

SUMMARY
Early detection of prostate cancer using the prostate-specific antigen test led to a sharp spike in the incidence of the disease accompanied by an equally sharp improvement in patient prognoses as evaluated at the point of advanced diagnosis. Observed outcomes represent age at diagnosis and stage, a categorical prognostic variable combining the actual stage and the grade of tumor. The picture is summarized by the stage-specific cancer incidence that represents a joint survival-multinomial response regressed on factors affecting the unobserved history of the disease before diagnosis (mixture). Fitting the complex joint mixed model to large population data is a challenge. We develop a stable and structured MLE approach to the problem allowing for the estimates to be obtained iteratively. Factorization of the likelihood achieved by our method allows us to work with only a fraction of the model dimension at a time. The approach is based on generalized self-consistency and the quasi-EM algorithm used to handle the mixed multinomial part of the response through Poisson likelihood. The model provides a causal link between the screening policy in the population and the stage-specific incidence.
Keywords: incidence, joint modeling, mixed multinomial logit model, screening, generalized self-consistency
1. INTRODUCTION
We based our analyses of prostate cancer on the data from Surveillance, Epidemiology and End Results database (SEER, http://seer.cancer.gov/). The data set includes 331 227 cases of prostate cancer diagnosed between 1973-2000 in the age range of 50-100 in nine states and metropolitan areas of the U.S. participating in the SEER program. Incidence data are represented by age at diagnosis, a, and year of birth, x, for each cancer case, the count of cancer cases, C(a, x), as well as the count P(a, x) representing the male population over the age of 50. The Prostate-specific antigen (PSA) test invented in the late 80s is a biomarker for the early detection of prostate cancer. Increased utilization of the test in the US male population induced a dramatic spike of the observed prostate cancer incidence
| (1) |
where da=1 year. Shown in Figure 1 is λobs(a|x) plotted by age a and year of diagnosis x+a. The effect gradually builds up with age and peaks at about 70. The probabilistic characteristic λ(a|x) corresponding to the empirical incidence (1) is essentially a hazard rate for the age at diagnosis of prostate cancer in the x-birth cohort. We call λ(a|x) marginal incidence to distinguish it from the joint distribution characterizing the disease presentation at diagnosis.
Figure 1.
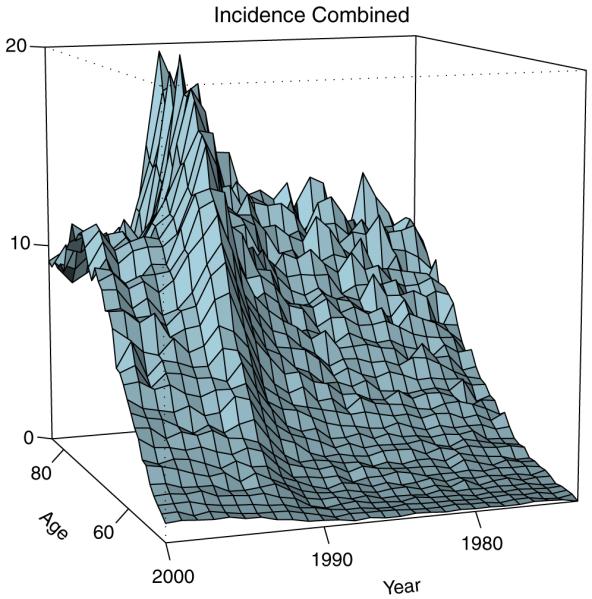
Observed marginal incidence λobs(a|x)×1000 of prostate cancer, λobs(a|x)=C(a, x)/P(a, x), by age a and year of diagnosis x+a. The empirical estimate corresponds to the ratio of the count of observed cases C to the size of the population P at risk measured in thousands. Single age-year bins are used. Figure is based on SEER data (http://seer.cancer.gov/).
Suppose, a categorical prognostic index Y is observed at diagnosis. Then we can consider the joint response represented by the random vector (A, Y). Here we treat the random age A at cancer diagnosis as a net competing risk, all other risks removed.
Here and in the sequel we will adopt the following notation rules unless noted otherwise. Capital letters are used for random variables and the corresponding lowercase letters for the values of those variables. In addition, λ will be a hazard function in its age a argument; f will be a probability density function (pdf); its discrete version will be denoted by π.
Two binary prognostic variables are considered: a stage and a grade of the disease. The stage is categorized as localized or regional (LR) vs distant (D). The grade represents a categorical assessment of the degree of differentiation of cancer cells, a measure of how different cancer cells are from the cells from which they originated. Poorer differentiation is caused by an increasing fraction of more rapidly dividing cells in a cancer tissue sample indicating a more aggressive cancer. In this paper, grade is dichotomized as well or moderately differentiated (WM) vs poor or undifferentiated (PU). While the exposition of the paper is general, in the examples Y is a multinomial response built from four possible combinations of the stage and grade of the disease as follows:
| (2) |
The categories of the prognostic index Y are ordered from the best prognosis to the worst. However, it is not clear whether a similar ordering exists in age as far as the natural history of the disease prior diagnosis is concerned. For example, it is not known whether tumor growth is accompanied by a progression of the grade, or the grade is constant in time given the subject. Therefore, we prefer not to make any ordering assumptions and treat Y as a multinomial rather than ordinal variable. On the population level it is clear that PSA screening leads to a shift to better categories of Y. Much of this shift is populated by subjects who would never be detected with cancer without PSA (overdiagnosis). The shift also occurs within the subject if the incidence of an advanced disease is prevented by the early detection of cancer while in a better prognostic category.
Let C(a, x, y) be the count of cases diagnosed in year x+a at the age of a and with prognostic index y. Then it is natural to break the empirical incidence (1) by y as
| (3) |
Clearly, , and . Plots of the empirical stage-specific incidence λobs(a, y|x) are shown in Figure 2. It is evident from Figure 2 that dynamics of the marginal incidence is dominated by the LR-stage-specific incidence. In addition, the incidence of the distant disease drops with the introduction of PSA indicating a favorable stage shift induced by the early detection of cancer on the population level.
