

Abstract
Recently, comparative genomic hybridization onto bacterial artificial chromosome (BAC) arrays (array-based comparative genomic hybridization) has proved to be successful for the detection of submicroscopic DNA copy-number variations in health and disease. Technological improvements to achieve a higher resolution have resulted in the generation of additional microarray platforms encompassing larger numbers of shorter DNA targets (oligonucleotides). Here, we present a novel method to estimate the ability of a microarray to detect genomic copy-number variations of different sizes and types (i.e. deletions or duplications). We applied our method, which is based on statistical power analysis, to four widely used high-density genomic microarray platforms. By doing so, we found that the high-density oligonucleotide platforms are superior to the BAC platform for the genome-wide detection of copy-number variations smaller than 1 Mb. The capacity to reliably detect single copy-number variations below 100 kb, however, appeared to be limited for all platforms tested. In addition, our analysis revealed an unexpected platform-dependent difference in sensitivity to detect a single copy-number loss and a single copy-number gain. These analyses provide a first objective insight into the true capacities and limitations of different genomic microarrays to detect and define DNA copy-number variations.
Key words: array CGH, molecular cytogenetics, microdeletion, copy-number variation, power analysis
1. Introduction
Conceptual and technological developments in molecular cytogenetic techniques are now enhancing the resolution power of conventional chromosome analysis from the megabase to the kilobase level. Array-based comparative genomic hybridization (array CGH), i.e. the application of CGH to an array of genomic fragments such as bacterial artificial chromosomes (BACs), has been the method of choice for genome-wide copy-number analysis in the last few years.1,2 The density of the various ‘whole-genome’ BAC clone sets commonly used varies from one clone per Mb3–5 to an overlapping clone set covering the entire human genome with one clone per 100 kb.6,7 Array CGH has rapidly become an important genome analysis tool in cancer research,8–10 in the identification of novel microdeletion syndromes and gene identification studies,11–15 in the diagnosis of patients with congenital malformation syndromes and/or unexplained mental retardation,5,16,17 and in prenatal diagnosis.18,19 Although disease-causing genomic alterations are thought to be rare, recent work using high-resolution microarray approaches has indicated that genomic copy-number variation without immediate phenotypic consequences is widespread throughout the entire human genome.17,20–23
Most currently used genomic copy-number profiling microarrays are produced in academic settings, and the resolution of these microarrays varies depending on the type and number of genomic targets selected, the protocols used, and the data-analysis tools employed. Only recently, private enterprises embarked on this novel microarray application, and several companies are now offering microarrays for genomic copy-number profiling. Most of these microarrays encompass 25–85-mer oligonucleotides targeting random genomic sequences24–26 or single-nucleotide polymorphisms (SNPs).27–31 The theoretical advantages of using such commercial platforms are numerous: (1) they provide a higher genome coverage than most microarrays generated in academia, (2) they can be produced in large quantities according to industrial quality standards, (3) they are available to all researchers, also those without dedicated microarray facilities, and (4) their widespread use will generate large comparable data sets that facilitate data comparison and cooperation between research groups. At present, however, little is known about the actual performance of these platforms, and first-time users will find limited guidance on which platform is most appropriate for their applications and requirements. Although various platform comparison studies have been reported for microarray-based expression profiling,32–35 as yet a comprehensive platform comparison for genomic profiling, including an adequate statistical power analysis, has not been reported.
A prerequisite for a performance comparison of genomic microarrays is the availability of a comprehensive method that accounts for specific requirements associated with genomic microarray data such as adjacency of probes and asymmetric y-axis measurements associated with deletions and/or duplications. Here, we introduce a method that adheres to these requirements, and that is based on statistical power calculations to compare the practical resolution of individual genomic microarray experiments obtained using different microarray platforms. The method is validated using simulated data sets as well as data sets obtained using our in-house tiling-resolution BAC arrays and commercially available 100k SNP, 250k SNP, and 385k oligonucleotide microarray platforms. From our results, we conclude that the increased probe density of the commercially available microarray platforms, although accompanied by a lower signal-to-noise ratio, results in a higher genome-wide copy-number detection resolution.
2. Methods
2.1. Patients and healthy donors
The platform comparison was performed using DNA from 13 patients harboring submicroscopic genomic copy-number variations previously identified by tiling-resolution array CGH.17 Genomic DNA was isolated from blood leukocytes by standard procedures. Male and female reference DNA pools previously used for tiling-resolution BAC array analysis were also used for hybridization to the NimbleGen oligonucleotide microarrays. These reference pools contain equal amounts of genomic DNA from 10 healthy donors (males or females). For the Affymetrix SNP oligonucleotide microarray experiments, using single color hybridizations, two male and two female reference pools were used for normalization purposes.
2.2. Tiling-resolution BAC array CGH
Previously, we reported an array CGH study17 using a tiling-resolution microarray encompassing 32,447 overlapping BAC clones selected to cover the entire human genome.6,7 Hundred patients with unexplained mental retardation were hybridized in duplicate against a sex-mismatched reference pool to this microarray. On the basis of these hybridizations, we selected 13 patients with validated submicroscopic copy-number variations, both single copy-number gains and losses, for hybridization to the other platforms.
2.3. Affymetrix 100k SNP arrays
The Affymetrix 100k SNP array contains 25-mer oligonucleotides representing a total of 116,204 SNPs, distributed over two microarrays. Array experiments were performed according to protocols provided by the manufacturer (Affymetrix, Inc., Santa Clara, CA) as described previously.27 Copy-number estimations were determined using the recently published software package CNAG (Copy Number Analyzer for Affymetrix GeneChip Mapping 100k arrays28). This algorithm strongly improves the signal-to-noise ratios of the copy-number data by (1) accounting for the length and GC content of the polymerase chain reaction products using quadratic regressions and by (2) normalizing the patient samples to reference samples run in parallel.
