

Abstract
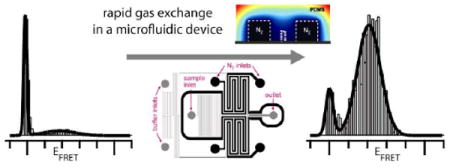
A microfluidic device made of PDMS addresses key limitations in single-molecule fluorescence experiments by providing high dye photostability and low sample sticking. Photobleaching is dramatically reduced by deoxygenation via gas diffusion through porous channel walls. Rapid buffer exchange in a laminar sheath flow followed by optical interrogation minimizes surface-sample contacts, and allows the in situ addition and combination of other reagents.
Single-molecule Förster resonance energy transfer (smFRET) experiments can elucidate biomolecular structure and dynamics at a resolution beyond the reach of common ensemble techniques.1 The quality of the smFRET signal depends on the number of emitted photons, which is generally limited by photobleaching of the fluorescent dyes. Because a substantial fraction of photobleaching pathways are mediated by molecular oxygen,2-6 a common remedy is the addition of an enzymatic oxygen scavenging system. However, such systems alter the chemical environment of the sample, can interfere with biological function and increase the autofluorescent background. In addition, they are incompatible with a variety of biological studies and experimental designs, e.g. protein folding investigations that require denaturants. Another limitation in single-molecule experiments is that proteins often have a strong tendency to adhere to surfaces, which can severely complicate experiments by depleting molecules or specific structural subpopulations. This problem is particularly pervasive because of long measurement times and the large ratio of surface area to the number of molecules in smFRET experiments on freely diffusing molecules,1 a popular measurement format that is utilized in this work. Adhesion problems can be mitigated by various surface coatings,7 but these typically need to be optimized for specific proteins and medium conditions.
Here we report a microfluidic device made of a single cast of silicone elastomer polydimethylsiloxane (PDMS) sealed to a microscope cover glass that addresses the above problems, providing a high-performance platform for smFRET measurements. Using diffusive mixing in a laminar sheath flow,8 the sample is rapidly transferred from the loading buffer into the measurement buffer and optically interrogated before the sample molecules encounter any surfaces. The sample solution and sheath buffer are deoxygenated in the device without the use of enzymes or other additives. Oxygen diffuses out of the solutions through porous PDMS walls into neighboring channels that are continuously ventilated with pure nitrogen. The microfluidic device (Figure 1) has a network of flow channels with depth h = 80 μm for the sample solution and buffers. The fluorescently tagged sample is fed into the sample inlet (Figure 1a). The measurement buffer is fed into the buffer inlets; its flow is evenly split into two channels and directed to a junction (Figure 1a,c), where the sample stream is sandwiched laterally between the two streams of the measurement buffer. The volumetric flow rate of the sample solution is substantially lower than that of the measurement buffer. Therefore, the sample solution stream is squeezed to a width wp of 1.8 μm. This small width leads to diffusive exchange of the buffer in the sample solution at a time scale tex= wp2/(8D) < 1 ms, for small molecules with a typical diffusion coefficient D = 5·10-6 cm2/s. The sample spends ∼100 ms traveling from the junction to a measurement point in a 300 μm wide interrogation channel. This time is more than sufficient for buffer exchange, but is substantially shorter than the time of diffusion of typical protein molecules to the side walls (∼2s).
Figure 1.

(a) Schematic of the microfluidic device. Channels for sample solution and buffers (grey), and for nitrogen (black). (b) Cross sectional view of a flow channel flanked by two nitrogen channels, with color-coded concentration of oxygen in PDMS. (c) 2D focusing and interrogation channel. Blue cone indicates measurement point.
Deoxygenation is based on the high gas permeability of PDMS9,10 and achieved by a continuous flow of nitrogen through two 160 μm deep, 200 μm wide gas channels flanking the flow channels upstream of the junction. In order to maximize the lateral diffusion of oxygen from the flow channels into the gas channels and to minimize the exposure of the flow channels to oxygen, the flow channels are made narrow (40 μm) and the distance between the flow and gas channels is made small (100 μm, Figure 1b). Experimental tests with an oxygen-sensitive fluorescent dye11,12 showed that at the typical flow velocities of smFRET experiments, [O2] in the optical interrogation area was <0.5% (with 20.9% corresponding to a solution saturated with air; see Supplementary Methods and Supplementary Figure 1-5 online for more details about the design, fabrication, operation, and testing of the device).