Figure 2.

Observed stage- and grade-specific incidence (per 1000 men) of prostate cancer (3) by age a and calendar time x+a, where x is year of birth. Single year age and calendar time bins are used (da=1); SEER data (http://seer.cancer.gov/). Stage: LR=localized-regional, D=distant; grade: WM=well or moderate differentiation, low grade, PU=Poor or no differentiation, high grade.
The joint hazard function of the random vector (A, Y) is expressed by the y-specific incidence λ(a, y|x) defined as
| (4) |
The joint distribution λ(a, y|x) is a hazard function in a, and a discrete probability distribution in y. Obviously,
Let π(y|a, x) be the conditional distribution of the index Y for the patient diagnosed at the age of a in year x+a. We have the following factorization for the stage-specific incidence:
| (5) |
where the conditional probability π(y|a, x) represents a multinomial regression model for a factor partitioning marginal incidence into its Y-specific components. From (3) we have an empirical estimate
| (6) |
The joint modeling of (A, Y) as a function of screening processes measured on the population level is the focus of this paper.
It is evident that A and Y represent random variables with a complex pattern of dependency. This dependency is explained by the unobserved random processes W summarizing the latent history of the disease and the unobserved random screening schedule in the subject. We keep W general for now, and will specify it and reduce it to a random vector surrogate later in this paper. The subject-specific processes W are shared by A and Y, inducing dependence
| (7) |
where f is the joint pdf. In this paper we will build a mechanistic model for (7). This model is intended to predict how the policy of cancer screening (yet to be formalized) affects the stage-specific incidence of the disease. We will identify with the distribution of the random schedule , a point process. is defined on the population level, while the effect of screening occurs on the individual level. Screening schedules in the U.S. population are not observed on the individual level. The problem thus is about building a link between a functional parameter and the multivariate response (A, Y). In the sequel, the dependence on will be suppressed for brevity when is not the focus of the exposition.
The estimation approach will consist of the following two stages.
Stage 1
- First we will set up the model for [A|x] using a marginal likelihood approach
(8) Second, we will determine the model for , a suitable surrogate of W. We will use posterior prediction based on the marginal model to obtain .
Stage 2
Assuming that the distribution represented by the pdf to be known from the previous stage, we will build and fit the model for π(y|a, x) using as a subject-specific random effect
| (9) |
Stage 1A has already been accomplished [1], and we will review this model in Section 3. Stage 1B will be dealt with in Section 4. In Section 5 multinomial mixed model will be formulated, and interpreted as an artificial mixture model. Using this interpretation we will devise the estimation procedures based on self-consistency [2, 3] in Section 7. An application of the methodology to SEER data and a simulation study will be done in Section 8.
A number of analytic marginal [1, 4, 5] and simulation models [6, 7] have addressed overdiagnosis and marginal incidence λ(a|x). Most analytical models of cancer screening are estimated based on screening trials data where the screening schedules are known. Simulation models are informed by a broad range of studies and the literature data. In both scenarios the population predictions of cancer incidence are generated based on a simulation of screening schedules in the real population, or using hypothetical schedules. Oftentimes, the process involves an ad hoc calibration of the model predictions to the observed population incidence. Our approach is different in that it is used to build inference procedures directly using population data such as the SEER cancer registries.
2. LIKELIHOOD
Likelihood construction is essentially similar to the argument used for martingales based on counting processes [8, 9], and in the age-period-cohort models [10]. Cases C(a, x, y) originate from a population at risk of cancer detection of the size P(a, x). In practice we will assume da=1 year to be sufficiently small for Taylor first-order approximations. In addition, the disease will be considered as rare enough to identify P with the population as reported in SEER. Empirical evidence to the validity of these assumptions is the fact that C⪡P as given by SEER data.
For a subject without prior cancer diagnosis of age a observed at time t=x+a, the conditional probability to be detected with cancer, marked by a prognostic index y, is given by λ(a, y|x)da as in (4). The marginal probability of being detected with cancer of any stage is . The conditional probability not to be detected is 1-λ(a|x)da. Therefore, the conditional likelihood is proportional to
| (10) |
Using the product-integral representation [11]
and dropping the terms that do not depend on the model parameters, the log-likelihood takes the form
| (11) |
Note that even though the likelihood (11) has Poisson form, the counts C do not represent a Poisson process. This is because P(a, x) are arbitrary, and the same subjects are contributing to multiple Ps inducing the dependence of Ps for different values of its arguments. Essentially, (11) is a consequence of the fact that any counting process behaves locally within a small interval da as a Poisson process, conditional on the prior history. In addition, (10) is a consequence of the number of events in the small interval being a Bernoulli random variable.
The factorization (5) yields a partition of the above likelihood ℓ=ℓI+ℓY into the one for the marginal incidence model estimated at the Stage 1 (8)
| (12) |
and the one for the multinomial response Y estimated at Stage 2 (9)
| (13) |
As we mentioned earlier in the Introduction, our approach will be to use the marginal likelihood ℓI to estimate the parameters of the conditional joint distribution of [W|a, x] (or its surrogate), and then to use these estimates to specify the mixed model likelihood for Y
| (14) |
based on (9). In doing so we will assume that at Stage 2 the distribution f (w|a, x) is known. This is justified by the fact that C⪡P. Note that the marginal incidence model of Stage 1 is fitted to the whole population P, while the Stage 2 model is fitted to cases C only.
3. THE MARGINAL INCIDENCE MODEL
The marginal incidence model [1] developed earlier as a mixed model for λ(a|x) will serve as a basis for the first stage (marginal modeling). The natural history of cancer is a version of the classical model [12]. A subject passes through the disease-free state and the pre-clinical state before being detected with cancer or censored without a diagnosis. The subject’s age at tumor onset AO represents the duration of the disease-free stage. It is assumed that AO has Weibull distribution with the hazard function of the form
where a is the age past 50. In the above expression, the Weibull distribution is parameterized through the mean μO and the shape parameter sO.
