

Abstract
We performed random sequencing of cDNAs from nine biologically or industrially important cultures of the industrially valuable fungus Aspergillus oryzae to obtain expressed sequence tags (ESTs). Consequently, 21 446 raw ESTs were accumulated and subsequently assembled to 7589 non-redundant consensus sequences (contigs). Among all contigs, 5491 (72.4%) were derived from only a particular culture. These included 4735 (62.4%) singletons, i.e. lone ESTs overlapping with no others. These data showed that consideration of culture grown under various conditions as cDNA sources enabled efficient collection of ESTs. BLAST searches against the public databases showed that 2953 (38.9%) of the EST contigs showed significant similarities to deposited sequences with known functions, 793 (10.5%) were similar to hypothetical proteins, and the remaining 3843 (50.6%) showed no significant similarity to sequences in the databases. Culture-specific contigs were extracted on the basis of the EST frequency normalized by the total number for each culture condition. In addition, contig sequences were compared with sequence sets in eukaryotic orthologous groups (KOGs), and classified into the KOG functional categories.
Key words: Aspergillus oryzae, cDNA, EST, solid-state culture, liquid culture
1. Introduction
Recently, expressed sequence tag (EST) analyses have been carried out in various organisms, including fungi,1–5 because this method involves essentially random sequencing of cDNA libraries and can be performed more easily than whole-genome sequencing. EST analysis will enable us to collect nucleotide sequence information of protein-coding regions rapidly and efficiently, and will be helpful for gene finding in genomes. It will also provide gene expression information for a particular type of cultivation or a particular growth phase. For these reasons, EST analysis is conducted as part of many genome projects.
The koji mold Aspergillus oryzae, which is a safe filamentous fungus,6,7 produces and secretes various enzymes into the medium. Consequently, it has long been used in industry, particularly in the manufacture of traditional Japanese fermented foods, such as sake, shoyu (soy sauce), and miso (bean paste), as well as commercially valuable enzyme production. High levels of enzyme productivity, the primary reasons for the industrial use of A. oryzae, are achieved through its cultivation on suitable cereals in solid-state culture (SC), not in culture in liquid medium, liquid culture (LC), or agar plate culture. Hence, SC is a major means of industrial cultivation of A. oryzae.8 To understand the superiority of SC, a number of genes transcribed specifically during SC have been identified.9,10 However, the molecular mechanisms responsible for the expression of the SC-specific characteristics of A. oryzae remain poorly understood. Secreted enzymes and the genes encoding them have been the major targets of molecular biological analysis of A. oryzae, and investigated actively because of their essential involvement in utilization process of A. oryzae,11,12 whereas there have been few studies of the intracellular physiology at the molecular level. Such studies are necessary for further advanced industrial utilization of A. oryzae, and to understand the behavior of A. oryzae during LC under various conditions because of critical importance of the LC method in modern bioindustries.
However, there are several technical difficulties in experimental systems for genetic studies. For example, teleomorphs are not observed through its life cycle, which makes genetic analysis by mating impossible. Vegetative cells and most conidia consistently contain multiple nuclei, and hence isolation of recessive mutants is problematic. However, recombinant DNA techniques for A. oryzae have already been developed, although the efficiency of gene targeting by homologous recombination is poor.11,13
Reverse-genetic and/or post-genomic approaches based on nucleotide sequence information are therefore vital for such investigations. However, the availability of sequence information needed for such approaches remains insufficient. Therefore, we performed large-scale EST analysis in A. oryzae, employing nine different cDNA libraries to obtain numerous independent sequences as efficiently as possible. In fact, large number of ESTs were obtained, and they worked as invaluable informational and material bases for subsequent studies; rough gene expression profiling from frequency information, reverse genetics, and DNA micro array preparation. Furthermore, the EST information substantially contributed for predicting ORFs in the A. oryzae whole-genome analysis.14
2. Materials and methods
2.1. Strain, medium, and cultivation
The wild-type A. oryzae strain RIB40 was used as the EST source.
