3B). Interestingly, massive induction of a number of cytokines—among them G-CSF, GM-CSF, and IL-17—was also detected (Supplemental Table 1). ELISA analysis of sTNFα in DKO at P1 and P2 showed the presence of sTNFα in multiple organs, suggesting that sTNFα can also act systemically and induce cachexia (Fig. 3C; Cheng et al. 1992; Sosic et al. 2003). In addition, serum from DKO pups induced a dramatic decrease in sTNFα-sensitive cell survival, which was rescued in the presence of a TNF-blocking antibody (Fig. 3D), indicating that the circulating sTNFα in DKO pups was biologically active. Similarly, increased levels of sTNFα were found in the epidermis of adult mice, which develop psoriasis-like skin disease after inducible deletion of JunB and c-Jun (DKO*) (Fig. 3E). The increased levels of procachectic cytokines found in the DKO—mainly, sTNFα—are likely responsible for the increased consumption of glycogen and lipids, the main energy stores of the body, which leads to cachexia in newborn mice. Furthermore, the presence of a pronounced inflammatory cell component in JunB/c-Jun epidermis certainly contributes to the amplification of the epidermis-derived cytokine cascade (Pasparakis et al. 2002; Perez-Moreno et al. 2006).
Figure 3.

Cytokine profiling of newborn DKO and adult DKO* mice lacking JunB and c-Jun in skin epidermis. (A) ELISA array of cytokines present in the epidermis of Co (n = 4) and DKO (n = 4) at P2. Data are shown as mean ± SD. (*) P < 0.05 (Student's t-test). (B) Cytokine ELISA array in serum from Co (n = 5) at P2 and DKO (n = 6) pups at P1 and P2. Data are shown as mean ± SD. (*) P < 0.05 (Student's t-test). (C) sTNFα ELISA in diverse tissues of Co (n = 3) at P2 and DKO (n = 3) pups at P1 and P2. Data are shown as mean ± SD. (*) P < 0.05 (Student's t-test). (D) MTT survival assay performed with 1:2 diluted serum from Co (n = 4) and DKO mice (n = 4) at P1 in the absence or presence of a TNFα-blocking antibody. PBS (1×) and 150 ng/mL recombinant TNFα were used as negative and positive controls, respectively. Data are shown as mean ± SD. (*) P < 0.05 (Student's t-test). (E) Comparison of epidermal sTNFα levels by ELISA in Co (n = 5) and DKO (n = 6) pups at P2, and Co (n = 3) and DKO* (n = 5) mice 2 wk after inducible deletion. Data are shown as mean ± SD. (*) P < 0.05 (Student's t-test).
Increased TNFα shedding in the absence of epidermal JunB and c-Jun
sTNFα is released by physiological shedding of membrane-anchored TNFα (mTNFα/pro-TNFα). To investigate the molecular mechanism leading to increased epidermal sTNFα levels, the expression levels of TNFα; TACE, the main protease responsible for TNFα shedding; and TIMP-3, a highly potent TACE inhibitor, were measured. Epidermal TNFα mRNA as well as sTNFα protein levels were increased in the DKO pups and DKO* mice, while mTNFα protein levels were reduced (Fig. 4A–D). In contrast, mRNA and protein levels of TACE were unchanged (Fig. 4A–D). Importantly, a prominent decrease of TIMP-3 mRNA and protein levels was observed specifically in the epidermis and not in the forestomach of DKO pups (Fig. 4A–D; data not shown). This indicates that Jun proteins are required for maintaining physiological TIMP-3 levels in the epidermis.
Figure 4.

Epidermal TIMP-3 down-regulation and increased TACE activity in newborns and adult mice lacking JunB and c-Jun (DKO and DKO*) in skin epidermis. (A) qRT–PCR analysis in DKO epidermis at P1. TNFα levels are increased, TACE levels are not changed, and TIMP-3 levels are reduced in DKO epidermis compared with Co epidermis (n = 4). Data are shown as mean ± SD relative to Actin. (*) P < 0.05 (Students t-test). (B) qRT–PCR analysis in DKO* epidermis at 17 d after the last tamoxifen injection. TNFα levels are increased, TACE levels are not changed, and TIMP-3 levels are reduced in DKO epidermis compared with Co epidermis (n = 4). Data are shown as mean ± SD relative to Actin. (*) P < 0.05 (Student's t-test). (C) Western blot analysis of TNF, TIMP-3, and TACE in control and DKO epidermis at P1. Actin indicates equivalent loading. (D) Western blot analysis of TNF and TIMP-3 in adult Co and DKO* epidermis. Actin indicates equivalent loading. (E) Increased TACE activity in DKO epidermis at P1 (n = 3). The same extracts were assayed in the presence of recombinant TIMP-3. Increased TACE activity, similar to DKO epidermis, in adult DKO* epidermis (n = 3). Data are shown as mean ± SD. (*) P < 0.05 (Student's t-test). (F) siRNA-mediated TACE knockdown (siTACE) in DKO keratinocytes demonstrates that TACE is the protease responsible for TNFα shedding in DKO keratinocytes. (G) Comparison of epidermal sTNFR1 by ELISA in Co (n = 5) and DKO (n = 6) pups at P2, and Co (n = 3) and adult DKO* (n = 5) mice 2 wk after deletion. Data are shown as mean ± SD. (*) P < 0.05 (Students t-test). (H) Comparison of epidermal sTNFR2 by ELISA in Co (n = 5) and DKO (n = 6) pups at P2, and Co (n = 3) and adult DKO* (n = 5) mice 2 wk after deletion. Data are shown as mean ± SD. (*) P < 0.05 (Student's t-test).
The down-regulation of TIMP-3 and the increase of the sTNFα/mTNFα ratio suggest a possible increase in epidermal TACE activity. An established fluorogenic activity assay in vitro demonstrated an approximately twofold increase in TACE activity in DKO epidermis, which was efficiently inhibited by the addition of recombinant TIMP-3 (rTIMP-3) (Fig. 4E). In DKO* adult epidermis, a similar increase in TACE activity was detected (Fig. 4D). Importantly, efficient siRNA knockdown of TACE (data not shown) in DKO keratinocytes demonstrated that the observed increase in TNFα shedding in DKO epidermis is specific to increased TACE activity (Fig. 4F). Furthermore, mRNA levels of ADAM10, another TNF-α sheddase, were unchanged in the epidermis of DKO and DKO* mice (Supplemental Fig. 4B). Consistent with increased TACE activity, increased levels of sTNFR1 and sTNFR2 were detected in the epidermis of mice after constitutive or inducible deletion of Jun proteins (Fig. 4G,H).