The duration of the pre-clinical stage is given by the delay time ξD=A-AO represented by the period between cancer onset at the age of AO and its diagnosis at the age of A. We assume that cancer diagnosis is a result of two competing risks, the time to detection by screening, ξSDx, and the time to the clinical diagnosis due to symptoms of the disease, ξCDx, so that ξD=min(ξSDx, ξCDx). The time ξCDx is referred to as the sojourn time. The Weibull distribution with mean μCDx and shape parameter sCDx is used to model the baseline sojourn time hazard hCDx(ξ), where ξ is the time since tumor onset. The sojourn time is affected by changes in the practice of cancer detection other than the studied modality of screening. For example, before PSA was introduced, prostate cancer was often detected incidentally as a result of surgery for benign prostate disorders (Transurethral Resection of the Prostate) [13]. These trends in the sojourn time hazard λCDx(ξ|t) are modeled using a multiplicative trend function TCDx(t) acting on the baseline sojourn time hazard hCDx
| (15) |
where ξ is the time since tumor onset (age of tumor), and t is the calendar time at this age. The trend function was assumed to be linear between the earliest year where data were available, 1973, and the last year before PSA was introduced, 1987, saturating in 1988. The trend function is set to a constant outside the observed data window, resulting in
Suppose are random ages when the subject is screened (a screening schedule). Given AO, the result of a screen at the age of a=AO+ξ is an independent Bernoulli random variable with the probability of success (detection) α(ξ), a non-decreasing function. The sensitivity of screening as a function of age is then
| (16) |
The detection by screening occurs at the age of AScr=τν, where ν is the number of the first successful screen in a series of so-called Poisson trials (Bernoulli trials with unequal probabilities). In order to specify AScr=τν, we also need to specify the process . The National Cancer Institute and collaborators have developed a generator of PSA schedules (a simulation model) for U.S. males [14]. Having experimented with the generator, we came up with the following analytical model approximating its output. Let λ1S(a, t) be the instantaneous risk (hazard) of the first PSA test for a man of age a in year t. Then the probability that a man born in year x will not be tested by the age of a is
| (17) |
We assume that in men who already had their first PSA tests, secondary tests {τ2, τ3 . . .} form a non-homogeneous Poisson process with intensity λ2S(a, t). Both intensities of PSA testing λ1S and λ2S are treated as known bivariate functions of age and calendar time, and are derived by a large sampling from the schedule generator. We identify the screening policy with the set of the two screening intensities defined as functions on the age by calendar time plane.
The survival function GSDx(ξ|x, AO) is the probability of no screening diagnosis for a subject with the following characteristics:
year of birth x;
projected age at tumor onset AO;
age of tumor (time since onset) ξ;
current age AO+ξ,
is given by (see [1] for the derivation)
| (18) |
where
| (19) |
and for any b≤a. The above expressions are a result of averaging over and ν, given AO.
Finally, using conditional independence of the competing risks of cancer diagnosis by screening and clinically, given age at tumor onset AO, we have
| (20) |
This model was fitted by maximizing the marginal likelihood (12) over the marginal incidence model parameters [1]. In the course of likelihood maximization, the screen sensitivity function specified as α(ξ)=1-exp(-βξ) converged to α=1(β→∞). Therefore, α was fixed at 1 for all subsequent analyses. The resultant estimates of model parameters are given in Table I.
Table I.
Estimates of model parameters and confidence intervals
| Parameter | Legend | Point estimate | 95 per cent CI |
|---|---|---|---|
| μCDx | Mean baseline sojourn time | 18.558 | (18.345, 18.775) |
| sCDx | Shape sojourn time | 1.541 | (1.5191, 1.5644) |
| c | Slope of trend for sojourn time | 0.09354 | (0.09068, 0.09641) |
| μO | Mean age past 50 at tumor onset | 72.732 | (72.498, 72.965) |
| sO | Shape of age past 50 at tumor onset | 1.6153 | (1.6067, 1.6239) |
| α | Screening sensitivity | 1.00 | — |
Time and age is measured in years.
4. A SURROGATE OF THE EFFECT OF SCREENING ON THE NATURAL HISTORY OF THE DISEASE
One could consider mechanistic mixed models of various complexity based on the mixing variables
associated with the natural history of the disease and screening. Screening intervention affects W, which determines the age at cancer diagnosis A=AO+min(τν, ξCDx). In addition, the stage Y is thought of as being regressed on W. With screening the distribution π(y|a, x) shifts toward values of y associated with better prognosis as a consequence of early detection.
From the point of view of computational tractability of the problem it would be unreasonable to use the mixing variable as complex as W. To simplify the modeling of π(y|a, x)=E{π(y|a, x, W)|a, x}, we replace W by a suitable surrogate of the unobserved processes that would allow us to reproduce the behavior of the observed response [Y|a, x]
| (21) |
A stronger assumption would be that of the perfect surrogacy [15] that implies (21) as
where the first equality is a formula of total probability while the second one is a consequence of the perfect surrogacy.
A combination of the delay time AD=A-AO between tumor onset and its diagnosis and the mode of cancer detection Iscr
is taken as a parsimonious surrogate of the effect of the tumor progression history, early detection and screening on the observed prognostic index Y,
The resultant model has the form
| (22) |
As we will see in Section 8, predictions based on the model (22) with the complete-data part specified according to the multinomial logit model show good agreement with the empirical estimates.
Given the age at tumor onset aO, the time to the diagnosis is a result of competing risks of clinical and screen detection. The two crude probability densities f (aD, iscr|aO, x), iscr=0, 1
| (23) |
represent the joint conditional distribution of , conditional on aO, x. Note that we can condition on age at diagnosis a instead of aD, since a=aO+aD, and aD is a part of
| (24) |
This pdf is used in the integral representing expectation (22), and in other similar expectations.
Shown in Figure 3 is the distribution (24) of the latent vector , a combination of the delay time, AD, and an indicator of screening diagnosis, Iscr, derived from the marginal incidence model [1] as specified in Section 4.
Figure 3.