2.4. Affymetrix 250k SNP arrays
Affymetrix provides two microarrays each containing approximately 250,000 SNPs, and together forming the 500k assay. For this study, we selected the Nsp 250k SNP array, which contains 262,264 25-mer oligonucleotides. For the 100k SNP array experiments, the 250k SNP array experiments were performed according to protocols provided by the manufacturer (Affymetrix, Inc., Santa Clara, CA). Copy-number estimates were determined using the CNAG software package,28 which was recently updated for the analysis of these arrays (version 2.0).
2.5. NimbleGen 385k oligonucleotide arrays
The NimbleGen whole genome oligonucleotide microarray contains 386,165 isothermal probes (45–75-mer), spanning the human genome at a mean probe spacing of 7 kb. Isothermal oligonucleotide design, array fabrication, DNA labeling, CGH experiments, data normalization, and log2(Cy3/Cy5) copy-number ratio calculations were performed by NimbleGen according to published procedures.26
2.6. Hidden Markov analysis
The normalized ratios were analyzed for loss and gain of regions by a standard Hidden Markov Model (HMM), which was optimized for each of the microarray platforms in order to maximize the detection of the known validated copy-number aberrations, while minimizing the false-positive rate, as described before.17
2.7. Statistical power analysis
For each of the four microarray platforms, we performed a statistical power analysis of adjacent targets surrounding a specific locus on a chromosome. This revealed the relationship between the genomic length of the copy-number variation, the noise contained in measurements, and, ultimately, the false-positive and false-negative detection rates for the microarray, and thus, provided a platform-independent discrimination statistic describing the ability of a microarray to detect single copy-number variations.
The statistical power analysis comprises the following steps:
(1) Determination of the distribution of the noise,
(2) Establishment of estimates for significant changes and the variance of noise within each experiment,
(3) Calculation of the number of data points required for detection of copy-number variations, and
(4) Determination of the resolution of a microarray platform.
2.7.1. Determination of the distribution of the noise
The method assumes a normal distribution of noise within the copy-number data. We used a χ2 goodness-of-fit test,36 using a p-value of less than 0.05, and could not reject this hypothesis, thereby justifying the application of the method used for calculating the statistical power.
2.7.2. Establishment of estimates for significant changes and variance of noise
To provide an estimation of a single copy-number loss, the mean log2 ratio is calculated over all targets on the X chromosome,37 excluding those mapped to the pseudo-autosomal regions. This provides an estimate of a significant change to be used in the power calculations, and requires that experiments used for the comparison are performed on the basis of sex mismatch (either in silico or in vitro, depending on the microarray platform used). From the estimate of a single loss, an estimate of a single gain (μ̂Gain) is calculated via
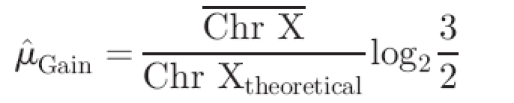 |
1 |
where 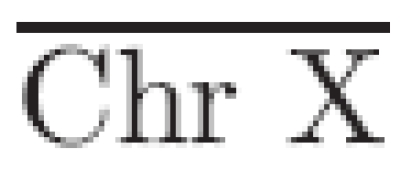 is the mean log2 ratio of targets located on chromosome X and Chr Xtheoretical the theoretical ratio of a single loss (see Supplementary Data). The standard deviation of all log2 ratios from autosomal targets, excluding those known to be involved in validated copy-number variations, is used as an estimate of the variance.
is the mean log2 ratio of targets located on chromosome X and Chr Xtheoretical the theoretical ratio of a single loss (see Supplementary Data). The standard deviation of all log2 ratios from autosomal targets, excluding those known to be involved in validated copy-number variations, is used as an estimate of the variance.
2.7.3. Calculation of the number of data points required for detection of genomic copy-number variations
We calculate the number of data points required to detect a genuine single copy-number variation (as estimated by the mean chromosome X values) given the autosomal standard deviation, with a confidence factor determined by the desired statistical power. This is done by determining the number of data points required to lie in the outer regions of the distribution of the copy-number ratios for it to be deemed unusual in terms of the expected (normal) distribution. We use the non-central T cumulative distribution in order to determine the number of sample points required to satisfy the desired power given estimates of significant changes and an estimate of the variance.38,39
In this study, we chose to use a power of 95% and a two-sided t-test, given the required significance level α. Note that the statistical power (1 − β) is the probability that a true aberration of n adjacent probes is detected (Type II error). The significance level α is the probability of observing a particular deviation between the mean of the n adjacent probes and the rest of the probes on the chromosome, when no actual copy-number variation is present (Type I error). Hence, we aim to solve the following series of equations for the desired power (1 − β) = 0.95. We first define the non-centrality parameter as
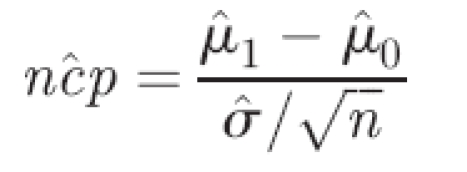 |
2 |
where μ̂1 is the estimate of the ratio pertaining to the copy-number variation, μ̂0 the mean of the autosomal ratios, σ̂ the standard deviation, estimated using the autosomal targets, and n the number of adjacent targets per aberration. We define two cut-offs, via the inverse of the Student's T cumulative (central) distribution function (http://mathworld.wolfram.com/NoncentralStudentst-Distribution.html), T−1, using the desired power, and the inverse of the power. The cut-offs C1 and C2 are defined as
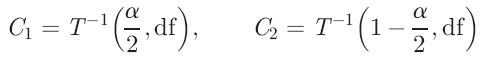 |
3 |
where the degrees of freedom df = n − 1 and α is the required significance level. The power is then calculated with the non-central cumulative T distribution function TNC as follows:
| 4 |
We then find the number of adjacent probes n [in Equation (2)] required to solve the function power in order to achieve our desired power.