A cell-autonomous mechanism regulates TNFα shedding in keratinocytes
To determine whether TNFα shedding occurs in epidermal keratinocytes in a cell-autonomous manner, deletion of JunB and c-Jun was performed in vitro using adenovirus-mediated delivery of Cre recombinase (AdCre). Western blot and qRT–PCR confirmed the efficient deletion of JunB and c-Jun (Fig. 5A; Supplemental Fig. 4A). TNFα and TACE levels were not changed, but TIMP-3 protein and mRNA levels were significantly down-regulated (Fig. 5A; Supplemental Fig. 4A). Consistent with the in vivo findings, mTNFα was almost completely converted to sTNFα (Fig. 5A). In addition, TACE activity was increased in the absence of JunB and c-Jun (Fig. 5B) and was inhibited by the addition of rTIMP-3, but not by the addition of rTIMP-1. It is worth noting that, according to non-cell-based assays, TIMP-1 does not inhibit TACE, but inhibits ADAM10 and all matrix metalloproteases (MMPs) known to have TNFα sheddase activity (Amour et al. 1998; Wisniewska et al. 2008). Furthermore, in the absence of JunB and c-Jun, the mRNA levels of other proteases known to induce TNFα shedding either were not changed, such as ADAM10 (Supplemental Fig. 4B), or were found reduced, such as MMP2, MMP-3, MMP7, and MMP-9 (Supplemental Fig. 4C). This indicates that increased activity of TACE is likely responsible for increased TNFα shedding in JunB- and c-Jun-deficient keratinocytes, which occurs in a cell-autonomous manner.
Figure 5.

Increased TNFα shedding in keratinocytes conditionally deficient for JunB and c-Jun in vitro and upon TIMP-1 and TIMP-3 knockdown. (A) Western blot analysis of c-Jun, JunB, TIMP-3, TACE, and mTNFα in AdGFP- or AdCre-infected JunBf/f; c-Junf/f keratinocytes. For sTNFα, the sample was obtained from cell culture supernatant. Actin indicates equivalent loading. (B) Increased TACE activity in AdCre-infected keratinocytes (n = 3). The same extracts were assayed in the presence of recombinant TIMP-3 and TIMP-1. Data are shown as mean ± SD. (*) P < 0.05 (Student's t-test). (C) MTT assay performed with culture supernatant from AdGFP-infected (n = 3) or AdCre-infected (n = 3) keratinocytes. Keratinocyte medium and recombinant TNFα were used as negative (Co) and positive (rTNFα, 150 ng/mL) controls, respectively. Data are shown as mean ± SD. (*) P < 0.05 (Student's t-test). (D) sTNFα ELISA upon siRNA knockdown of mouse TIMP-3 (siT3) in wild-type keratinocytes. siGLO and siNo-T were used as transfection and target specificity controls, respectively. (E) Increased TACE activity in wild-type keratinocytes upon TIMP-3 knockdown (siT3). TACE siRNA (siTACE) inhibited the increase in TACE activity. Data are shown as mean ± SD. (*) P < 0.05 (Student's t-test). (F) Increased sTNFα shedding as measured by ELISA upon siRNA knockdown of mouse TIMP-1 (siT1), TIMP-3 (siT3), and TIMP-1/TIMP-3 (siT1 + siT3) in wild-type keratinocytes. The increased shedding was inhibited upon concomitant knockdown of TACE (siTACE). Data are shown as mean ± SD. (*) P < 0.05 (Student's t-test). (G) Schematic illustration of the mouse TIMP-3 promoter showing the six putative AP-1-binding sites. Due to ChIP constraints, the six sites were grouped into three different domains that could be amplified by different sets of primers. In the right panel, ChIP and PCR amplification demonstrates c-Jun and JunB binding to the TIMP-3 promoter in epidermal samples from Co, but not from DKO, pups at P1. When JunB and c-Jun were expressed, binding was detected in all three domains. (H) ChIP analysis of c-Jun and JunB binding to the TIMP-3 promoter in Co- and DKO-derived keratinocytes. (I) ChIP analysis of c-Jun and JunB binding to the TIMP-3 promoter in cultured AdGFP- and AdCre-infected JunBf/f; c-Junf/f keratinocytes. Isotype IgG was used as a control (IgG). (J) TIMP-3 Western blot analysis of AdGFP or AdCre JunBf/f, c-Junf/f or JunBf/f; c-Junf/f infected keratinocytes. Actin indicates equivalent loading.
In addition, a marked decrease in sTNFα-sensitive cell survival in the presence of supernatant of AdCre-infected keratinocytes was measured (Fig. 5C), and this was efficiently inhibited by TNFα-blocking antibodies (Supplemental Fig. 4D). Moreover, sTNFα-mediated cytotoxicity was also efficiently prevented by using a TACE inhibitor, TAPI-1 (Supplemental Fig. 4E).
Importantly, specific knockdown of TIMP-3 in wild-type keratinocytes induced an increase in TNFα shedding and TACE activity (Fig. 5D; Supplemental Fig. 4F). This activity was inhibited upon simultaneous siRNA-mediated TACE knockdown (Fig. 5E). An increase in TIMP-1 expression was also observed in TIMP-3 knockdown cells (Supplemental Fig. 4G). In addition, siRNA-mediated TIMP-1 knockdown in wild-type keratinocytes induced a TACE-dependent increase in TNFα shedding (Fig. 5F), as well as an increase in TIMP-3 expression (Supplemental Fig. 4G). Interestingly, concomitant TIMP-1 and TIMP-3 siRNA knockdown induced a synergistic increase in TACE-dependent TNFα shedding as compared with single knockdowns (Fig. 5F; Supplemental Fig. 4G). This is due to increased TACE activity, since it is inhibited upon siRNA-mediated TACE knockdown (Fig. 5F).
These data demonstrate that the specific decrease of TIMP-1 or TIMP-3 alone is sufficient to increase TACE activity and TNFα shedding in epidermal keratinocytes. These data also show that depletion of both TIMP-1 and TIMP-3 induces a synergistic increase in TACE activity and TNFα shedding.