Distribution (23) of the mixing vector derived from the marginal incidence model. Top left: marginal distribution of the mode of detection Iscr; probability of screening diagnosis. Top right: crude distribution of delay time AD for disease diagnosed by screening in year 1995. Bottom left: same as top right for clinical diagnoses in year 1995. Bottom right: same as bottom left in year 1980. Note the shift of probability mass toward smaller delay times with the introduction of screening, particularly for screen-detected cancers.
The marginal distribution of the mode of diagnosis shown in the top left corner of the figure indicates a rapid increase in the fraction of screen-detected prostate cancers in the late 80-ties. It is also clear that a pool of subjects around 70 years of age is most likely to be screen-detected. Lower detection rates in younger patients can be explained by lower prevalence of the latent disease in this group, while lower rates in very old men are likely a consequence of reduced screening rates in this population associated with perceived high chance of over-diagnosis and short residual lifetime expectation.
The dynamics of the joint distribution of the delay time and the mode of detection with calendar time shown in the rest of Figure 3 is quite remarkable. Note that the support for the distribution of age at onset of prostate cancer is assumed to start at the age of 50, as very few cases of prostate cancer are seen before that age. Thus, the delay time cannot be larger than age-50, which makes parts of the plots in Figure 3 corresponding to delay times exceeding the boundary equal to 0. Comparing year 1980 before PSA (bottom right) with year 1995 we see that the probability mass moves toward shorter delay times indicating early detection. We also see that a high chance of the screen detection around 70 years of age shows as a peak of probability mass for the crude density of the delay time for the screen detected cases (top right) and as the corresponding niche carved out in the crude density of the delay time for cases missed by screening (bottom left).
In what follows we will suppress the ‘∼’ accent from the surrogate , will consider W a random variable (r.v.) conditional on a, x, and will remove this conditioning from expectations for brevity.
5. THE MULTINOMIAL MIXED MODEL
5.1. Multinomial logit model
Let Yi∈{1, 2, . . . , K} be the ith subject’s multinomial response in one of the K possible categories. Let
a random variable, and
its realization in the ith subject. Let zi be a vector of observed covariates and Wi be a vector of unobserved (random) covariates with known joint distribution that may depend on zi.
The marginal category-specific probabilities averaged over the unobserved covariates are given by
| (25) |
where n is the sample size. Connecting the various notations introduced earlier we may write
On the complete-data level the probabilities Pr{Δik=1|zi, wi} are given by the multinomial logit model
| (26) |
where the functions θ are parameterized using regression coefficients αk and βk associated with unobserved and observed covariates, respectively. We will use the following parameterization:
For identifiability, regression coefficients corresponding to the baseline first category are set to βi1=αi1=0, hence the summation in the denominator of (26) starts from c=2. Note that θ factors into into and ,
| (27) |
6. GENERALIZED SELF-CONSISTENCY
Maximum likelihood estimation in a variety of settings can often be simplified by substituting complex multinomial likelihoods by Poisson likelihoods as an approximation [5] or augmentation of the original model [16]. Multinomial-Poisson (MP) transformation has been a popular technique simplify maximum likelihood estimation in a variety of models yielding multinomial likelihoods [16]. The approach works by substituting a Poisson likelihood for the multinomial likelihood at the cost of augmenting the model parameters by axillary ones and restricting the problem to discrete covariates. The MP transformation can be justified through the method Lagrange multipliers [17]. In this paper we use an alternative approach based on the generalized self-consistency methodology [2, 3] that allows us to use Poisson likelihood with arbitrary covariate structures. The proposed procedure is very stable numerically as it does not include matrix inverses and works with only a fraction of dimension at a time. We extend the framework of [3] to the mixed model considered in this paper.
Let us artificially represent π(y|z, w) as a marginal probability based on the mixture
| (28) |
where U is a ‘missing’ variable, and π(·|·, U) are complete-data probabilities conditional on U. In other words, an artificial mixture model is considered such that one gets the original target model when the missing data are integrated out.
In the multinomial case, exponentially distributed random variable U ∝Exp(1) yields an artificial mixture formulation such that π on the left of (28) corresponds to the multinomial distribution, while π on the right of (28) gives a Poisson complete-data likelihood. The MLE procedure is an EM algorithm with the E-step solving the problem of imputation of U and W, while the M-step dealing with maximizing a log-likelihood obtained from the complete-data model π(y|z, w, u).
The multinomial probabilities (26) can be written in the artificial mixture form
| (29) |
where
Note that the complete-data model π0=π(y|z, w, u) obtained by dropping the ‘E’ symbol from (29) does not make sense as a probability. It suffices to note that the expression under E is not normalized. We call the resultant algorithm Quasi-EM to acknowledge this issue.
The multinomial logistic model (26) is constructed by linking response probabilities to positive predictors θ. Then θs are normalized to model probabilities. The alternative procedure (29) achieves normalization by means of averaging over artificial missing data.
Note that the log-complete-data ‘probabilities’ resulting from (29) give rise to a Poisson-form complete-data likelihood. Indeed, collecting the terms of the complete-data log-likelihood specific to category k, we get
| (30) |
where ℓk are Poisson likelihoods parameterized using a subset of the parameter matrix β specific to the category k of the response,
| (31) |
and U={Ui}, W={Wi}. Factorization (27) of θ will ensure that the Poisson form (31) be preserved after the averaging over the random effects W. In addition, ℓk is linear in U. The above observations make the EM-like approach to the problem particularly attractive.
Generalized self-consistency argument replaces E in (28) by a quasi-expectation operator QE acting on π(·|·, u) as a function of u so that it would no longer be required that the complete-data model π(y|z, w, u) makes sense as a probability. An argument similar to [3] justifies the Quasi-EM algorithm to be constructed despite the fact that the complete data model is not real.