2.7.4. Determination of the resolution of a microarray platform
To calculate the resolution of a microarray platform, the outcome of the power analysis is used in conjunction with the genomic coverage of the platform. The distribution of the microarray probes throughout the genome is determined by the size of the gaps between the microarray targets. For our calculation, we take into account the uneven genomic distribution of the microarray targets and assume that copy-number variations can occur randomly throughout the whole genome. Next, we create a window with size equal to the number of data points required to detect a copy-number variation, as given by the power calculation, and determine the number of instances within the genome where the window has a size less than the size of the variation to be detected. This is compared to the total possible number of windows that occur within the genome. By doing so, we create a genome-wide probability that a copy-number variation with a particular size independent of its genomic location will be detected by a microarray platform. We calculated the resolution for single copy-number variations with genomic sizes ranging from 10 kb to 1 Mb, separately for gains and losses.
3. Results
3.1. Study design
In this study, we assessed the capacity of various genomic microarray platforms to detect submicroscopic single copy-number variations, including deletions and duplications. We selected samples from 13 patients in which we have previously identified and validated copy-number variations using our in-house produced tiling-resolution 32k BAC arrays.17 These samples were hybridized onto 100k Affymetrix SNP arrays, 250k Affymetrix SNP arrays, and 385k NimbleGen oligonucleotide arrays. As an example, Fig. 1 shows the chromosome profile obtained for the various platforms in a patient with a 0.54-Mb sized deletion at 9q33.1. We applied a standard HMM algorithm to automatically detect copy-number variations onto the different platforms. Next, we developed and tested a novel method based on statistical power analysis for an objective comparison of the detection resolution of the different platforms.
Figure 1.

Detection of known and validated submicroscopic copy-number variations by high-density BAC, SNP, and oligonucleotide arrays. Individual chromosome plots are shown for Patient 8 (chromosome 9), with the log2 T/R (test-over-reference) values plotted on the y-axis versus the genomic position on chromosome 9 on the x-axis. Results are shown for the tiling-resolution 32k BAC array (A), the 100k SNP array (B), the 250k SNP array (C), and the 385k oligonucleotide array (D). A known and validated microdeletion of 0.54 Mb on 9q33.1 is detected by all four genomic microarray platforms (see black arrow). In addition, a previously undetected microduplication is clearly visible on the chromosome profile obtained by the 250k SNP array (see grey arrow). This figure also shows the different levels of microarray noise present for the different microarray platforms.
3.2. Automatic detection of copy-number aberrations by HMM
In order to obtain independent information on the performance of the different microarray platforms in identifying submicroscopic copy-number variations, we applied a single automated HMM algorithm to the experiments performed in this study (see Table 1). The known and validated copy-number changes were previously identified on the 32k BAC microarray platform,17 and ranged in sizes from 230 kb to 8.9 Mb. Samples from 10 patients were tested on the 385k NimbleGen oligonucleotide microarray platform, and all of the previously identified and validated copy-number variations were detected by the automated HMM algorithm. In contrast, two of the previously identified and validated copy-number variations out of the 13 tested were not automatically detected on the Affymetrix 100k SNP array platform. One of these was a 350 kb deletion on chromosome 7q11.21 (Patient 5), and the other was a 1.65 Mb deletion on chromosome 15q24 (Patient 11). The HMM algorithm correctly detected 10 out of 11 previously identified and validated copy-number variations on the Affymetrix 250k SNP microarray. Again, the 350 kb deletion on 7q11.21 could not be detected automatically, whereas the 1.65 Mb deletion on 15q24 was readily detected on this platform. In addition to the known and validated copy-number variations, a large number of additional copy-number variations was detected but not validated.
Table 1.
Detection of known and validated submicroscopic copy-number variations onto high-density BAC, SNP and oligonucleotide microarrays
| Patient | Copy number | Chromosome | Size (Mb) | No. of targets in region | Average ratio targets in region | Detected by HMMa | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 32k | 100k | 250k | 385k | 32k | 100k | 250k | 385k | 100k | 250k | 385k | ||||
| BACs | SNPs | SNPs | Oligos | BACs | SNPs | SNPs | Oligos | SNPs | SNPs | Oligos | ||||
| 1 | Loss | 1 | 3.93 | 42 | 125 | 230 | n.d. | −0.41 | −0.51 | −0.45 | n.d. | Yes | Yes | n.d. |
| 2 | Gain | 1 | 2.12 | 21 | 50 | 120 | 176 | 0.44 | 0.49 | 0.77 | 0.59 | Yes | Yes | Yes |
| 3 | Loss | 2 | 0.92 | 11 | 47 | 64 | 127 | −0.59 | −0.43 | −0.52 | −0.45 | Yes | Yes | Yes |
| 4 | Gain | 5 | 1.24 | 16 | 40 | n.d. | n.d. | 0.29 | 0.30 | n.d. | n.d. | Yes | n.d. | n.d. |
| 5 | Loss | 7 | 0.35 | 18 | 1 | 13 | 30 | −0.23 | −0.52 | −0.40 | −0.24 | No | No | Yes |
| 6 | Gain | 9 | 0.23 | 5 | 23 | 38 | 40 | 0.35 | 0.30 | 0.47 | 0.27 | Yes | Yes | Yes |
| 7 | Loss | 9 | 2.85 | 30 | 145 | 320 | n.d. | −0.39 | −0.45 | −0.44 | n.d. | Yes | Yes | n.d. |
| 8 | Loss | 9 | 0.54 | 6 | 22 | 70 | 88 | −0.50 | −0.44 | −0.44 | −0.37 | Yes | Yes | Yes |
| 9 | Loss | 11 | 9.15 | 80 | 551 | 923 | 1299 | −0.35 | −0.47 | −0.47 | −0.50 | Yes | Yes | Yes |
| 10 | Gain | 12 | 2.30 | 39 | 69 | n.d. | 353 | 0.32 | 0.26 | n.d. | 0.23 | Yes | n.d. | Yes |
| 11 | Loss | 15 | 1.65 | 16 | 4 | 40 | 204 | −0.33 | −0.36 | −0.50 | −0.35 | No | Yes | Yes |
| 12 | Gain | 17 | 2.89 | 28 | 64 | 151 | 420 | 0.37 | 0.26 | 0.46 | 0.29 | Yes | Yes | Yes |
| 12 | Gain | 17 | 1.43 | 14 | 18 | 91 | 198 | 0.36 | 0.19 | 0.44 | 0.31 | Yes | Yes | Yes |
| 12 | Gain | 17 | 2.88 | 30 | 205 | 279 | 442 | 0.41 | 0.29 | 0.45 | 0.28 | Yes | Yes | Yes |
| 12 | Gain | 17 | 1.48 | 24 | 21 | 64 | 189 | 0.33 | 0.26 | 0.52 | 0.31 | Yes | Yes | Yes |
| 13 | Loss | 22 | 2.66 | 35 | 36 | 130 | 306 | −0.41 | −0.47 | −0.41 | −0.23 | Yes | Yes | Yes |
aAll copy-number variations were initially detected by an automated HMM on the 32k BAC array.