JunB and c-Jun directly control the TIMP-3 promoter
The mouse TIMP-3 promoter was studied and found to contain six putative AP-1 binding sites clustered in three domains: A, B, and C (Fig. 5G; Kim et al. 1997). Chromatin immunoprecipitation (ChIP) experiments performed with extracts from isolated epidermal keratinocytes showed that both JunB and c-Jun efficiently bound each of the domains in controls, whereas the binding was almost undetectable in DKO (Fig. 5H). This result was confirmed in vitro after AdCre-mediated deletion of JunB and c-Jun (Fig. 5I). Interestingly, single deletion of either JunB or c-Jun alone did not affect TIMP-3 protein levels, indicating that both JunB and c-Jun are required for proper TIMP-3 expression (Fig. 5J). These data indicate that both JunB and c-Jun proteins directly control the transcription of TIMP-3. These results demonstrate that deletion of JunB and c-Jun in epidermal keratinocytes leads to a major transcriptional down-regulation of TIMP-3, thereby specifically increasing TACE activity, which consequently increases cell-autonomous TNFα shedding.
Complete genetic rescue of the DKO phenotype in a TNFR1-deficient background
To genetically study the contribution of sTNFα and its downstream signaling through TNFR1 to the sTNFα -dependent phenotype, triple knockout (TKO) mice were generated constitutively lacking JunB/c-Jun in the epidermis and TNFR1 in every tissue. Interestingly, TKO mice were viable and showed no overt phenotype in either skin or any other organ at P2 and throughout life (Fig. 6A,B). Furthermore, immunohistological analyses of TKO at P2 revealed a normal skin when compared with DKO TNFR1+/− pups (Supplemental Fig. 5A). Epidermal and serum cytokine measurements of TKO mice revealed reduced levels of all cytokines found to be up-regulated in the DKO (Fig. 6C; Supplemental Fig. 5B; Supplemental Table 2). Detailed analysis of sTNFα serum levels in TKO pups showed an initial increase, which disappeared over time and reached comparable concentrations with control littermates around P3 as well as in adulthood (Supplemental Fig. 5C). Detailed analyses of inflammatory cells from TKO mice showed cell counts similar to controls (Supplemental Fig. 5D,E). We therefore hypothesize that signaling through TNFR1 is required to maintain TNFα expression in the DKO, likely via NF-κB signaling (Kollias 2005). In TKO mice, the absence of this positive feedback loop from TNFR1 to TNFα transcription leads to an attenuation of TNFα synthesis (Fig. 6D) and release. As a consequence, the TNFα-dependent signaling cascade is perturbed, followed by the absence of an inflammatory component and leading to a complete rescue of the lethal phenotype. These data indicate that, in mouse epidermis, sTNFα acts as a key regulator of the inflammatory response through TNFR.
Figure 6.

Rescue of DKO lethality in mice lacking TNFR1 (TKO mice) and upon TIMP-3 overexpression. (A) Pups at P2. (B) Co and TKO adult mice (6 wk old). (C) ELISA of cytokines present in the epidermis of Co (n = 4), DKO TNFR+/− (n = 4), and TKO (n = 4) at P2. Fold induction shown in logarithmic scale. Data are shown as mean ± SD. (*) P < 0.05 (Student's t-test). Co is set to 1. (D) qRT–PCR analysis of TNFα in Co, DKO TNFR1+/−, and TKO at P2 (n = 3). Data are shown as mean ± SD relative to Actin. (*) P < 0.05 (Student's t-test). (E) TIMP-3/DAPI immunofluorescence in DKO and DKO pups following subcutaneous injections of AdTIMP-3 at P3. Western blot analysis of TIMP-3 in DKO and DKO pups following subcutaneous injections of AdTIMP-3 at P3 and P5. Actin indicates equivalent loading. (D) Dermis; (E) epidermis. (F) Kaplan-Meier survival curves for DKO and DKO pups following subcutaneous injections of AdTIMP-3 (DKO, n = 7; DKO + Ad-TIMP-3, n = 7). (G) Decreased TACE activity in DKO + AdTIMP-3 epidermis at P3 (n = 7) compared with DKO. Data are shown as mean ± SD. (*) P < 0.05 (Student's t-test). (H) TNFα ELISA in Co, DKO, and DKO + AdTIMP-3 (n = 7). Data are shown as mean ± SD. (*) P < 0.05 (Student's t-test).
Ectopic TIMP-3 expression rescues the DKO phenotype
To investigate whether TIMP-3 down-regulation is solely responsible for the sTNFα-dependent lethal cachectic phenotype of DKO pups, TIMP-3 was ectopically expressed using subcutaneous injections of Adeno-TIMP-3 into DKO newborn pups. TIMP-3 protein was found to be expressed efficiently, in particular in the epidermis of mutant pups and not in other tissues such as the dermis, liver, skeletal muscle, or inflammatory cells (Fig. 6E; data not shown). This expression pattern is due to the increased levels of Coxsackie-Adenovirus Receptor present in the epidermis (Raschperger et al. 2006). Strikingly, all DKO pups ectopically expressing TIMP-3 survived up to P3 or P5, when they were sacrificed for analysis (Fig. 6F). Epidermal TACE activity and sTNFα in serum as well from epidermis were reverted to control levels (Fig. 6G,H; data not shown). As a consequence, inflammatory cells such as macrophages and granulocytes were absent in the epidermal/dermal compartment of TIMP-3-overexpressing DKO pups (Supplemental Fig. 6A–C). This indicates that TIMP-3 is the essential Jun target gene responsible for the sTNFα-dependent phenotypes observed in mice lacking epidermal JunB and c-Jun.
Discussion
To define the role of epidermal Jun proteins in skin homeostasis, we generated mice in which epidermal JunB and c-Jun proteins were deleted either in a constitutive or inducible manner. While inducible deletion of Jun proteins in the epidermis of adult mice (DKO*) leads to a psoriasis-like disease, including psoriatic arthritis (Zenz et al. 2005), constitutive deletion of Jun proteins (DKO) leads to perinatal death from cachexia. Here we show that these phenotypes are a consequence of massive epidermal cytokine production regulated by a TIMP-3/TACE/sTNFα-dependent pathway. In addition, TIMP-3 is a direct transcriptional target of epidermal Jun proteins. Reduced levels of TIMP-3 caused a specific increase in TACE activity, which induced increased shedding of TNFα from the epidermis and subsequent induction of sTNFα-dependent inflammatory cytokines. This resulted in skin inflammation, a systemic increase in sTNFα and sTNFα-dependent cytokines and cachexia in newborn mice (Fig. 7). Importantly, these phenotypes were genetically rescued by additionally removing TNFR1 in all cells; the adult DKO* psoriasis-like disease phenotype was drastically improved and the lethal phenotype of the DKO mice was completely rescued, demonstrating that these phenotypes are dependent on sTNFα signaling. Furthermore, adenovirus-mediated TIMP-3 re-expression in the epidermis of DKO pups rescued the lethality, indicating that the observed phenotype is strictly TIMP-3 dependent. These data show for the first time that JunB and c-Jun are important physiological regulators of TNFα shedding.