Mycelia from nine different cultures were used for cDNA library construction. Culture was performed as follows:
LR library (liquid nutrient-rich culture): Culture was carried out in YPD medium (1% yeast extract, 2% Bacto-peptone, 0.04% adenine sulfate, 2% glucose) at 30°C for 22 h. Vigorously growing mycelia were harvested.
LH library (liquid nutrient-rich culture at higher temperature): Culture was carried out in YPD medium at 37°C for 24 h. Vigorously growing mycelia were harvested.
LM library (liquid maltose-inductive culture): Preculture was performed in ACM medium (2% malt extract, 0.1% Bacto-peptone, 2% glucose) at 37°C for 24 h. Subsequently, mycelia were washed with and transferred into maltose-inductive medium (1% Bacto-peptone, 0.5% KH2PO4, 0.1% NaNO3, 0.05% MgSO4•7H2O), then incubated at 37°C for 4 h. Transcription in harvested mycelia would be affected by maltose induction.
LS library (liquid carbon-starved culture): Preculture was performed in YPD medium at 30°C for 22 h. Subsequently, mycelia were washed with water and transferred into carbon-lacking CD medium (0.2% NaNO3, 0.1% KH2PO4, 0.05% MgSO4•7H2O, 0.001% FeSO4), and then incubated at 30°C for 8 h. Carbon-starved mycelia were harvested.
-
LG library (liquid germinated conidia and conidia): A suspension of matured conidia was inoculated into SP medium (3.5% soluble starch, 2% Bacto-peptone, 0.5% KH2PO4, 0.5% MgSO4•7H2O). After incubation at 30°C for 12 h, germinated conidia were harvested and mixed with non-precultured conidia, then used for RNA preparation.
The cultures described in (i)–(v) were performed in liquid media with rotary shaking.
PA library (alkaline pH culture on agar plates): Culture was performed on a cellophane sheet covering alkaline CD agar plate medium (0.2% NaNO3, 0.1% KH2PO4, 0.05% MgSO4•7H2O, 0.001% FeSO4, 1% glucose, 2% agar, adjusted to pH 10) at 30°C for 3 days. Mycelia adapted to alkaline conditions were harvested.
SW library (solid-state wheat bran culture): A conidial suspension was inoculated onto autoclaved wheat bran, and incubated at 30°C for 28 h. Vigorously growing mycelia that were differentiating into submerged hyphae and aerial hyphae were harvested.
SS library (solid-state soybean culture): Water-absorbed defatted soybeans were mixed with roasted and crushed wheat. They were autoclaved and inoculated with conidial suspension. Then, they were incubated at 30°C for 34 h, and subsequently at 25°C for 3 h. Mycelia that were developing asexually were harvested.
SR library (solid-state rice culture): Polished rice retaining the inner 70% of the whole and α-preprocessed was autoclaved. The conidial suspension was inoculated onto the rice, and then incubated at 37°C for 28 h. Mycelia that were growing vigorously and differentiating into submerged hyphae and aerial hyphae were harvested.
2.2. Construction of cDNA libraries
Total RNA was prepared from mycelia growing under various culture conditions as described in Section 2.1. From the LC (including PA), i.e. 2.1(i)−2.1(vi), total RNA was prepared with Isogen (Nippon Gene Co. Ltd), TRIzol reagent (Invitrogen Corp.), or an RNeasy kit (Qiagen Inc.), according to the manufacturer's instructions. From the SC, i.e. 2.1(vii)−2.1(ix), total RNA was prepared using Cathala's method with some modifications.10,15 Poly(A)+ RNA was isolated from the total RNA sample using Oligotex dT30 super (Takara Inc.) or an Oligo(dT) cellulose column (Amersham Pharmacia Biotech Inc.), according to the manufacturer's instructions.