3.3. Verification of power calculation using simulated data
In order to verify the power calculation, we created a step function of a single copy-number loss based on our model for an aberration and corrupted it with a noise signal that had a normal distribution to simulate a single copy-number loss (Supplementary Fig. 1A). The results of the power analysis on this data set are displayed in Supplementary Fig. 1B. This analysis shows that a minimum of four data points with log2 ratios outside the normal distribution is required for a single copy-number loss to be detected with the desired power (95%). Subsequently, a Monte Carlo simulation was used to test the behavior of the power calculation. We artificially generated 400 samples of size 4 under the null hypothesis with a mean of 0, and another 400 with a mean resembling a loss, which represents the alternate hypothesis. The results of this analysis are illustrated in Supplementary Fig. 1C, where the null hypothesis converges to the expected 5% and the alternative hypothesis to 95%. This analysis shows that the power calculation is effective in determining the required data points for the successful detection of a copy-number variation.
3.4. Power calculation on experimental data
After having verified the power calculation, and having confirmed that the distribution of the experimental noise was normal (Supplementary Fig. 2), we applied this method to the experimental data set described above. For each sex-mismatched experiment, we calculated the mean of all unique chromosome X log2 ratios and the standard deviation of the autosomal log2 ratios to provide an initial insight into the performance of each microarray platform and the values to be used in the power calculations (Table 2). The average log2 ratios of these chromosome X targets were similar for the BAC array platform and the Affymetrix SNP array platform (∼0.47), whereas the NimbleGen oligonucleotide platform exhibited a lower average of approximately 0.38. The average standard deviation of the log2 ratios of the autosomal targets varied twofold between the different microarray platforms. The 32k BAC platform exhibited the lowest standard deviation, and the 385k NimbleGen oligonucleotide platform the highest. In addition, as all BAC array hybridizations were performed in duplicate, we were able to determine the influence of replicate analyses on the noise level. As can be seen in Table 2, the autosomal standard deviation is reduced by almost 50% after averaging data from two experiments.
Table 2.
Signal-to-noise parameters of the four genomic copy-number profiling platforms
| Patient | 32k BAC array | Duplicate 32k BAC array | Affymetrix 100k SNP array | Affymetrix 250k SNP array | NimbleGen 385k Oligonucleotide array | ||||
|---|---|---|---|---|---|---|---|---|---|
| Mean Xa | Auto STDb | Auto STD | Mean X | Auto STD | Mean X | Auto STD | Mean X | Auto STD | |
| 1 | 0.47 | 0.10 | 0.06 | 0.48 | 0.15 | 0.49 | 0.18 | n.d. | n.d. |
| 2 | 0.49 | 0.14 | 0.08 | 0.48 | 0.13 | 0.48 | 0.14 | 0.42 | 0.20 |
| 3 | 0.49 | 0.12 | 0.07 | 0.48 | 0.16 | 0.45 | 0.16 | 0.35 | 0.25 |
| 4 | 0.42 | 0.13 | 0.07 | 0.47 | 0.14 | n.d. | n.d. | n.d. | n.d. |
| 5 | 0.40 | 0.09 | 0.05 | 0.45 | 0.16 | 0.46 | 0.18 | 0.29 | 0.24 |
| 6 | 0.45 | 0.10 | 0.05 | 0.47 | 0.17 | 0.43 | 0.13 | 0.38 | 0.28 |
| 7 | 0.46 | 0.12 | 0.06 | 0.48 | 0.14 | 0.43 | 0.13 | n.d. | n.d. |
| 8 | 0.47 | 0.11 | 0.06 | 0.46 | 0.13 | 0.42 | 0.15 | 0.35 | 0.24 |
| 9 | 0.40 | 0.12 | 0.06 | 0.48 | 0.24 | 0.47 | 0.16 | 0.51 | 0.21 |
| 10 | 0.49 | 0.11 | 0.07 | 0.48 | 0.13 | n.d. | n.d. | 0.39 | 0.22 |
| 11 | 0.43 | 0.09 | 0.06 | 0.45 | 0.16 | 0.49 | 0.14 | 0.41 | 0.25 |
| 12 | 0.56 | 0.13 | 0.07 | 0.48 | 0.16 | 0.44 | 0.14 | 0.43 | 0.21 |
| 13 | 0.50 | 0.10 | 0.06 | 0.48 | 0.17 | 0.48 | 0.17 | 0.32 | 0.20 |
| Average | 0.46 | 0.11 | 0.06 | 0.47 | 0.16 | 0.46 | 0.15 | 0.38 | 0.23 |
aMean log2-transformed test-over-reference ratio of the X chromosome, excluding the pseudo-autosomal regions, obtained from calculations in sex-mismatched hybridization experiments. For the BAC and the NimbleGen platforms, data were obtained within each two-color experiment, for the Affymetrix SNP platform, data were combined in silico from different one-color experiments. For this analysis, four reference pool samples (two of each sex) were processed in parallel with the patient samples.
bStandard deviation calculated over the log2-transformed test-over-reference ratios for all autosomal targets, excluding the genomic regions known to harbor submicroscopic copy-number variations.