7. THE ALGORITHM
7.1. E-step
Based on [3], for any function φ(V) of a random vector V=(U, W), the imputed value φ‾ of φ(V) has the form
| (32) |
where L(β, α|V)=eℓ(β, α|V) is the complete-data likelihood given by (30). The contribution of the ith subject with the response in category yi=k to the complete-data likelihood follows from (29) and is represented by complete-data ‘probabilities’
| (33) |
We consider an iterative sequence of parameters α(m), β(m), m=1, 2, . . . . With the M-step of the algorithm in mind, φ in (32) is parameterized by the next-iteration (m+1)-parameters, while L in (32) is parameterized using the previous iteration (m) version. Applying the imputation operator (32) to the complete-data log-likelihood (30) φ(V)=ℓ(β(m+1), α(m+1)|V) we get after a little algebra
| (34) |
where
In the derivation of (34) we used the fact that
obtained by the differentiation of the Laplace transform of the exponential distribution
| (35) |
with respect to s, and a change of the order of summation with respect to subjects i and categories k.
7.2. M-step
The problem to be solved at the M-step is
| (36) |
Note that the β(m+1)-kernel of ℓ‾ represented by the second line of (34) has the form of a Poisson regression likelihood, and moreover that, given , the estimates of , k=1, . . . , K, can be obtained independently using Poisson likelihoods
| (37) |
When the Poisson likelihood is interpreted as a likelihood for rates, δik plays the role of a ‘count of events’, and the expectation in the right part of (37) plays the role of ‘person years’.
Likewise, the solution for can be obtained independently for each category k=2, . . . , K, given using the -likelihood kernel
| (38) |
Therefore, it is convenient to solve the M-step problem (36) by a Gauss-Zeidel procedure alternating between
| (39) |
given and
| (40) |
given , within each category k=2, . . . , K. One could use a unidimensional line search along the vector
after each iteration of Gauss-Zeidel to speed up the convergence, which would make this procedure similar to conjugate gradient methods [18]. The category-specific M-step likelihoods (37) and (38) are maximized by a Newton-Raphson procedure with respect to the low-dimensional fixed effect coefficients and the random effect coefficients , respectively.
7.3. Standard errors
Standard error estimates are based on the inverse of the observed information matrix
| (41) |
where μ={β, α} is the combined vector of the model parameters, and
is the model log-likelihood maximized as a result of the Quasi-EM algorithm. Here as in (30) we understand that U and W are collections of independent subject-specific random vectors, and that ℓ is a sum of contributions to the likelihood over subjects.
The observed information matrix is derived by an application of the missing information principle representing the observed information as the difference between expected complete-data information and the missing information, given observed data [19, 20]. Specifically, let I1(μ|W) be the observed information matrix conditional on W (the so-called complete-data information). This matrix corresponds to the standard multinomial logit model and is derived straightforwardly by differentiation of the log of the right part of (26) with k=yi, and summing up over subjects . Let S1(μ|W) be the corresponding complete-data score vector, the gradient of the complete-data likelihood . Then,
| (42) |
where we used notation for the conditional expectation similar to (32). Here
is the conditional expectation of (·) given observed data,
is the expected complete-data information, and
is the expected missing information. The above expectations are taken numerically for each subject based on the subject-specific contribution to the score vector and to the information matrix, and then summed up over subjects. Note that the observed information representation given above is only valid at the MLE point and will generally not hold true for any Hessian matrix unless the gradient of the observed likelihood is zero. Alternatively, a bootstrap estimate of standard errors could be done. In the bootstrap approach, parametric bootstrap was used. Replicates of the observed data were simulated from the fitted joint model λ(a, y|x) with the parameters set at the MLEs (Tables I and II). Empirical standard errors of the MLEs computed over a large number of replicates (1500) were used as bootstrap estimates.
Table II.
Parameter estimates for the mixed multinomial logit model fit to Prostate Cancer SEER Data. Each estimate shows MLE and the empirical mean (in brackets) of the MLEs obtained by parametric bootstrap (simulation from the fitted model). Standard errors (SE) and confidence intervals (CI) shown are based on parametric bootstrap. Upper part of the table has fixed effects, lower part shows random effects. Response categories: 1: Stage=LR, Grade=WM; 2: Stage=LR, Grade=PU; 3: Stage=D, Grade=WM; 4: Stage=D, Grade=PU
| Contrasts |
|||
|---|---|---|---|
| Variables | 2 vs 1 | 3 vs 1 | 4 vs 1 |
| Intercept | -0.509 (-0.500) | 2.301 (2.283) | 2.901 (2.890) |
| SE | 0.0125 | 0.0257 | 0.0236 |
| CI | (-0.524, -0.475) | (2.233,2.334) | (2.844,2.936) |
| Age | 0.0347 (0.0346) | 0.0638 (0.0636) | 0.0749 (0.0749) |
| SE | 0.00108 | 0.00201 | 0.00171 |
| CI | (0.0325,0.0368) | (0.0597,0.0676) | (0.0716,0.0783) |
| Year | -0.0144 (-0.0145) | -0.0560 (-0.0558) | -0.0585 (-0.0584) |
| SE | -0.000264 | 0.000488 | 0.000542 |
| CI | (-0.0150, -0.0140) | (-0.0567, -0.0548) | (-0.0595, -0.0573) |
| DT | -0.0375 (0.0376) | -0.153 (-0.152) | -0.249 (-0.249) |
| SE | 0.0340 | 0.0100 | 0.0128 |
| CI | (-0.0442, -0.0309) | (-0.171, -0.132) | (-0.274, -0.224) |
| ScrDx | -0.108 (-0.107) | -3.394 (-3.428) | 4-3.741 (-3.761) |
| SE | 0.0270 | 0.415 | 0.466 |
| CI | (-0.160, -0.0539) | (-4.241, -2.614) | (-4.674, -2.848) |
8. DATA ANALYSIS AND SIMULATIONS
Data for C(a, x, y) come from 331 227 prostate cancer cases diagnosed in nine areas of the U.S. (San Francisco-Oakland, Connecticut, Detroit, Hawaii, Iowa, New Mexico, Seattle, Utah, Atlanta) participating in the SEER program. Population counts P(a, x) come from the SEER population files corresponding to those areas. We use the age by year box corresponding to age interval [50, 100] and calendar year interval [1973-2000]. Of the total number of cases, the vast majority, 229 726, are detected in the early localized or regional (LR) stage with low grade representing well or moderately differentiated (WM) cells. 60 272 LR cases have high grade representing predominantly poorly differentiated or undifferentiated (PU) tumors. Of the total 41 229 distant (D) stage cancers, 17 957 were low grade (WM), and 23 272 were high grade (PU).