We first tested the microfluidic device using a FRET construct consisting of a common dye pair, Alexa488 (donor) and Cy5 (acceptor), attached 9 base pairs apart on a DNA duplex. The excitation power was set to a relatively high level (Supplementary Figure 6), at which photostability is a major limitation for smFRET. An smFRET histogram of this sample without additives measured in a standard cuvette exposed to atmospheric air exhibits two peaks (Figure 2a, left panel). The low amplitude peak at a FRET efficiency EFRET = 0.39 (FRET peak) originates from FRET in DNA molecules dual-labeled with active dyes. The peak at EFRET = 0 (zero peak) originates from events without acceptor signal, which can occur due to inactivation of acceptors by photobleaching, transition into dark states, or insufficient labeling. The sample was then tested in the device, which was operated at a flow speed of 1 mm/s at the measurement point with the gas channels ventilated with ambient air. The flow through the detection volume reduced the zero peak and enhanced the FRET peak (Figure 2a, middle panel), due to a reduction e.g. in the reentry of previously bleached molecules into the detection volume. Ventilation of the gas channels with N2 resulted in a further substantial decrease of the zero peak (Fig. 2a, right-hand panel). This signal gain was due to suppression of oxygen mediated bleaching pathways, in agreement with previous studies that used enzymatic scavenger systems.4,5,13,14
Figure 2.
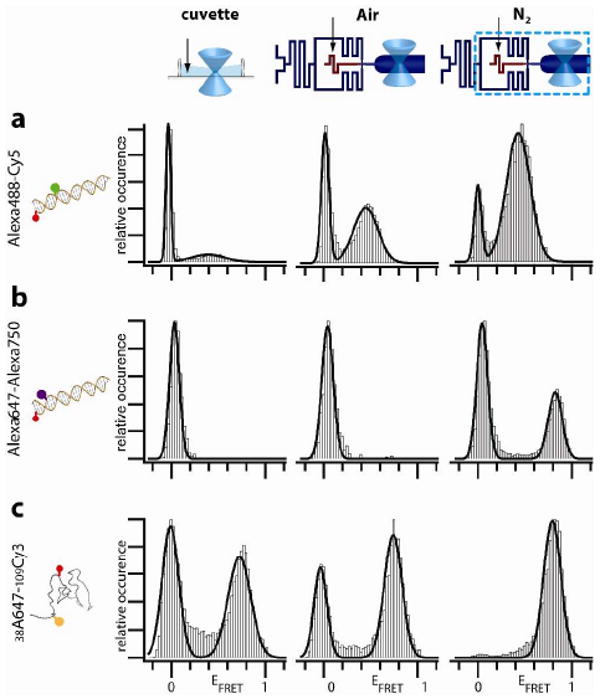
EFRET histograms of dual-labeled samples measured in a cuvette (left panels), in the microfluidic device with ambient air (middle panels) or N2 (right panels) flowing through the gas channels. (a) DNA labeled with Alexa488-Cy5. (b) DNA labeled with the near-IR dye pair Alexa647-Alexa750. (c) T4Lysozyme labeled with Cy3 and Alexa647.
We then tested the proposed additive-free deoxygenation method for enhancement of the signal of dye pairs with emission in the near-IR. The enhancement of the smFRET spectral range provided by such dye pairs is of great interest for advanced multicolor smFRET studies, which enable monitoring of multiple distances within a molecule and visualization of the coupling between conformational changes and binding.15 However, the use of IR dyes such as Alexa 750 in smFRET measurements is hampered by their rapid photobleaching, which is especially problematic for multicolor measurements. Indeed, for a dsDNA sample labeled with Alexa 647 (donor) four base pairs apart from Alexa 750 (acceptor), virtually no EFRET population was detectable in the cuvette and in the microfluidic device with air in the gas channels (Figure 2b, left and middle panels). Ventilation of the gas channels with N2 had a dramatic effect, producing a strong peak at EFRET ∼ 0.8.