Next, the statistical power analysis was used to determine the minimum number of adjacently located autosomal targets required for the reliable detection of a single copy-number loss or gain (Table 3, Supplementary Table 1). An average of four adjacently located BAC clones showing a single copy-number loss provided 95% confidence of representing a true copy-number variation. A similar power for detection of a copy-number loss required, on average, five consecutive SNPs on the 100k platform, four SNPs on the 250k platform, and eight consecutive oligonucleotides on the 385k platform. The reliable detection of a single copy-number gain required more consecutive targets, as could be expected based on the theoretical log2 ratio difference between a single copy-number loss (−1) and gain (0.66). For the 32k BAC and for the 100k and 250k SNP array platforms, this increase was moderate, with one to three additional targets being required, respectively. For the 385k oligonucleotide platform, this increase was considerable, i.e. at least twice as many targets were required for reliable detection of a single copy-number gain (Fig. 2).
Table 3.
Result of the statistical power analysis: How many consecutive targets are required to detect a single copy-number loss or gain?
| Patient | 32k BAC array | Duplicate 32k BAC array | Affymetrix 100k SNP array | Affymetrix 250k SNP array | NimbleGen 385k Oligonucleotide array | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Loss | Gain | Loss | Gain | Loss | Gain | Loss | Gain | Loss | Gain | |
| 1 | 4 | 4 | 3 | 3 | 4 | 6 | 5 | 7 | n.d. | n.d. |
| 2 | 4 | 11 | 3 | 4 | 4 | 6 | 4 | 6 | 5 | 13 |
| 3 | 4 | 5 | 3 | 3 | 4 | 7 | 5 | 7 | 10 | 19 |
| 4 | 4 | 6 | 3 | 4 | 4 | 6 | n.d. | n.d. | n.d. | n.d. |
| 5 | 4 | 5 | 3 | 3 | 5 | 8 | 5 | 8 | 11 | 31 |
| 6 | 3 | 5 | 3 | 3 | 5 | 8 | 4 | 6 | 9 | 27 |
| 7 | 4 | 5 | 3 | 3 | 4 | 6 | 4 | 6 | n.d. | n.d. |
| 8 | 4 | 5 | 3 | 3 | 4 | 6 | 5 | 8 | 8 | 24 |
| 9 | 4 | 5 | 3 | 4 | 7 | 9 | 4 | 7 | 5 | 8 |
| 10 | 3 | 5 | 3 | 4 | 4 | 6 | n.d. | n.d. | 7 | 14 |
| 11 | 3 | 5 | 3 | 4 | 5 | 7 | 4 | 6 | 7 | 18 |
| 12 | 3 | 5 | 3 | 4 | 5 | 8 | 4 | 5 | 5 | 16 |
| 13 | 3 | 4 | 3 | 4 | 5 | 7 | 4 | 7 | 9 | 14 |
| Average | 4 | 5 | 3 | 4 | 5 | 7 | 4 | 7 | 8 | 18 |
For this analysis, we used a power of 95%.
Figure 2.

Result of the power analysis of the four genomic microarray platforms for detection of a single copy-number gain or loss contained by different numbers of consecutive targets. The resulting power for a single copy-number gain (dotted) and a single copy-number loss (line) are displayed for the 32k BAC array platform (A), the 100k SNP array (B), the 250k SNP array (C), and 385k oligonucleotide array platform (D). The increase in number of targets has a varying impact on the resulting power across the four different microarray platforms. In addition, the number of consecutive targets required to detect single copy-number gains differs considerably from the number of targets needed to detect a single copy-number loss, and this difference appears to be platform-dependent.
These power analysis results can be translated into genome-wide copy-number detection resolutions by combining these results with the genomic coverage of the different microarray platforms (Table 4, Supplementary Table 2, Supplementary Fig. 3). This resulted for each platform in a probability to detect a single copy-number gain or a loss throughout the genome with a size range from 10 kb to 1 Mb and a desired power of 95%. From this analysis, it can be concluded that (1) high-density oligonucleotide/SNP-based platforms are significantly better in detecting copy-number variations below 1 Mb than the BAC array platform, (2) copy-number variations smaller than 100 kb remain difficult to detect even onto these high-density platforms, despite an average target spacing of 7, 12, or 30 kb, and (3) small-sized single copy-number gains are much more difficult to detect than single copy-number losses of the same size.
Table 4.
Probability to detect a single copy-number gain or loss with different genomic sizes onto the four platforms
| 32k BAC array | Affymetrix 100k SNP array | Affymetrix 250k SNP array | NimbleGen 385k Oligonucleotide array | |||||
|---|---|---|---|---|---|---|---|---|
| Loss | Gain | Loss | Gain | Loss | Gain | Loss | Gain | |
| 10 kb | 0.00 | 0.00 | 0.03 | 0.01 | 0.12 | 0.02 | 0.00 | 0.00 |
| 50 kb | 0.01 | 0.00 | 0.28 | 0.14 | 0.68 | 0.38 | 0.28 | 0.00 |
| 100 kb | 0.02 | 0.01 | 0.56 | 0.39 | 0.88 | 0.75 | 0.94 | 0.01 |
| 200 kb | 0.11 | 0.05 | 0.81 | 0.71 | 0.94 | 0.91 | 0.95 | 0.93 |
| 300 kb | 0.32 | 0.16 | 0.88 | 0.83 | 0.95 | 0.94 | 0.95 | 0.94 |
| 400 kb | 0.60 | 0.36 | 0.91 | 0.88 | 0.95 | 0.94 | 0.95 | 0.94 |
| 500 kb | 0.83 | 0.60 | 0.93 | 0.91 | 0.95 | 0.95 | 0.95 | 0.95 |
| 1 Mb | 0.95 | 0.95 | 0.94 | 0.94 | 0.95 | 0.95 | 0.95 | 0.95 |
This table combines the results from Table 3 with those of Supplementary Table 2. For this analysis, we used a power of 95%.