Multinomial response probabilities are based on the following parameterization of the predictors:
specific to subject i=1, . . . , n and category k∈{1, 2, 3, 4}. The response category k is coded as specified in the Introduction by (2). For the ith subject, the fixed effects z are represented by zi, 1=age-50, and zi, 2=calendar year-1900. Both covariates z are treated as continuous. Fixed-effects regression parameters βkr, r=0, 1, 2 are specific to the response category k. Identifiability restriction implies θ=1 when k=1, so that β1r=0, r=0, 1, 2. The set of intercepts βk0 models the baseline distribution of the stage and grade combined response Y, where the baseline group of subjects represents men 50 years of age at the beginning of the 20th century. Mixed effects are represented by two random intercept terms, the delay time Wi1=ξD, and the indicator of screening mode of detection Wi2=Iscr. The posterior distribution of the random vector W predicted from the marginal incidence model in Section 4 is shown in Figure 3. The random effects coefficients αkr, r=1, 2, are restricted to α1r=0 for the baseline response category for identifiability. Note that since the distribution of the random vector W is treated as known, αkr remain otherwise unrestricted.
The model-predicted marginal probabilities are computed by averaging over the random effects W
| (43) |
The EM algorithm of Section 7 is used to fit the model. The maximum likelihood estimates and their standard errors are shown in Table II.
To study the properties of the algorithm by simulations, we built a simulation counterpart of the model. Conditioning on the total number of cancer cases, we simulated their frequency by age, stage and grade at diagnosis from the joint model λ(a, y|x). Simulated data include 331 227 prostate cancer cases diagnosed in nine SEER areas of the U.S. The parameters of the simulation model were fixed at the MLEs given in Table II. For each of the 1500 replicates of the simulated data, the model (43) was fitted by the self-consistency algorithm of Section 7. Empirical means of the estimated regression coefficients show excellent correspondence with the original MLEs used in the place of the ‘true’ values (Table II). Table II also shows standard errors and 95 per cent confidence intervals derived from the simulation. The Q-Q plots for all 15 model parameters (not shown) suggest that MLEs are asymptotically normal.
The coefficients associated with the mode of detection (Table II) indicate that screening leads to an increase of the chance of detection in the best prognostic category of LR stage and WM grade, and a drop in the distant stage contrasts.
The negative coefficients associated with the calendar year indicate a secular trend of slightly improved prognosis over time. Age in Table II, however, has an adverse effect. Note that age has two causal paths according to the model: the direct effect as shown in the table, and the effect exerted through the age-dependent delay time distribution (Figure 3). Age effects are a result of a complex interplay of selection effects and the unobserved heterogeneity of tumor growth.
A tumor detected in an older person without screening is generally associated with a longer sojourn time because the sojourn time in this situation is bounded from above by age. If longer sojourn time is an indication that the tumor had more time for the latent growth, an advanced stage could be expected, leading to an adverse effect of age. If tumors are heterogeneous in the sense that faster growing tumors get detected earlier and present themselves in more advanced stages, age would exert a favorable selection effect on the tumor stage at diagnosis.
Screening adds another layer of complexity through the length bias effect. Length bias is a phenomenon known from point processes [12]. When a needle is thrown on the trajectory of a point process, the sojourn time between two successive events, conditional on the needle falling between them, is generally larger than the unconditional time between the events. The source of bias is the fact that the needle is more likely to catch a larger interval than a smaller one. Applied to cancer screening this means that the sojourn time for cancers detected by screening is generally larger than the unconditional duration of the pre-clinical stage in the population.
Age is clearly a surrogate of screening as the early detection generally leads to tumors detected at an earlier age. Based on the length bias phenomenon, one would therefore expect that slower growing tumors caught by screening would shift to earlier ages at diagnosis, while the more aggressive tumors missed by screening would become more pronounced in older men, a point in the direction of the adverse effect of age.
Larger sojourn and delay times may be indicative of slow tumor growth. Not surprisingly then we observe negative coefficients associated with the delay time that indicate an improved prognosis associated with slow tumor growth.
Without incorporating a mechanistic description of the tumor progression through stages, the model currently has a limited potential to measure attributions of the above mentioned effects to the dynamics of the response (A, Y). Our strategy has been to be inclusive about the possible tumor progression mechanisms, and to keep restrictions on the model to a minimum through the multinomial distribution assumption. The model was designed to construct a causal link between the screening policy in the population and the presentation of the disease at diagnosis summarized by the stage- and grade-specific incidence. This causal link would be impossible if the model simply conditioned on intermediate outcomes such as age at diagnosis, because such outcomes are affected by the policy.
Shown in Figure 4 is the model-based prediction of the stage- and grade-specific incidence (5) of prostate cancer that shows good agreement with its empirical counterpart (Figure 2). As evident from the figure, the model reproduces the spike of the incidence of the early stage cancer induced by screening and the corresponding drop in the incidence of the distant disease associated with a fraction of distant stages being prevented by the early detection.
Figure 4.

Model-based expected stage- and grade-specific incidence of prostate cancer by age and calendar time. LR=localized-regional stage; WM=well or moderate differentiation, low grade; D=distant stage; PU=poor or no differentiation, high grade.
9. LINK FUNCTIONS
In this section we study sensitivity of the results to the link function misspecification in the multinomial logit model. In the exercise that follows we compared the multinomial logit and the generalized probit models. In doing so we noticed that the idea behind the multinomial probit model leads to a general functional form for multinomial models with arbitrary link functions. This line of thought could be quite useful in model building and sensitivity analyses. Deferring a detailed study to future research, the idea is briefly discussed in this section.