Figure 7.
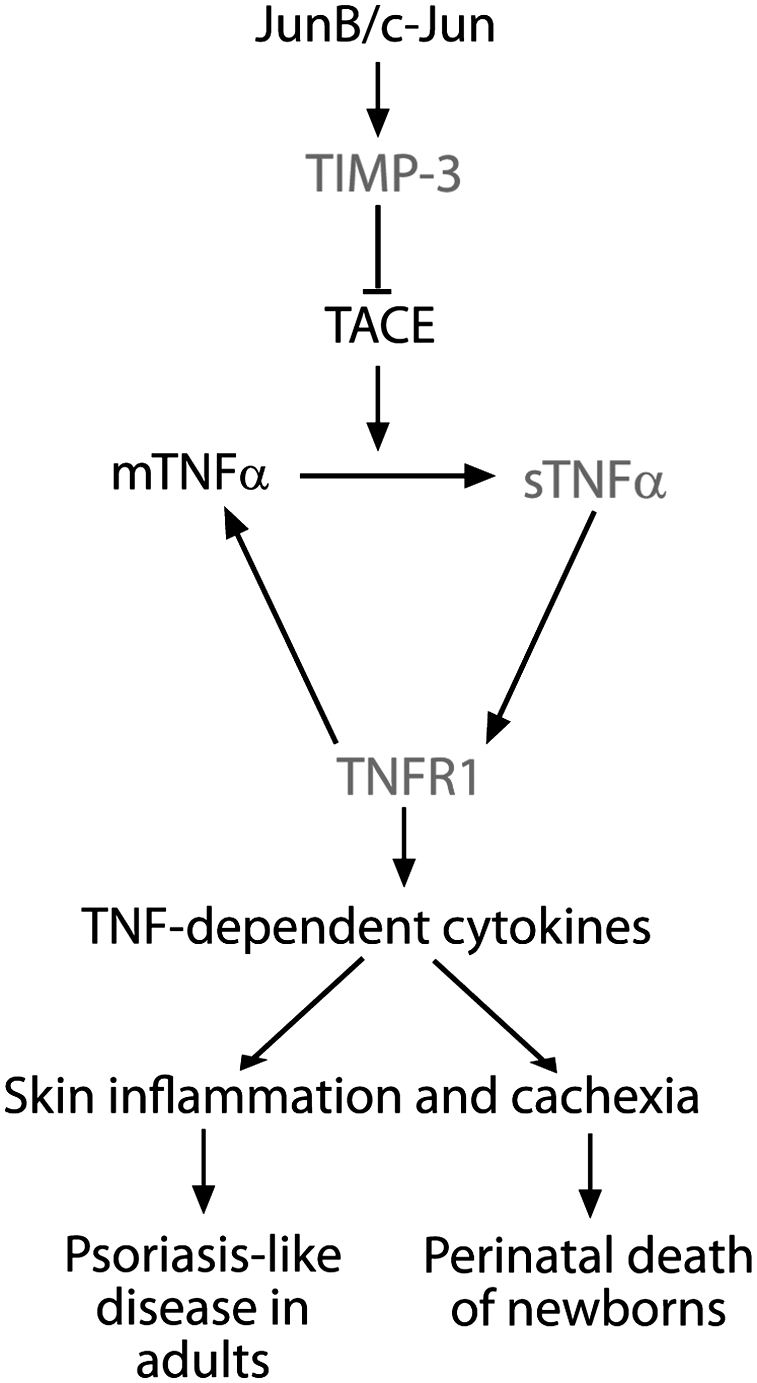
Schematic model illustrating how Jun proteins regulate the TIMP-3, TACE, and TNFα pathway: Jun proteins transcriptionally activate TIMP-3 expression, which is responsible for TACE inhibition. The absence of JunB and c-Jun leads to an increase in TACE activity, followed by increased levels of sTNFα, which signals through TNFR1, inducing more TNFα RNA and TNFα-dependent cytokines. This cascade causes skin inflammation leading to a psoriasis-like disease in adult mice and to skin inflammation and cachexia with perinatal death in newborns.
DKO pups had no obvious epidermal barrier defect or suckling behavioral defects, and perinatal death was likely the consequence of the sTNFα-dependent exhaustion of energy reservoirs. The elevated concentrations of sTNFα and sTNFα-dependent procachectic cytokines such as IL-1α, IL-1β, IL-6, and KC detected in sera of DKO pups critically contributed to the observed metabolic changes and ultimately led to death (see also Cheng et al. 1992; Sosic et al. 2003). We propose that keratinocytes produce high cytokine levels—in particular, sTNFα—and the inflammatory cells act as amplifiers of the cytokine cascade, possibly due to inflammasome activation (Martinon et al. 2009). Increased cytokine levels can reach the bloodstream and act in a paracrine fashion via TNFR1 (Pasparakis et al. 2002). Systemically, increased levels of procachectic cytokines interfere with glucose metabolism in the liver and skeletal muscle, and with lipid metabolism in the liver and subcutaneous fat to induce cachexia. Consistent with these findings, it has been shown recently that sTNFα-driven cachectic symptoms are improved in psoriatic patients undergoing anti-TNFα therapies (Saraceno et al. 2008).
In TKO mice, which lack sTNFα signaling through TNFR1, the phenotype was completely rescued and no infiltrating inflammatory cells could be detected. In these mice, cytokine levels were normal in epidermis and serum, except for a transient increase in sTNFα. The initial increase of sTNFα in TKO mice is likely due to TACE-mediated proteolytic cleavage of mTNFα. In the absence of TNFR1, downstream pathways cannot be activated, and therefore the positive regulatory feedback loop to TNFα synthesis is suppressed and the induction of sTNFα-dependent cytokines is absent (Kruppa et al. 1992; Chen and Goeddel 2002). As a result, the slight increase in sTNFα decayed within hours, and its levels became comparable with controls. Therefore, TNFR1 signaling is essential for the induction of the sTNFα and sTNFα-dependent epidermal cytokine cascade (Chen and Goeddel 2002).