Next, the performance of the device was tested at conditions relevant for protein folding studies, with a denaturant in the medium. Experiments were performed with the protein T4 lysozyme, which was site-specifically labeled with Cy3 and Alexa647 at amino acids 5 and 38 using a recently reported labeling strategy based on incorporation of the genetically encoded non-natural amino acid p-acetylphenylalanine.16 SmFRET experiments on a solution of the protein in a denaturing buffer (4M GdmCl in the solution and in the buffer fed to the buffer inlet; Figure 2c) showed that the zero peak was essentially eliminated by performing the measurements in the microfluidic device with deoxygenation with N2. We note that deoxygenation with an enzymatic system is not possible here because the essential enzymes are not functional in the presence of 4M GdmCl.17 On another note, the absence of any detectable zero peak after deoxygentaion also shows that the strategy of orthogonal labeling via genetically encoded unnatural amino acids results in nearly 100% labeling efficiency.16
To test the capability of the device to facilitate smFRET measurements on proteins in sticky native conformations, the experiments described in the previous paragraph were repeated with a physiological buffer (without GdmCl) fed to the buffer inlets. The removal of GdmCl from a protein solution in the buffer exchange channel (Figure 1) led to folding of the protein, as evidenced by a shift of the FRET peak from EFRET ∼0.1 to ∼0.4 (Figure 3a right panel vs. middle panel and Supplementary Figure 7). Importantly, the removal of the denaturant did not result in appreciable sample loss as compared with the experiments described in the previous paragraph. In contrast, smFRET measurements for GdmCl concentrations lower than 1M in an optical cuvette were severely complicated by continuous and unpredictable loss of the sample (Figure 3a and Supplementary Figure 7). As a more general strategy to prevent sample loss due to surface sticking, the sample of proteins or other biological species can be dissolved in a buffer promoting biomolecule solubility (e.g. with detergents), which is exchanged in the device to an appropriate measurement buffer immediately upstream of the measurement point.
Figure 3.
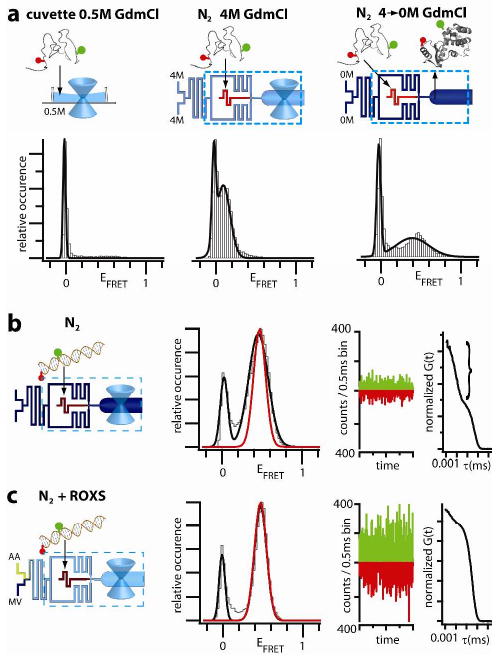
(a) T4Lysozyme dual-labeled with Alexa488-Cy5 measured in a cuvette in 0.5M GdmCl (left) and in the device with N2 flow in 4M GdmCl buffer (middle) and after the buffer is exchanged to 0M GdmCl, which induces folding (right). (b,c) DNA labeled with Alexa488-Alexa647 measured in the device with N2 and N2 plus ROXS. EFRET histograms (left panels; fit to the N2+ROXS peak overlayed in red), real time traces of photon counts per 0.5 ms (middle panels), and FCS curves (right panels, triplet component in bracket) are shown.