The idea of building a link function by first making multinomial probabilities proportional to individually parameterized predictors
and then normalizing them, implies the logit link function (26). The logit link based on the normalization does not generalize to alternative link functions. Therefore, a different approach is needed. In the context of the probit models, a latent variable approach has been instrumental [21-23]. A set of linear models specific to categories k=1, 2, . . . , K
| (44) |
is the starting point. The categorical response Y is identified as the index of the first-order statistics based on
| (45) |
It is straightforward to show that if ε follows a Gumbel type I extreme value distribution
| (46) |
where F is a cumulative distribution function (cdf), then (45) is equivalent to the multinomial-logistic model (26) used in this paper. Alternatively, a standard normal choice of ε, F(x)=Φ(x), gives the so-called multinomial-probit models. In the case of a binary response K=2, the two choices reduce to the logit and probit regression.
Let us derive a general expression for a multinomial model based on an arbitrary link function induced by the latent variable approach outlined above. We have
The probability of the response falling into category c is
where the expectations are taken over . Writing the last expectation as an integral over the variable y, and changing it to the new variable u=F(y-logθc), we finally get
| (47) |
where F-1 is the inverse of F, and F is supposed to be an absolutely continuous cdf defined on (-∞, ∞). The general form (47) allows one to build multinomial models based on an arbitrary link function specified by any continuous cdf F defined on the real line. The general model (47) deserves a separate careful study that goes beyond the scope of the present paper.
We used (47) here to study the stability of the model fit with respect to a misspecified link function. Shown in Table III are predicted probabilities under the correct (generalized logit) link function, and their counterparts under a misspecified (generalized probit) link function. Estimates of the model parameters for both link functions are fixed at the MLEs given in Table II.
Table III.
Predictions for response probabilities based on the fitted multinomial logistic model under logit and probit link functions. The predictor is computed from the MLE estimates (Table II). Response categories 1-4 are same as in Table II
| Response probabilities |
||||||
|---|---|---|---|---|---|---|
| Link | Age a | Year t | y=1 | y=2 | y=3 | y=4 |
| Probit | 60 | 1985 | 0.697 | 0.115 | 0.054 | 0.126 |
| Logit | 0.595 | 0.149 | 0.097 | 0.159 | ||
| Probit | 70 | 0.516 | 0.126 | 0.100 | 0.250 | |
| Logit | 0.449 | 0.159 | 0.138 | 0.253 | ||
| Probit | 80 | 0.322 | 0.117 | 0.149 | 0.403 | |
| Logit | 0.305 | 0.153 | 0.178 | 0.364 | ||
| Probit | 60 | 1992 | 0.769 | 0.116 | 0.032 | 0.077 |
| Logit | 0.661 | 0.150 | 0.073 | 0.117 | ||
| Probit | 70 | 0.613 | 0.138 | 0.067 | 0.174 | |
| Logit | 0.526 | 0.168 | 0.109 | 0.197 | ||
| Probit | 80 | 0.423 | 0.141 | 0.113 | 0.314 | |
| Logit | 0.379 | 0.172 | 0.149 | 0.300 | ||
| Probit | 60 | 2000 | 0.828 | 0.112 | 0.015 | 0.039 |
| Logit | 0.723 | 0.146 | 0.051 | 0.080 | ||
| Probit | 70 | 0.706 | 0.144 | 0.038 | 0.105 | |
| Logit | 0.605 | 0.173 | 0.080 | 0.142 | ||
| Probit | 80 | 0.536 | 0.162 | 0.075 | 0.219 | |
| Logit | 0.465 | 0.188 | 0.117 | 0.230 | ||
It is evident from Table III that the differences in the parameter estimates between the two models are small, and the basic diversity of responses and the shape of the distribution of stage and grade at diagnosis of prostate cancer are robust to the link function misspecification.
10. DISCUSSION
Cancer screening exerts complex dynamic effects in the target population. These effects are rooted in the cancer heterogeneity that largely remains unobserved, and its interaction with the screening intervention. Overdiagnosis, lead time (early detection) and length bias lead to a shift in age, stage and grade at diagnosis toward better prognosis. For the same reason post-diagnosis survival shows a profound improvement, and an increased apparent cure rate. These improvements work against an equally dramatic increase in the cancer incidence to produce a slowly changing mortality in the population. All these changes would still be happening if screening were of no benefit to mortality. In the counteraction of the many selection effects and the associated cancer treatment strategies lies the proof of the benefit of screening and treatment. Quantitative evidence of such benefits requires careful joint modeling of the latent processes and the observed outcome. In this paper we presented one such model that reproduces the complex dynamics of the disease presentation at diagnosis induced by introduction of a screening intervention.
Interaction of screening and the latent history of the disease is modeled with the help of a surrogate mixing random vector representing the delay in cancer diagnosis and the mode of detection. It should be noted that the delay time alone showed itself as a poor surrogate resulting in the distant stage predictions showing a slight bump in the cancer incidence after introduction of PSA not seen in the empirical estimates (not shown). Lack of perfect surrogacy of the delay time is likely a consequence of the heterogeneity of tumor growth rates and the length bias of cancer screening [24]. Indeed, if tumor progression was deterministic, its prognostic index at the time of diagnosis would be well explained by the delay time (age of tumor). On the other hand, with the growth rates being heterogeneous, and with screening predominantly detecting slower growing tumors (length bias), the mode of detection provides additional information essential for the goodness of fit.
The use of artificial mixtures is a technical trick that allowed us to simplify the complex estimation problems. Using this approach we achieved a transformation and factorization of the likelihood into a set of problems of reduced dimension.
Even though the Poisson complete-data likelihood does not correspond to any valid model, the algorithm is justified by invoking Quasi-Expectation and Quasi-EM techniques developed earlier.
The joint model presented in this paper is a necessary step toward providing a causal link between the policy of cancer screening defined on the population level and the dynamics of cancer mortality. Note that traditional survival models that condition on age, stage and grade as covariates measured at diagnosis, cannot be used directly to study the effect of screening on mortality, because much of this effect is expressed through changes in the covariates. The idea of the latent surrogate variables used as mixed effects to model the causal link could be extended to model mortality. Based on the model presented in this paper, a surrogate variable could be developed for use as a mixed effect in the survival model (frailty model). For the survival model, the distribution of frailty needs to be updated conditionally on age, stage and grade at diagnosis. As a result, a joint model for stage-grade-specific incidence and survival would emerge. This leads to a causal model of mortality as a function of screening policy integrating dramatic changes in incidence, disease presentation at diagnosis and survival, occurring with screening.