Double-stranded cDNA was synthesized from each mRNA sample using commercially available cDNA synthesis kits, such as Superscript II plasmid system (Gibco BRL Co.), Timesaver cDNA synthesis kit (Amersham Pharmacia Biotech Inc.), or SMART cDNA library construction kit (Clontech), according to the manufacturer's instructions. In all preparations, oligo-dT primers were used for first-strand cDNA synthesis. The resultant cDNAs were inserted unidirectionally into a vector—pSPORT1 (Gibco BRL Co.), pBluescriptII SK(+) (Stratagene), or λTripEx2 (Clontech)—then introduced into Escherichia coli strain DH5α, JM109, or XL1Blue. The E. coli clones were arbitrarily picked and then plasmids carrying the A. oryzae cDNA were prepared.
2.3. Nucleotide sequencing and sequence analyses
Nucleotide sequences of cDNA clones were determined using automated DNA sequencing machines by standard sequencing methods. Vector sequences and low-quality sequences were removed by hand. Sequence assembly of the raw EST data was performed using the program Phrap (http://www.phrap.org/phredphrapconsed.html). Assembly conditions used were: minimum length of matching word, 50; minimum alignment score, 30; repeat stringency, 0.95.
Similarity searches against public non-redundant protein databases were carried out using BLASTX on the National Center for Biotechnology Information web server (http://www.ncbi.nlm.nih.gov/BLAST/). The EST contigs were classified into KOG categories according to the results of BLASTX searches against amino acid sequences in the KOG set (http://www.ncbi.nlm.nih.gov/COG/).16 These sequence similarities were judged to be significant when the expected-value (E-value) was less than 1E-9 at the amino acid sequence level.
2.4. General techniques for nucleic acid manipulation
Nucleic acid manipulations not described earlier were performed using standard methods, as described by Sambrook et al.17
2.5. Cluster analysis
A hierarchical cluster analysis of culture conditions was performed with the nearest neighbor-joining method, with STATISTICA software (StatSoft, Inc.), in which the normalized frequency values of the contigs were used as variations.
3. Results and discussion
3.1. Construction of the cDNA libraries and generation of ESTs
The major objectives of this study were as follows: (i) to accumulate gene resources for screening valuable genes and providing a substantial informational basis for post-genomic analyses; (ii) to construct a basis of gene finding in whole-genome analyses; and (iii) to collect frequency information of each EST for comparison of expression profiles among culture conditions. Characteristics of A. oryzae are known to differ markedly according to type of culture, such as LC or SC. Consequently, it was important to collect EST information from cDNA libraries obtained under various culture conditions. In this study, we selected nine diverse cultures that varied in many aspects: nutrients, temperature, pH, and physical phase of media (liquid or solid-state). The biological and industrial importance of the cultures examined is outlined in what follows.
In the laboratory, LC is the most important type of culture in regular use for ease of treatment. Nevertheless, LC is inferior to SC used in industrial applications because of the low levels of enzyme productivity.8 However, the use of LC is preferable to SC from the viewpoint of culture engineering. Physical simplicity of the systems is a salient advantage of LC. The conditions of LC (medium composition, degree of aeration, and temperature) can be controlled easily according to specific requirements.18 In future advanced utilization of A. oryzae, LC would therefore be preferred to SC if the weak points of LC could be resolved. ‘Liquid nutrient-rich culture’ (cultivation in YPD medium at 30°C) was designed as a standard LC condition, and provided the LR library. Here, we set three other LC conditions as modified variations of LR to obtain EST information in LCs at higher temperature (LH library), under conditions of maltose induction (LM library), and under conditions of carbon starvation (LS library). In particular, induction of various amylolytic enzymes by maltose has been focused as a model system for regulation of gene expression, and in addition, applied to artificial gene expression systems.8,11,12 EST analyses of these variations were expected to yield valuable information regarding basic metabolism, adaptation to different conditions, etc.
Collecting ESTs from the LG library that originated in conidia and germinating conidia would yield information regarding the gene expression profile in the early stages of growth of filamentous fungi. Such information may be useful for elucidating the biological events during germination and for the development of techniques for controlling germination in industrial applications.