Depending on the experimental conditions and properties of the dyes and labeling sites, removal of oxygen can substantially increase dye blinking (switching between fluorescent bright and dark states), 18 thus reducing the brightness of the fluorophores. For molecules in solution, this is reflected in a large amplitude of the triplet fraction in fluorescence correlation spectroscopy (FCS) data, as we observed for Alexa488-Alexa647-DNA (Figure 3b, right panel, indicated by a curly bracket) and Cy3b-Alexa647-siRNA (Supplementary Figure 9).
We tested the effect of a well-studied triplet quencher ROXS 5,13,14 to improve signal quality under such conditions. ROXS was made by on-chip mixing of the oxidizer methyl viologen (MV) and reductant ascorbic acid (AA), which were fed to the two buffer inlets. Their streams merged and proceeded through a long channel, where they mixed by diffusion, before splitting into the two sheath buffer streams (Figure 1a). This pre-mixing of ROXS just prior to optical interrogation was beneficial for reducing the autofluorescent background from degrading ROXS (Supplementary Figure 8). We note that due to differences in the optical setups and detection schemes, fluorescent background of the solution is a much larger problem in smFRET experiments with diffusing than with immobilized samples. The use of ROXS in addition to deoxygenation led to substantial improvements for the DNA (Figure 3b,c) and siRNA data (Supplementary Figure 9). In fact, a major increase in the photon-counts per event was evident in the raw time-trace data, and the triplet amplitude in the FCS curve was substantially reduced as compared to the experiment without ROXS (cf. Figure 3b and 3c). This enhancement of photon flux (brightness) increased the signal-to-noise ratio and reduced the width of the FRET peak from 0.17 to 0.10 for the DNA (results from Gaussian fits of the N2 + ROXS peaks are overlaid in Figure 3b and c in red to highlight this reduction). Thus, the enhanced photon flux produces narrower EFRET peaks (also see Supplementary Figure 10a), which will permit improved separation of populations with similar FRET efficiencies, improved time resolution, and faster acquisition of FRET histograms (Supplementary Figure 10b). An improved time-resolution in single molecule flow experiments should prove very valuable in mapping the time-evolution of transient structural subpopulations or intermediates during rapid protein folding or other biological processes.
The presented device offers several benefits for smFRET experiments as compared to standard techniques. Most important, efficient inhibition of oxygen-mediated photobleaching is achieved independent of solution composition (denaturant, ionic strength, or pH) and without additional chemicals. A triplet quencher (or another additive) for further signal improvement can be supplied to the measurement buffer and a product of a binary reaction, such as ROXS, can be formed in situ. The short time interval between buffer exchange and optical interrogation also minimizes potential negative effects of cross-reactivity between additives and the biological specimen. Loss and degradation of potentially highly adherent folded or other conformational states of molecules are minimized by rapid transfer from non-adherent conditions immediately followed by smFRET measurement, which makes the device also a potential tool for kinetic studies of protein folding.19,20 Importantly, the brighter signals achieved here also translate into better peak- and time-resolution. Using these features, we have demonstrated substantial improvements for DNA, protein, and RNA samples, with different dye pairs, highlighting the broad applicability of this technology. We note that the good spectral separation of the Alexa647-Alexa750 dye-pair from common visible smFRET dye-pairs makes it an excellent candidate for use in future 4-color smFRET technology. The sample consumption is extremely low, (e.g., ∼2 μL of a 0.1 μM protein solution is sufficient per day), and the novel architecture of the device, with deoxygenation in a single layer of microchannels, makes it robust, simple to fabricate, and easy to operate and reuse. Based on these multiple benefits, we anticipate that this experimental platform will substantially extend the reach of single-molecule studies in a broad range of equilibrium and kinetic investigations, including folding, assembly, and function of proteins and other molecular machines of the cell.
Supplementary Material
Acknowledgments
We thank Lubica Supekova for assistance with the siRNA. This work was supported by grants PHY0750049 and OCE-0428900 from the NSF (A.A.D., V.V., and A.G.), GM073104 from the NIGMS, NIH (A.A.D.), NIH GM62159 (P.G.S.) the Humboldt Foundation (E.A.L.), the LJIS program (Y.G.) and Taiwan Merit Scholarships Program (H-W.L.).