Jun proteins control TNFα shedding through the regulation of TIMP-3 transcription. We show that binding of JunB and c-Jun to the promoter is necessary for the induction of TIMP-3 expression, since only the absence of both proteins leads to down-regulation of TIMP-3. In agreement with our data, EMSA studies indicated that c-Jun may bind to the TIMP-3 promoter (Kim et al. 1997). These data indicate that Jun proteins are key regulators of TIMP-3 expression.
Besides TACE, several MMPs and ADAM10 have also been shown to have TNFα sheddase activity (Seals and Courtneidge 2003; Edwards et al. 2008). However, the enzymatic in vitro experiments using a fluorogenic peptide indicate that TACE is the protease likely responsible for shedding of TNFα upon JunB and c-Jun deletion. In JunB/c-Jun-deficient keratinocytes, increased protease activity is attributed to TACE, since the increased activity is inhibited by TIMP-3, a highly potent TACE inhibitor, when compared with other TIMPs (Amour et al. 1998; Mohammed et al. 2004; Wisniewska et al. 2008). In addition, specific siRNA knockdown of TACE demonstrated that TACE is the protease responsible for increased TNFα shedding in DKO keratinocytes. Furthermore, the increased TACE activity is not inhibited by TIMP-1, which in non-cell-based assays has been shown to inhibit all other TNFα sheddases, with the exception of TACE (Amour et al. 1998; Wisniewska et al. 2008). Therefore, TIMP-3 down-regulation primarily affects TACE activity.
While the contribution of the TACE pathway in several diseases is widely accepted (Malemud 2006), little is known regarding the physiological and tissue-specific roles of TIMP-3-regulated and TACE-mediated TNFα shedding. Mice lacking TACE protease activity are not viable (Peschon et al. 1998). Moreover, mice without TIMP-3 exhibit increased TACE activity and develop sTNFα-dependent liver and heart disease, but no spontaneous skin abnormalities (Fedak et al. 2004; Mohammed et al. 2004). We speculate that, in the context of constitutive broad inactivation of the TIMP-3 gene, a compensatory mechanism is activated in these mice to suppress TACE activation and epidermal inflammation. The specific knockdown experiments of both TIMP-1 and TIMP-3 demonstrate that the decrease of TIMP-1 or TIMP-3 alone in epidermal keratinocytes is sufficient to increase TACE activity and TNFα shedding. These data also show that, in epidermal keratinocytes, TIMP-1 is induced and can partially compensate for the lack of TIMP-3 and vice versa, in particular since depletion of both proteins induced a synergistic increase in TACE activity and TNFα shedding. Consistent with these in vitro findings, preliminary analyses of TIMP-3 knockout epidermis showed increased expression of TIMP-1, whereas TIMP-1 levels were not altered in keratinocytes lacking JunB and c-Jun (data not shown). To our knowledge, no report has yet described the in vivo role of epidermal TIMP-1, with respect to TACE, in the presence or absence of TIMP-3. In addition, a reduction in TACE protein levels was observed in the TIMP-3-deficient mice (Mohammed et al. 2004). Therefore, an altered TIMP/TACE expression pattern might also account for the lack of a skin phenotype in TIMP-3 knockout mice. A conditional, epidermis-specific deletion of TIMP-3 might unravel the importance of compensatory mechanisms existing in the complete TIMP-3 knockout mice.
In conclusion, we describe a novel mechanism of how JunB/c-Jun proteins regulate physiological TNFα shedding and inflammation by regulating epidermal TIMP-3 expression. Since sTNFα plays an essential role in the initiation and progression of many human inflammatory skin diseases, the manipulation of the Jun/AP-1–sTNFα connection in epidermal keratinocytes may offer a new target area to treat inflammatory skin diseases. This study also emphasizes the importance of the epidermis as an endocrine-like organ regulating innate inflammatory skin responses and energy metabolism.
Materials and methods
Mice: generation of DKO and TKO mice
JunBf/f (Kenner et al. 2004) and c-Junf/f (Behrens et al. 2002) were crossed to K5-Cre mice (Tarutani et al. 1997) to obtain JunBf/f c-Junf/f, referred to as Co, and JunBf/f c-Junf/f K5-Cre, referred to as DKO; JunBf/+ c-Junf/+ K5-Cre were also used and were indistinguishable from Co. DKO* mice were described previously (Zenz et al. 2005). DKO mice were crossed to mice deficient in TNFR1 (TNFR1−/−) (Rothe et al. 1993) to obtain JunBf/f c-Junf/f TNFR1+/− K5-Cre, referred to as DKO TNFR1+/−, and JunBf/f c-Junf/f TNFR1−/− K5-Cre, referred to as TKO. The genetic background of the mice was C57Bl/6 × 129 for c-junf/f, JunBf/f, TNFR1−/−, and a mixed C57B6/C3H/129 for K5-Cre. TIMP-3−/− mice were kindly provided by R. Khokha. All mouse experiments were performed in accordance with local and institutional regulations/licenses.
Cytokine array and ELISA
Multiple cytokine measurements (23-plex panel) were performed using a Bio-Plex array (Bio-Rad) following the manufacturer's instructions. Single sTNFα, sTNFRI, and sTNFRII ELISA were performed using TNFα Quantikine ELISA System (R&D Systems).
TACE enzymatic assay
Epidermis and dermis were separated, and cells were plated as specified in keratinocyte culture procedures. Twelve hours later, TACE lysis buffer was added (Mohammed et al. 2004), and activity from 25 μg of total protein were measured for 5 min at 37°C, 10 min after the addition of 50 μM TACE peptide (Peptides International). Fluorescence was measured with an excitation wavelength of 320 nm and an emission wavelength of 420 nm. Incubation with 100 nM recombinant proteins of either TIMP-1 or TIMP-3 was performed 10 min prior to adding the peptide. rTIMP-1 and rTIMP-3 were from R&D Systems.