A. oryzae is adaptable to a wide range of pH. In traditional procedures of industrial conidia preparation, which are dependent on alkaline adaptability of A. oryzae, the pH of the culture medium is shifted to alkaline (ca. pH 10) by sprinkling with wood-ash thereby preventing microbial contaminants, and conidia formation is facilitated by alkaline conditions.11 However, gene expression behavior under conditions of alkaline pH remains poorly understood. Alkaline plate culture was employed to construct a PA library as a model for alkaline adaptation to obtain EST information.
Generally, A. oryzae mycelia cultured on agar plates show properties intermediate between those from LC and SC. On agar plates, asexual development occurs in late culture phase as in SC, whereas the levels of enzyme productivity are low as in LC.19 However, enzyme productivity is a more important mycelial property for industrial use, and consequently, we treated PA cultures as a variation of LC in this study.
SC is the traditional culture method, and remains the typical method for industrial utilization of A. oryzae because of its superiority over LC in the productivity of secreted enzymes.8 A. oryzae has recently been considered a strong host candidate for heterologous protein production both because it is a eukaryote and because of its safety and remarkable behavior, i.e. high productivity of secreted enzymes.20,21 The EST information from SCs would provide some insights into the molecular mechanisms involved in the high levels of productivity of secreted proteins. It would also be available as an abundant gene resource for rapid screening of novel proteins, such as valuable enzymes. In SC, optimal cereal materials are chosen as substrates according to the cultivation objective.8,18 The present study employed three major solid-state substrates as media for EST collection: wheat bran (SW library), defatted soybeans with wheat kernel flour (SS library), and polished rice (SR library), which are used in manufacturing processes of commercial enzymes, shoyu (soy sauce), and Japanese sake, respectively. As noteworthy nutrient features, SS medium contains protein in high ratio, and SR medium contains starch in high ratio.
Cultivation was performed as described in Section 2.1. Poly(A)+RNAs were isolated from them, followed by preparation of cDNA libraries, which were inserted unidirectionally into a vector. None of the cDNA libraries were normalized, thereby maintaining the original abundance ratio of transcripts to the greatest possible degree. Resultant E. coli colonies were picked arbitrarily and subjected to single-pass sequencing from the 5′ end of the cDNA insert. Ultimately, 21 446 partial cDNA sequences were accumulated as ESTs.
3.2. General features of the ESTs
Subsequently, 21 446 ESTs were assembled to 2854 non-redundant consensus sequences (contigs) and 4735 singletons, which appeared in the total ESTs with frequency 1. Singletons can be recognized as lone contigs showing no overlap with other ESTs, and hence we treated all of these 7589 independent sequences including the singletons as contigs in this study.
Nevertheless, when each contig was assigned to the genomic region of A. oryzae,14 2635 contigs were originated from 1157 protein-encoding genes. We assumed several reasons for this redundancy as follows: (i) since it is generally difficult to obtain full-length cDNA from an oligo-dT primed cDNA library, multiple non-overlapping contigs can be generated from the same gene by difference of 5′ end truncation among cDNA clones; (ii) Diversity of transcription initiation site in the same gene may also cause multiple non-overlapping contigs same as (i); (iii) cDNAs alternatively spliced or containing introns failed to be spliced may cause multiple contigs from the same gene. If the incompleteness of reverse transcription and splicing were stringently considered, the total number of the genes corresponding to EST contigs was less than 7589, and at approximately 6100 minimum. However, we did not have enough information to accurately evaluate the actual reason for the multiple transcripts in each case, and accordingly, all the multiple contigs from the same gene were treated as independent contigs in this report.
The sequence length distributions of raw ESTs and contigs were analyzed. The average length and total length of raw ESTs were 569 bases and 12.20 Mb, respectively. These values for the contigs were 658 bases and 5.00 Mb, respectively (Fig. 1). Overall length distribution shifted upwards after assembling raw ESTs, and approximately 180 contigs longer than 1.5 kb were generated. However, probably because of the high ratio (62.4%) of singletons within all contigs, mode values of raw ESTs and contigs both belonged to the same class, i.e. 501–600 bases. The total length of contigs (5.00 Mb) and number of contigs (7589) corresponded to 13.5% of the whole-genome size (37.05 Mb) and 54.0% of the number of predicted ORFs (14 063), respectively (Fig. 1, Table 1).14
Figure 1.