4. Discussion
We have developed a novel method for establishing the practical resolution of a genomic microarray to detect copy-number variation and applied this method, based on statistical power analysis, to three commercially available microarray platforms and to our in-house BAC microarray platform. For each platform, we calculated the number of adjacent targets required to reliably detect a single copy-number variation (gain or loss), given the required minimum rate of false-positives and false-negatives. On the basis of this calculation, we determined the probability of detecting copy-number variations of different sizes onto a genomic microarray, taking into account the number and genomic position of all targets on the microarray platform used. This unbiased resolution statistic is an important performance measure for genomic microarray platforms as well as for individual microarray experiments, which had not been established for genomic microarrays before. Previously, the resolution of a genomic microarray could only be judged by the mean spacing of targets, a measure that solely reflects the overall genomic coverage. The results of our power analysis, however, clearly demonstrate that the level of noise and the sensitivity of copy-number measurements co-determine the practical resolution. In addition, our analysis revealed an unexpected platform-dependent difference in sensitivity to detect a single copy-number loss and a single copy-number gain. Accurate performance measures are important for researchers to gage the sensitivity and specificity of individual experiments or different platforms. Also, in a diagnostic setting, where microarray-based genome profiling is rapidly being introduced,17,30 it will be essential to have a robust estimate of the practical resolution of the genome-wide scan.
Several platform comparisons have been performed for gene expression microarrays.32–35 The statistical methods described in these studies, however, cannot be used for genomic microarrays as they do not account for various intrinsic aspects of genomic microarrays such as the adjacency of targets and the difference between detecting a single copy-number loss and a single copy-number gain. Several statistical methods have been developed for different aspects of genomic microarray analysis, such as preprocessing (normalization17,28), automatic detection of copy-number variations,40,41 and the analysis of Type I errors across genomic microarrays obtained from multiple experiments and samples.42 Here, we report on a resolution statistic for genomic microarrays that uses an approach based on hypothesis testing and statistical power calculations. The method is based on the following variables: (1) the genomic coverage of the platform, (2) an estimate of the noise in the microarray experiment (the standard deviation of the autosomal targets), (3) an estimate of a single copy-number loss (ratio of the chromosome X unique regions of sex-mismatch experiments),37 and (4) the desired statistical power. The method requires a normal distribution of the noise, which was confirmed by a χ2-test, thereby allowing the use of the T distributions. We validated our method using a Monte Carlo technique to simulate the Type I and Type II detection errors for a single copy-number loss, requiring a statistical power of 95%. These simulations were in agreement with the calculated Type I and Type II errors resulting from a two-sided t-test. The calculations yielded the minimal number of required adjacent targets at each locus in order to detect a single copy-number variation, taking into account the required statistical power.36,43 By combining the genomic location of targets with the minimally required sample size, we obtained an objective genome-wide resolution statistic.
We used our method to characterize the detection performance of the four genomic microarray platforms using experimental data from 13 samples with submicroscopic copy-number variations hybridized to the different platforms. Automatic copy-number analyses detected the large majority of known submicroscopic copy-number variations on all genomic microarray platforms. Two genomic variations were not detected by the 100k SNP array platform, due to poor SNP coverage for these regions, a problem reported also by others44 (see also Supplementary Fig. 3). This can be reduced by simply adding more targets for these regions. Indeed, one of the two variations was identified automatically by the 250k SNP array. This analysis also revealed considerable and reproducible differences in signal-to-noise ratios between the different platforms. Signal-to-noise ratios were highest for the BAC array platform, which may be due to a more robust hybridization performance of larger genomic fragments as compared to the smaller targets used for the other platforms. The Affymetrix SNP arrays containing only 25-mers, however, showed signal-to-noise ratios which were only slightly lower than the BAC array platform after data normalization using the CNAG package.28 It should be noted that no such data preprocessing was performed for the NimbleGen oligonucleotide platform which displayed the lowest signal-to-noise ratios. This may indicate that preprocessing of the data can have a significant effect on the detection resolution of an individual genomic microarray experiment and argues for a significant effort to be made in this field of genomic microarray data analysis.45 In addition, the noise in a genomic microarray experiment can be significantly reduced using replicate analyses, as was shown for the BAC array platform.
The statistical power analysis indicated that, on average, four consecutive BACs are required for the reliable detection of a single copy-number loss, five for the 100k SNP array platform, four for the 250k SNP array platform, and eight consecutive oligonucleotide probes for the NimbleGen 385k oligonucleotide platform. These numbers are markedly different for the detection of single copy-number gains (see Fig. 2). This is caused by the fact that the theoretical ratio of a single copy-number gain (three vs. two copies) is much closer to the random noise level than a single copy-number loss (one vs. two copies). Therefore, it is relatively difficult to discriminate between a true copy-number gain and random experimental noise. This poses a serious problem for those platforms that display a high noise level. The estimate for a single copy-number loss on the NimbleGen oligonucleotide platform is − 0.38 and that for a single copy-number gain is 0.22, within one standard deviation of the mean (0.23 for this platform, see Table 2 and Supplementary Table 1). As a consequence, reliable detection of a single copy-number gain on this platform requires 10 consecutive oligonucleotides more (18) than detection of a single copy-number loss (8). In contrast, the detection of a single copy-number gain on the BAC array platform with the lowest noise level requires only one consecutive clone more (5) than that of a single copy-number loss (4). These results demonstrate the impact of the different detection limits regarding single losses and gains, resulting in more targets being required in the latter case.46 It is important to account for this asymmetric detection limit caused by the different signal-to-noise ratios associated with gains and losses.
The commercially available microarrays contain 3 to 12 times as many targets as our tiling-resolution BAC microarray, and this can compensate for the lower signal-to-nose level obtained on these platforms. In addition, the targets on these microarrays are much smaller in size as compared to BAC clones with an average insert size of 170 kb, thereby theoretically allowing the detection of aberrations below 100 kb. Table 4 shows the probability of detecting a single copy-number gain or loss with different genomic sizes onto the four platforms. This table clearly shows that the commercial platforms outperform the BAC array platform for the detection of aberrations below 1 Mb in size. The Affymetrix 250k SNP array appeared most sensitive for the detection of copy-number variations below the 100-kb level, especially for copy-number gains. However, even on this platform, the probability of detecting a single copy-number gain with a genomic size of 50 kb was only 38% (68% for a single copy-number loss). A similar analysis was performed in silico for the 500k SNP array platform by assuming that the 250k Sty array shows a similar sensitivity and noise distribution as the 250k Nsp array used in this study. This calculation indicated for the 500k SNP array that the probability of detecting a single copy-number loss or gain with a genomic size of 50 kb was 87 and 72%, respectively (Supplementary Table 2). This shows that even these high-density platforms will have significant difficulties in detecting single copy-number variations smaller than 100 kb. As stated above, the use of replicate measurements and/or improvements in data preprocessing can significantly improve the sensitivity of the different genomic microarray platforms.