Histology, immunohistochemistry, and immunofluorescence
Tissues were fixed in PBS-buffered 3.7% formalin or frozen in OCT at −80°C. Formalin-fixed tissues were dehydrated, and 4-μm sections were used. Seven-micron cryosections were cut from OCT-fixed and frozen tissues. Hematoxilin and eosin (H&E) and periodic acid Schiff (PAS) stainings were performed according to standard procedures. Ki67 (Novocastra) immunohistochemistry was performed using an automated Discovery XT immunohistochemistry system (Ventana Medical Systems). Immunofluorescence was performed as described (Perez-Moreno et al. 2006) using keratin 14, keratin 6, and keratin 10 (Covance); Mac-1 and Gr-1 hybridomas (kindly provided by Gordon Keller); isotype control antibodies (Pharmingen; data not shown); Alexa Fluor 488 and Alexa Fluor 647 dye-labeled secondary antibodies (Invitrogen); and DAPI counterstaining (Sigma). Oil Red O (Sigma) and Nile Red (Sigma) stainings were performed on cryosections according to standard procedures.
Cell culture and adenoviral infection and injection
Isolation and culture of primary keratinocytes were performed as described (Zenz et al. 2003). Briefly, the epidermis was separated from the dermis by incubation in 0.75% trypsin (Gibco) at 37°C. One hour later, the epidermis was incubated in medium containing DNase (Sigma) for 30 min. Cells were spun down and plated in coated dishes (Cascade Biologicals) with 8% chelated fetal calf serum (FCS) Eagle MEM (Sigma). Adenoviruses Adeno-Cre (AdCre) or Adeno-green fluorescent protein (AdGFP) were purchased from the University of Iowa. Primary keratinocytes were infected with 300 particles per cell in medium with 4% chelated FCS. Two hours later, normal keratinocyte medium (8% chelated FCS) was added. Cells were collected 96 h post-infection. WEHI-13VAR cells (American Type Culture Collection) were grown with DMEM and 10% FBS. AdTIMP-3, kindly provided by A. Baker, was subcutaneously injected immediately after DKO pups were born and every 48 h for 3 or 5 d. Fifty microliters of 3 × 109 pfu/mL solution were injected.
siRNA
Dharmacon ON-TARGETplus SMARTpool was used for the knockdown of mouse TIMP-1 and TIMP-3 (siT1 and siT3, respectively) in wild-type keratinocytes (C57B6/C3H/129 background) and for the TACE (siTACE) knockdown in DKO keratinocytes following the manufacturer's instructions. As controls, siGLO and ON-TARGETplus Non-Targeting Pool (siNo-T) were used. The analysis was performed after 96-h Lipofectamine 2000 (Invitrogen) transfection.
MTT assays
TACS MTT (R&D Systems) TNFα cytotoxicity assays were performed following the manufacturer's instructions in the presence of ActinomycinD (Sigma). WEHI-13VAR (Khabar et al. 1995) viability was assayed 16 h after the addition of either serum-free keratinocyte culture supernatant or mouse serum (diluted 1:2 in PBS). When indicated, recombinant TNFα (R&D Systems), TNFα-blocking antibody, or isotype control IgG2A (Pharmingen) was added. TAPI-1 (133 μM) or DMSO was added 16 h before collecting keratinocyte supernatants.
Protein isolation
Backskin epidermis was separated from dermis after incubation in 0.75% trypsin during 45 min at 37°C and was rinsed in ice-cold PBS (1×). Protein isolation for Western blot was performed in RIPA buffer containing a protease inhibitor cocktail (Sigma), 0.1 mM Na3VO4, 40 mM B-glycerophospate, 40 mM NaPPi, and 1 mM NaF, and tissues were disrupted using Precellys 24 (Bertin Technologies). For TNFα Western blot, proteins were isolated as described (Mohammed et al. 2004). For ELISA measurement of bioavailable cytokines, proteins were isolated as described (Mohammed et al. 2004). Proteins from cell culture supernatants were precipitated using the TCA (trichloroacetic acid) method.
Western blot
Western blot analysis was performed according to standard procedures using the following antibodies: TNFα (Antigenics America); TIMP-3 and TACE (Chemicon); Actin (Sigma); c-Jun (Transduction Laboratories); and JunB (Santa Cruz Biotechnologies). HRP-linked secondary antibodies were from Amersham and DAKO. Western blots were developed with ECLplus and ECL Hyperfilm (Amersham).
ChIP
ChIP was performed with a ChIP Assay Kit (Chemicon) according to the manufacturer's instructions. Antibodies used were c-Jun (Transduction Laboratories) and JunB (Santa Cruz Biotechnologies). Cells from Co and DKO newborns were plated as for keratinocyte culture in order to exclude any other cell type in the sample. The isotype antibody used was IgG2a (BD Biosciences).
qRT–PCR
RNA was isolated using Trizol reagent (Invitrogen). cDNA synthesis was performed using 1 μg of total RNA using “Ready-To-Go You-Prime-It First-Strand-Beads” (Amersham Pharmacia Biotech) and random primers (Invitrogen). PCR products were quantified by real-time PCR analysis using Ep-Realplex (Eppendorf) and the CT method. Primer sequences are available upon request.
Flow cytometric analysis
Dermal and epidermal cell suspensions were isolated as described for keratinocyte culture and stained with the following antibodies: anti-CD11c-biotin, anti-CD44-PE, anti-CD80-PE, anti-CD86-FITC, and anti-MHCII-FITC (BD Pharmingen); and anti-CD11b-FITC, anti-F4/80, andanti-Gr-1-PE (CALTAG). Biotinylated antibodies were identified using Streptavidin APC-Cy7 (DAKO), and dead cells were excluded by 7AAD staining. Data were acquired on a FACSAria flow cytometer (BD Biosciences) and were analyzed using CellQuest (BD Biosciences).
Toluidine blue staining
Euthanized newborn pups were rinsed in PBS (1×) and immersed in 25%, 50%, 75%, and 100% methanol for 1 min each successively; pups were then rehydrated in PBS (1×) and stained in 0.1% toluidine blue for 10 min at room temperature. They were washed briefly in PBS and photographed immediately.
TEWL measurements
Measurements in pups at P0 and P1 were performed with Tewameter TM300 following the manufacturer's instructions. Units are given in gram per hour per square meter.
Glucose measurements
Glucose concentration in serum (n = 5) was measured with Reflotron (Roche Diagnostics)
Statistical analysis
All experiments were performed at least three times. Data in bar graphs represent mean ± SD of triplicates. Statistical analysis was performed using nondirectional two-tailed Student's test. P < 0.05 is considered as significant.
Acknowledgments
We are very grateful to Drs. P. Angel, P. Hasselblatt, J. Hess, L. Hui, D. Migliorini, M. Pérez-Moreno, and E. Tschachler for critical reading of the manuscript and helpful discussions; V. Komnenovic for technical assistance; and H. Tkadletz and T. Kulcsar for help with preparing the illustrations.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.543109.