ACKNOWLEDGEMENTS
This research is supported by National Cancer Institute’s Cancer Intervention and Surveillance modeling NeTwork (CISNET) grant U01 CA97414 ‘Population Impact of Cancer Control Processes’. The work of Solomon Chefo is a part of his PhD thesis.
Contract/grant sponsor: National Cancer Institute and Surveillance modeling NeTwork (CISNET); contract/grant number: U01 CA97414
REFERENCES
- 1.Tsodikov A, Szabo A, Wegelin J. A population model of prostate cancer incidence. Statistics in Medicine. 2006;25:2846–2866. doi: 10.1002/sim.2257. [DOI] [PubMed] [Google Scholar]
- 2.Tsodikov A. Semiparametric models: a generalized self-consistency approach. Journal of the Royal Statistical Society, Series B: Statistical Methodology. 2003;65(3):759–774. doi: 10.1111/1467-9868.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsodikov A, Chefo S. Generalized self-consistency: multinomial logit model and Poisson likelihood. Journal of Statistical Planning and Inference. 2008;138:2380–2397. doi: 10.1016/j.jspi.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davidov O, Zelen M. Overdiagnosis in early detection programs. Biostatistics. 2004;5:603–613. doi: 10.1093/biostatistics/kxh012. [DOI] [PubMed] [Google Scholar]
- 5.Shen R, Zelen M. Parametric estimation procedures for screening programmes: stable and nonstable disease models for multimodality case finding. Biometrika. 1999;86:503–515. [Google Scholar]
- 6.Etzioni R, Penson D, Legler J, Tommaso Dd, Boer R, Gann P, Feuer E. Prostate-specific antigen screening: lessons from U.S. prostate cancer incidence trends. Journal of the National Cancer Institute. 2002;13:981–990. doi: 10.1093/jnci/94.13.981. [DOI] [PubMed] [Google Scholar]
- 7.Draisma G, Boer R, Otto S, van der Cruijsen I, Damhuis R, Schroder F, de Koning H. Lead times and overdetection due to prostate-specific antigen screening: estimates from the European Randomized Study of Screening for Prostate Cancer. Journal of the National Cancer Institute. 2003;95:868–878. doi: 10.1093/jnci/95.12.868. [DOI] [PubMed] [Google Scholar]
- 8.Andersen P, Borgan O, Gill R, Keiding N. Statistical Models based on Counting Processes. Springer; New York: 1993. [Google Scholar]
- 9.Keiding N. Statistical inference in the Lexis diagram. Philosophical Transaction of the Royal Statistical Society of London, Series A. 1990;332:487–509. [Google Scholar]
- 10.Holford T. Analysing the temporal effects of age, period and cohort. Statistical Methods in Medical Research. 1992;1:317–337. doi: 10.1177/096228029200100306. [DOI] [PubMed] [Google Scholar]
- 11.Gill R, Johansen S. A survey of product integration with a view toward application in survival analysis. The Annals of Statistics. 1990;18:1501–1555. [Google Scholar]
- 12.Zelen M, Feinleib M. On the theory of screening for chronic diseases. Biometrika. 1969;56:601–614. [Google Scholar]
- 13.Merrill R, Feuer E, Warren J, Schussler N, Stephenson R. Role of transurethral resection of the prostate in population-based prostate cancer incidence rates. American Journal of Epidemiology. 1999;150(8):848–860. doi: 10.1093/oxfordjournals.aje.a010090. [DOI] [PubMed] [Google Scholar]
- 14.Mariotto A, Etzioni R, Krapcho M, Feuer E. Reconstructing prostate-specific antigen (PSA) testing patterns among black and white men in the US from Medicare claims and the National Health Interview Survey. Cancer. 2007;109:1877–1886. doi: 10.1002/cncr.22607. [DOI] [PubMed] [Google Scholar]
- 15.Prentice R. Surrogate endpoints in clinical trials: definition and operational criteria. Statistics in Medicine. 1989;8:431–440. doi: 10.1002/sim.4780080407. [DOI] [PubMed] [Google Scholar]
- 16.Baker S. The multinomial-Poisson transformation. The Statistician. 1994;43:495–504. [Google Scholar]
- 17.Lang J. On the comparison of multinomial and Poisson log-linear models. Journal of the Royal Statistical Society, Series B: Statistical Methodology. 1996;58:253–266. [Google Scholar]
- 18.Press W, Flannery B, Teukolsky S, Vetterling W. The Art of Scientific Computing. Cambridge University Press; New York, NY: 1994. Numerical recipies in Pascal. [Google Scholar]
- 19.Dempster A, Laird N, Rubin D. Maximum likelihood from incomplete data via the em algorithm. Journal of the Royal Statistical Society B. 1977;39:1–38. [Google Scholar]
- 20.Louis TA. Finding the observed information matrix when using the EM algorithm. Journal of the Royal Statistical Society, Series B: Statistical Methodology. 1982;4(2):226–233. [Google Scholar]
- 21.McFadden D. A method of simulated moments for estimation of discrete response models without numerical integration. Econometrica. 1989;57:995–1026. [Google Scholar]
- 22.Chan JSK, Kuk AYC. Maximum likelihood estimation for probit-linear mixed models with correlated random effects. Biometrics. 1997;53:86–97. [Google Scholar]
- 23.Gueorguieva R, Agresti A. A correlated probit model for joint modeling of clustered binary and continuous responses. Journal of the American Statistical Association. 2001;96:1102–1112. [Google Scholar]
- 24.Zelen M. Forward and backward recurrence times and length biased sampling: age specific models. Lifetime Data Analysis. 2004;10:325–334. doi: 10.1007/s10985-004-4770-1. [DOI] [PubMed] [Google Scholar]