Next to performance, many other factors, including the availability and consistency in quality of microarrays over time, the amount and quality of input DNA required, the price, and the access to a microarray facility or service company, may affect the choice for a certain microarray platform. An important advantage of using a widely available commercial platform is that it facilitates data exchange between research groups. In addition, the production of arrays containing more than a hundred thousand targets is not practically achievable for academic groups, especially since most currently available microarray spotters have a practical limitation of ∼60,000 spots per slide. An important bonus of using SNP arrays is that it allows genotyping together with CGH. This provides additional information such as copy-number neutral loss-of-heterozygosity. Initial SNP selection against regions with a high frequency of copy-number variation in the population, however, has recently been shown to impact the detection of this specific form of copy-number variation on these platforms.44 Besides Affymetrix and NimbleGen, companies such as Agilent and Illumina have also developed high-density genomic microarrays that can be used for CGH applications.24,31
In conclusion, we present a straightforward statistical method for establishing the practical resolution of an individual genomic microarray experiment. Application of this method to different genomic microarray platforms clearly shows that these platforms vary in their capacity to reliably detect copy-number variations of different sizes and different types. This should be taken into account for estimating the practical resolution of a platform to detect genomic copy-number variations.
Acknowledgements
The authors thank Rolph Pfundt, Bert de Vries, and Han Brunner for useful discussions and critical proofreading of the manuscript. This work was supported by grants from the Netherlands Organization for Health Research and Development (to J.A.V., ZonMW 912-04-047 and 917-66-363).
Supplementary Data
Supplementary data are available online at www.dnaresearch.oxfordjournals.org.
References
- 1.Pinkel D., Albertson D. G. Comparative genomic hybridization. Annu. Rev. Genomics Hum. Genet. 2005;6:331–354. doi: 10.1146/annurev.genom.6.080604.162140. [DOI] [PubMed] [Google Scholar]
- 2.Speicher M. R., Carter N. P. The new cytogenetics: blurring the boundaries with molecular biology. Nat. Rev. Genet. 2005;6:782–792. doi: 10.1038/nrg1692. [DOI] [PubMed] [Google Scholar]
- 3.Snijders A. M., Nowak N., Segraves R., et al. Assembly of microarrays for genome-wide measurement of DNA copy number. Nat. Genet. 2001;29:263–264. doi: 10.1038/ng754. [DOI] [PubMed] [Google Scholar]
- 4.Fiegler H., Carr P., Douglas E. J., et al. DNA microarrays for comparative genomic hybridization based on DOP-PCR amplification of BAC and PAC clones. Genes Chromosomes Cancer. 2003;36:361–374. doi: 10.1002/gcc.10155. [DOI] [PubMed] [Google Scholar]
- 5.Vissers L. E., de Vries B. B., Osoegawa K., et al. Array-based comparative genomic hybridization for the genome-wide detection of submicroscopic chromosomal abnormalities. Am. J. Hum. Genet. 2003;73:1261–1270. doi: 10.1086/379977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ishkanian A. S., Malloff C. A., Watson S. K., et al. A tiling resolution DNA microarray with complete coverage of the human genome. Nat. Genet. 2004;36:299–303. doi: 10.1038/ng1307. [DOI] [PubMed] [Google Scholar]
- 7.Krzywinski M., Bosdet I., Smailus D., et al. A set of BAC clones spanning the human genome. Nucleic Acids Res. 2004;32:3651–3660. doi: 10.1093/nar/gkh700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davies J. J., Wilson I. M., Lam W. L. Array CGH technologies and their applications to cancer genomes. Chromosome Res. 2005;13:237–248. doi: 10.1007/s10577-005-2168-x. [DOI] [PubMed] [Google Scholar]
- 9.Jonsson G., Naylor T. L., Vallon-Christersson J., et al. Distinct genomic profiles in hereditary breast tumors identified by array-based comparative genomic hybridization. Cancer Res. 2005;65:7612–7621. doi: 10.1158/0008-5472.CAN-05-0570. [DOI] [PubMed] [Google Scholar]
- 10.Pinkel D., Albertson D. G. Array comparative genomic hybridization and its applications in cancer. Nat. Genet. 2005;37(suppl.):S11–S17. doi: 10.1038/ng1569. [DOI] [PubMed] [Google Scholar]
- 11.Vissers L. E., van Ravenswaaij C. M., Admiraal R., et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 12.Vissers L. E., Veltman J. A., Geurts van Kessel A., Brunner H. G. Identification of disease genes by whole genome CGH arrays. Hum. Mol. Genet. 2005;14:R215–R223. doi: 10.1093/hmg/ddi268. [DOI] [PubMed] [Google Scholar]
- 13.Koolen D. A., Vissers L. E., Pfundt R., et al. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat. Genet. 2006;38:999–1001. doi: 10.1038/ng1853. [DOI] [PubMed] [Google Scholar]
- 14.Shaw-Smith C., Pittman A. M., Willatt L., et al. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat. Genet. 2006;38:1032–1037. doi: 10.1038/ng1858. [DOI] [PubMed] [Google Scholar]
- 15.Sharp A. J., Hansen S., Selzer R. R., et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat. Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 16.Shaw-Smith C., Redon R., Rickman L., et al. Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J. Med. Genet. 2004;41:241–248. doi: 10.1136/jmg.2003.017731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Vries B. B. A., Pfundt R., Leisink M., et al. Diagnostic genome profiling in mental retardation. Am. J. Hum. Genet. 2005;77:606–616. doi: 10.1086/491719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rickman L., Fiegler H., Shaw-Smith C., et al. Prenatal detection of unbalanced chromosomal rearrangements by array-CGH. J. Med. Genet. 2006;43:353–361. doi: 10.1136/jmg.2005.037648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roa B. B., Pulliam J., Eng C. M., Cheung S. W. Evolution of prenatal genetics: from point mutation testing to chromosomal microarray analysis. Expert Rev. Mol. Diagn. 2005;5:883–892. doi: 10.1586/14737159.5.6.883. [DOI] [PubMed] [Google Scholar]