Supplemental material is available at http://www.genesdev.org.
References
- Aggarwal BB. Signalling pathways of the TNF superfamily: A double-edged sword. Nat Rev Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- Amour A, Slocombe PM, Webster A, Butler M, Knight CG, Smith BJ, Stephens PE, Shelley C, Hutton M, Knauper V, et al. TNF-α converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 1998;435:39–44. doi: 10.1016/s0014-5793(98)01031-x. [DOI] [PubMed] [Google Scholar]
- Angel P, Szabowski A, Schorpp-Kistner M. Function and regulation of AP-1 subunits in skin physiology and pathology. Oncogene. 2001;20:2413–2423. doi: 10.1038/sj.onc.1204380. [DOI] [PubMed] [Google Scholar]
- Barton BE. IL-6-like cytokines and cancer cachexia: Consequences of chronic inflammation. Immunol Res. 2001;23:41–58. doi: 10.1385/IR:23:1:41. [DOI] [PubMed] [Google Scholar]
- Behrens A, Sibilia M, David JP, Mohle-Steinlein U, Tronche F, Schutz G, Wagner EF. Impaired postnatal hepatocyte proliferation and liver regeneration in mice lacking c-jun in the liver. EMBO J. 2002;21:1782–1790. doi: 10.1093/emboj/21.7.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler B, Greenwald D, Hulmes JD, Chang M, Pan YC, Mathison J, Ulevitch R, Cerami A. Identity of tumour necrosis factor and the macrophage-secreted factor cachectin. Nature. 1985;316:552–554. doi: 10.1038/316552a0. [DOI] [PubMed] [Google Scholar]
- Black RA. Tumor necrosis factor-α converting enzyme. Int J Biochem Cell Biol. 2002;34:1–5. doi: 10.1016/s1357-2725(01)00097-8. [DOI] [PubMed] [Google Scholar]
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- Chen G, Goeddel DV. TNF-R1 signaling: A beautiful pathway. Science. 2002;296:1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- Cheng J, Turksen K, Yu QC, Schreiber H, Teng M, Fuchs E. Cachexia and graft-vs.-host-disease-type skin changes in keratin promoter-driven TNF α transgenic mice. Genes & Dev. 1992;6:1444–1456. doi: 10.1101/gad.6.8.1444. [DOI] [PubMed] [Google Scholar]
- Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med. 2008;29:258–289. doi: 10.1016/j.mam.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eferl R, Wagner EF. AP-1: A double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- Ezhkova E, Pasolli HA, Parker JS, Stokes N, Su IH, Hannon G, Tarakhovsky A, Fuchs E. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell. 2009;136:1122–1135. doi: 10.1016/j.cell.2008.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedak PW, Smookler DS, Kassiri Z, Ohno N, Leco KJ, Verma S, Mickle DA, Watson KL, Hojilla CV, Cruz W, et al. TIMP-3 deficiency leads to dilated cardiomyopathy. Circulation. 2004;110:2401–2409. doi: 10.1161/01.CIR.0000134959.83967.2D. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Horsley V. More than one way to skin. Genes & Dev. 2008;22:976–985. doi: 10.1101/gad.1645908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhardt C, Nemeth J, Angel P, Hess J. S100A8 and S100A9 in inflammation and cancer. Biochem Pharmacol. 2006;72:1622–1631. doi: 10.1016/j.bcp.2006.05.017. [DOI] [PubMed] [Google Scholar]
- Gelin J, Moldawer LL, Lonnroth C, Sherry B, Chizzonite R, Lundholm K. Role of endogenous tumor necrosis factor α and interleukin 1 for experimental tumor growth and the development of cancer cachexia. Cancer Res. 1991;51:415–421. [PubMed] [Google Scholar]
- Jones PH, Simons BD, Watt FM. Sic transit gloria: Farewell to the epidermal transit amplifying cell? Cell Stem Cell. 2007;1:371–381. doi: 10.1016/j.stem.2007.09.014. [DOI] [PubMed] [Google Scholar]
- Kenner L, Hoebertz A, Beil T, Keon N, Karreth F, Eferl R, Scheuch H, Szremska A, Amling M, Schorpp-Kistner M, et al. Mice lacking JunB are osteopenic due to cell-autonomous osteoblast and osteoclast defects. J Cell Biol. 2004;164:613–623. doi: 10.1083/jcb.200308155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khabar KS, Siddiqui S, Armstrong JA. WEHI-13VAR: A stable and sensitive variant of WEHI 164 clone 13 fibrosarcoma for tumor necrosis factor bioassay. Immunol Lett. 1995;46:107–110. doi: 10.1016/0165-2478(95)00026-2. [DOI] [PubMed] [Google Scholar]
- Kim H, Pennie WD, Sun Y, Colburn NH. Differential functional significance of AP-1 binding sites in the promoter of the gene encoding mouse tissue inhibitor of metalloproteinases-3. Biochem J. 1997;324:547–553. doi: 10.1042/bj3240547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollias G. TNF pathophysiology in murine models of chronic inflammation and autoimmunity. Semin Arthritis Rheum. 2005;34:3–6. doi: 10.1016/j.semarthrit.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Kruppa G, Thoma B, Machleidt T, Wiegmann K, Kronke M. Inhibition of tumor necrosis factor (TNF)-mediated NF-κB activation by selective blockade of the human 55-kDa TNF receptor. J Immunol. 1992;148:3152–3157. [PubMed] [Google Scholar]
- Malemud CJ. Matrix metalloproteinases (MMPs) in health and disease: An overview. Front Biosci. 2006;11:1696–1701. doi: 10.2741/1915. [DOI] [PubMed] [Google Scholar]
- Martinon F, Mayor A, Tschopp J. The inflammasomes: Guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- Matthys P, Dijkmans R, Proost P, Van Damme J, Heremans H, Sobis H, Billiau A. Severe cachexia in mice inoculated with interferon-γ-producing tumor cells. Int J Cancer. 1991;49:77–82. doi: 10.1002/ijc.2910490115. [DOI] [PubMed] [Google Scholar]
- Mehic D, Bakiri L, Ghannadan M, Wagner EF, Tschachler E. Fos and jun proteins are specifically expressed during differentiation of human keratinocytes. J Invest Dermatol. 2005;124:212–220. doi: 10.1111/j.0022-202X.2004.23558.x. [DOI] [PubMed] [Google Scholar]