Footnotes
Supporting Information Available. General procedures, design fabrication and characterization, supporting figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Alex Groisman, Email: agroisman@ucsd.edu.
Ashok A. Deniz, Email: deniz@scripps.edu.
References
- 1.Deniz AA, Mukhopadhyay S, Lemke EA. J Royal Soc Interface. 2008;5:15–45. doi: 10.1098/rsif.2007.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Donnert G, Eggeling C, Hell SW. Nat Methods. 2007;4:81–86. doi: 10.1038/nmeth986. [DOI] [PubMed] [Google Scholar]
- 3.Eggeling C, Widengren J, Brand L, Schaffer J, Felekyan S, Seidel CAM. J Phys Chem A. 2006;110:2979–2995. doi: 10.1021/jp054581w. [DOI] [PubMed] [Google Scholar]
- 4.Rasnik I, McKinney SA, Ha T. Nat Methods. 2006;3:891–893. doi: 10.1038/nmeth934. [DOI] [PubMed] [Google Scholar]
- 5.Vogelsang J, Kasper R, Steinhauer C, Person B, Heilemann M, Sauer M, Tinnefeld P. Angew Chem-Int Ed. 2008;47:5465–5469. doi: 10.1002/anie.200801518. [DOI] [PubMed] [Google Scholar]
- 6.Kong XX, Nir E, Hamadani K, Weiss S. J Am Chem Soc. 2007;129:4643–4654. doi: 10.1021/ja068002s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rasnik I, Mckinney SA, Ha T. Acc Chem Res. 2005;38:542–548. doi: 10.1021/ar040138c. [DOI] [PubMed] [Google Scholar]
- 8.Knight JB, Vishwanath A, Brody JP, Austin RH. Phys Rev Lett. 1998;80:3863–3866. [Google Scholar]
- 9.Merkel TC, Bondar VI, Nagai K, Freeman BD, Pinnau I. J Polymer Sci Part B. 2000;38:415–434. [Google Scholar]
- 10.Vollmer AP, Probstein RF, Gilbert R, Thorsen T. Lab On A Chip. 2005;5:1059–1066. doi: 10.1039/b508097e. [DOI] [PubMed] [Google Scholar]
- 11.Sud D, Mehta G, Mehta K, Linderman J, Takayama S, Mycek MA. J Biomed Opt. 2006;11 doi: 10.1117/1.2355665. [DOI] [PubMed] [Google Scholar]
- 12.Mehta G, Mehta K, Sud D, Song JW, Bersano-Begey T, Futai N, Heo YS, Mycek MA, Linderman JJ, Takayama S. Biomed Microdevices. 2007;9:123–134. doi: 10.1007/s10544-006-9005-7. [DOI] [PubMed] [Google Scholar]
- 13.Vogelsang J, Cordes T, Forthmann C, Steinhauer C, Tinnefeld P. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0811875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cordes T, Vogelsang J, Tinnefeld P. J Am Chem Soc. 2009;131:5018–9. doi: 10.1021/ja809117z. [DOI] [PubMed] [Google Scholar]
- 15.Clamme JP, Deniz AA. Chemphyschem. 2005;6:74–7. doi: 10.1002/cphc.200400261. [DOI] [PubMed] [Google Scholar]
- 16.Brustad EM, Lemke EA, Schultz PG, Deniz AA. J Am Chem Soc. 2008;130:17664–5. doi: 10.1021/ja807430h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akhtar MS, Ahmad A, Bhakuni V. Biochemistry. 2002;41:3819–27. doi: 10.1021/bi0116700. [DOI] [PubMed] [Google Scholar]
- 18.Vogelsang J, Cordes T, Tinnefeld P. Photochem Photobiol Sci. 2009;8:486–96. doi: 10.1039/b822318c. [DOI] [PubMed] [Google Scholar]
- 19.Hamadani KM, Weiss S. Biophys J. 2008;95:352–365. doi: 10.1529/biophysj.107.127431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lipman EA, Schuler B, Bakajin O, Eaton WA. Science. 2003;301:1233–1235. doi: 10.1126/science.1085399. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.