- 20.Iafrate A. J., Feuk L., Rivera M. N., et al. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 21.Sebat J., Lakshmi B., Troge J., et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 22.Sharp A. J., Locke D. P., McGrath S. D., et al. Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feuk L., Carson A. R., Scherer S. W. Structural variation in the human genome. Nat. Rev. Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- 24.Barrett M. T., Scheffer A., Ben-Dor A., et al. Comparative genomic hybridization using oligonucleotide microarrays and total genomic DNA. Proc. Natl. Acad. Sci. USA. 2004;101:17765–17770. doi: 10.1073/pnas.0407979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van den Ijssel P., Tijssen M., Chin S. F., et al. Human and mouse oligonucleotide-based array CGH. Nucleic Acids Res. 2005;33:e192. doi: 10.1093/nar/gni191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Selzer R. R., Richmond T. A., Pofahl N. J., et al. Analysis of chromosome breakpoints in neuroblastoma at sub-kilobase resolution using fine-tiling oligonucleotide array CGH. Genes Chromosomes Cancer. 2005;44:305–319. doi: 10.1002/gcc.20243. [DOI] [PubMed] [Google Scholar]
- 27.Huang J., Wei W., Zhang J., et al. Whole genome DNA copy number changes identified by high-density oligonucleotide arrays. Hum. Genomics. 2004;1:287–299. doi: 10.1186/1479-7364-1-4-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nannya Y., Sanada M., Nakazaki K., et al. A robust algorithm for copy number detection using high-density oligonucleotide single-nucleotide polymorphism genotyping arrays. Cancer Res. 2005;65:6071–6079. doi: 10.1158/0008-5472.CAN-05-0465. [DOI] [PubMed] [Google Scholar]
- 29.Slater H. R., Bailey D. K., Ren H., et al. High-resolution identification of chromosomal abnormalities using oligonucleotide arrays containing 116,204 SNPs. Am. J. Hum. Genet. 2005;77:709–726. doi: 10.1086/497343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friedman J. M., Baross Á., Delaney A. D., et al. Oligonucleotide microarray analysis of genomic imbalance in children with mental retardation. Am. J. Hum. Genet. 2006;79:500–513. doi: 10.1086/507471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peiffer D. A., Le J. M., Steemers F. J., et al. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16:1136–1148. doi: 10.1101/gr.5402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bammler T., Beyer R. P., Bhattacharya S., et al. Standardizing global gene expression analysis between laboratories and across platforms. Nat. Methods. 2005;2:351–356. doi: 10.1038/nmeth754. [DOI] [PubMed] [Google Scholar]
- 33.Barnes M., Freudenberg J., Thompson S., Aronow B., Pavlidis P. Experimental comparison and cross-validation of the Affymetrix and Illumina gene expression analysis platforms. Nucleic Acids Res. 2005;33:5914–5923. doi: 10.1093/nar/gki890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irizarry R. A., Warren D., Spencer F., et al. Multiple-laboratory comparison of microarray platforms. Nat. Methods. 2005;2:345–350. doi: 10.1038/nmeth756. [DOI] [PubMed] [Google Scholar]
- 35.Larkin J. E., Frank B. C., Gavras H., Sultana R., Quackenbush J. Independence and reproducibility across microarray platforms. Nat. Methods. 2005;2:337–344. doi: 10.1038/nmeth757. [DOI] [PubMed] [Google Scholar]
- 36.Siegel S. Nonparametric Statistics for the Behavioral Sciences. 2nd ed. New York: McGraw-Hill; 1988. [Google Scholar]
- 37.Loo L. W. M., Grove D. I., Williams E. M., et al. Array comparative genomic hybridization analysis of genomic alterations in breast cancer subtypes. Cancer Res. 2004;64:8541–8549. doi: 10.1158/0008-5472.CAN-04-1992. [DOI] [PubMed] [Google Scholar]
- 38.Neyman J., Tokarska B. Errors of the second kind in testing “Student's” hypothesis. J. Am. Stat. Assoc. 1936;31:318–326. [Google Scholar]
- 39.Johnson N. L., Welch B. L. Applications of the non-central t-distribution. Biometrika. 1940;31:362–389. [Google Scholar]
- 40.Olshen A. B., Venkatatraman E. S., Lucito R., Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 41.Fridlyand J., Snijders A. M., Pinkel D., Albertson D. G., Jain A. N. Hidden Markov models approach to the analysis of array CGH data. J. Multivariate Anal. 2004;90(1):132–153. [Google Scholar]
- 42.Diskin S. J., Eck T., Greshock J., et al. STAC: a method for testing the significance of DNA copy number aberrations across multiple array-CGH experiments. Genome Res. 2006;16:1149–1158. doi: 10.1101/gr.5076506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murphy K. R. Statistical Power Analysis: A Simple and General Model for Traditional and Modern Hypothesis Tests. 2nd ed. London: Lawrence Erlbaum Associates; 2004. [Google Scholar]
- 44.Wirtenberger M., Hemminki K., Burwinkel B. Identification of frequent chromosome copy-number polymorphisms by use of high-resolution single-nucleotide-polymorphism arrays. Am. J. Hum. Genet. 2006;78:520–522. doi: 10.1086/500793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hsu L., Self S. G., Grove D., et al. Denoising array-based comparative genomic hybridization data using wavelets. Biostatistics. 2005;6:211–226. doi: 10.1093/biostatistics/kxi004. [DOI] [PubMed] [Google Scholar]
- 46.Wilcox R. R. Fundamentals of Modern Statistical Methods: Substantially Improving Power and Accuracy. New York: Springer; 2001. [Google Scholar]