- Meixner A, Zenz R, Schonthaler HB, Kenner L, Scheuch H, Penninger JM, Wagner EF. Epidermal JunB represses G-CSF transcription and affects haematopoiesis and bone formation. Nat Cell Biol. 2008;10:1003–1011. doi: 10.1038/ncb1761. [DOI] [PubMed] [Google Scholar]
- Mohammed FF, Smookler DS, Taylor SE, Fingleton B, Kassiri Z, Sanchez OH, English JL, Matrisian LM, Au B, Yeh WC, et al. Abnormal TNF activity in Timp3−/− mice leads to chronic hepatic inflammation and failure of liver regeneration. Nat Genet. 2004;36:969–977. doi: 10.1038/ng1413. [DOI] [PubMed] [Google Scholar]
- Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-α. Nature. 1997;385:733–736. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- Murphy G, Murthy A, Khokha R. Clipping, shedding and RIPping keep immunity on cue. Trends Immunol. 2008;29:75–82. doi: 10.1016/j.it.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Overall CM, Blobel CP. In search of partners: Linking extracellular proteases to substrates. Nat Rev Mol Cell Biol. 2007;8:245–257. doi: 10.1038/nrm2120. [DOI] [PubMed] [Google Scholar]
- Pasparakis M, Courtois G, Hafner M, Schmidt-Supprian M, Nenci A, Toksoy A, Krampert M, Goebeler M, Gillitzer R, Israel A, et al. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature. 2002;417:861–866. doi: 10.1038/nature00820. [DOI] [PubMed] [Google Scholar]
- Perez-Moreno M, Davis MA, Wong E, Pasolli HA, Reynolds AB, Fuchs E. p120-catenin mediates inflammatory responses in the skin. Cell. 2006;124:631–644. doi: 10.1016/j.cell.2005.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, et al. An essential role for ectodomain shedding in mammalian development. Science. 1998;282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- Raschperger E, Thyberg J, Pettersson S, Philipson L, Fuxe J, Pettersson RF. The coxsackie- and adenovirus receptor (CAR) is an in vivo marker for epithelial tight junctions, with a potential role in regulating permeability and tissue homeostasis. Exp Cell Res. 2006;312:1566–1580. doi: 10.1016/j.yexcr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- Rothe J, Lesslauer W, Lotscher H, Lang Y, Koebel P, Kontgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature. 1993;364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- Saraceno R, Schipani C, Mazzotta A, Esposito M, Di Renzo L, De Lorenzo A, Chimenti S. Effect of anti-tumor necrosis factor-α therapies on body mass index in patients with psoriasis. Pharmacol Res. 2008;57:290–295. doi: 10.1016/j.phrs.2008.02.006. [DOI] [PubMed] [Google Scholar]
- Seals DF, Courtneidge SA. The ADAMs family of metalloproteases: Multidomain proteins with multiple functions. Genes & Dev. 2003;17:7–30. doi: 10.1101/gad.1039703. [DOI] [PubMed] [Google Scholar]
- Sosic D, Richardson JA, Yu K, Ornitz DM, Olson EN. Twist regulates cytokine gene expression through a negative feedback loop that represses NF-κB activity. Cell. 2003;112:169–180. doi: 10.1016/s0092-8674(03)00002-3. [DOI] [PubMed] [Google Scholar]
- Spiegelman BM, Hotamisligil GS. Through thick and thin: Wasting, obesity, and TNFα. Cell. 1993;73:625–627. doi: 10.1016/0092-8674(93)90243-j. [DOI] [PubMed] [Google Scholar]
- Sunnarborg SW, Hinkle CL, Stevenson M, Russell WE, Raska CS, Peschon JJ, Castner BJ, Gerhart MJ, Paxton RJ, Black RA, et al. Tumor necrosis factor-α converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J Biol Chem. 2002;277:12838–12845. doi: 10.1074/jbc.M112050200. [DOI] [PubMed] [Google Scholar]
- Szabowski A, Maas-Szabowski N, Andrecht S, Kolbus A, Schorpp-Kistner M, Fusenig NE, Angel P. c-Jun and JunB antagonistically control cytokine-regulated mesenchymal-epidermal interaction in skin. Cell. 2000;103:745–755. doi: 10.1016/s0092-8674(00)00178-1. [DOI] [PubMed] [Google Scholar]
- Tarutani M, Itami S, Okabe M, Ikawa M, Tezuka T, Yoshikawa K, Kinoshita T, Takeda J. Tissue-specific knockout of the mouse Pig-a gene reveals important roles for GPI-anchored proteins in skin development. Proc Natl Acad Sci. 1997;94:7400–7405. doi: 10.1073/pnas.94.14.7400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale MJ. Cachexia in cancer patients. Nat Rev Cancer. 2002;2:862–871. doi: 10.1038/nrc927. [DOI] [PubMed] [Google Scholar]
- Werner S, Smola H. Paracrine regulation of keratinocyte proliferation and differentiation. Trends Cell Biol. 2001;11:143–146. doi: 10.1016/s0962-8924(01)01955-9. [DOI] [PubMed] [Google Scholar]
- Wisniewska M, Goettig P, Maskos K, Belouski E, Winters D, Hecht R, Black R, Bode W. Structural determinants of the ADAM inhibition by TIMP-3: Crystal structure of the TACE-N–TIMP-3 complex. J Mol Biol. 2008;381:1307–1319. doi: 10.1016/j.jmb.2008.06.088. [DOI] [PubMed] [Google Scholar]
- Zenz R, Wagner EF. Jun signalling in the epidermis: From developmental defects to psoriasis and skin tumors. Int J Biochem Cell Biol. 2006;38:1043–1049. doi: 10.1016/j.biocel.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Zenz R, Scheuch H, Martin P, Frank C, Eferl R, Kenner L, Sibilia M, Wagner EF. c-Jun regulates eyelid closure and skin tumor development through EGFR signaling. Dev Cell. 2003;4:879–889. doi: 10.1016/s1534-5807(03)00161-8. [DOI] [PubMed] [Google Scholar]
- Zenz R, Eferl R, Kenner L, Florin L, Hummerich L, Mehic D, Scheuch H, Angel P, Tschachler E, Wagner EF. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature. 2005;437:369–375. doi: 10.1038/nature03963. [DOI] [PubMed] [Google Scholar]