Sequence length distribution of raw ESTs and contigs. Hatched bar, raw EST (before assembly); Solid bar, contigs (after assembly).
Table 1.
Brief summary of the features of the A. oryzae ESTs from various culture conditions
| Source cDNA libraries (cultures) | Number of raw ESTs | Number of contigs | Average frequency | Singletons | Unique contigsa | Contigs with no similarity to databaseb | Research organizations in charge | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Number | Ratio (%)c | Number | Ratio (%)c | Number | Ratio (%)c | |||||
| Total ESTs | 21446 | 7589 | 2.83 | 4735 | 62.4 | 5491d | 72.4 | 3843 | 50.6 | |
| ESTs from LCs (including plate culture: PA) | 9262 | 4181 | 2.22 | 2233 | 53.4 | 2742e | 65.6 | 2022 | 48.4 | |
| LR library (nutrient-rich culture) | 2611 | 1518 | 1.72 | 616 | 40.6 | 656 | 43.2 | 561 | 37.0 | AIST, UT, NU |
| LH library (nutrient-rich culture at higher temperature) | 2049 | 1086 | 1.89 | 371 | 34.2 | 427 | 39.3 | 442 | 40.7 | NFRI |
| LM library (maltose-inductive culture) | 926 | 653 | 1.42 | 247 | 37.8 | 278 | 42.6 | 262 | 40.1 | NU |
| LS library (carbon-starved culture) | 1940 | 1217 | 1.59 | 422 | 34.7 | 471 | 38.7 | 434 | 35.7 | AIST |
| LG library (germinated conidia and conidia) | 1000 | 701 | 1.43 | 376 | 53.6 | 389 | 55.5 | 428 | 61.1 | TUAT |
| PA library (alkaline pH agar plate culture) | 736 | 519 | 1.42 | 201 | 38.7 | 225 | 43.4 | 233 | 44.9 | UT |
| ESTs from solid-state cultures | 12184 | 4847 | 2.51 | 2502 | 51.6 | 3408e | 70.3 | 2236 | 46.1 | |
| SW library (wheat bran culture) | 7725 | 3707 | 2.08 | 1731 | 46.7 | 2177 | 58.7 | 1637 | 44.2 | NRIB, THU |
| SS library (soybean culture) | 991 | 486 | 2.04 | 184 | 37.9 | 204 | 42.0 | 194 | 39.9 | AIST |
| SR library (rice culture) | 3468 | 1701 | 2.04 | 587 | 34.5 | 664 | 39.0 | 654 | 38.4 | NRIB |
AIST, National Institute of Advanced Industrial Science and Technology; NFRI, National Food Research Institute; NRIB, National Research Institute of Brewing; NU, Nagoya University; THU, Tohoku University; TUAT, Tokyo University of Agriculture and Technology; UT, University of Tokyo.
aContigs composed of ESTs from only LC, SC, or a particular library (including singletons).
bContigs of which the E-values from the BLAST search against the most similar amino acid sequence were not less than 1E-9.
cRatio to ‘Number of contigs’ in each line.
dSum of unique contigs from each library. The contigs unique to LC or SC were not included.
eContigs obtained only from LC or SC. Redundancies among the libraries were not removed.
General features of the raw ESTs and contigs are summarized by source cDNA library in Table 1. Frequency distributions of the contigs are shown in Fig. 2A–C. Of the total 7589 contigs, 4735 (62.4%) were singletons. Contigs with frequencies of no more than five accounted for over 90% of the total number of contigs (Fig. 2A). The average frequency of total ESTs was 2.83 (Table 1, Fig. 2A). Average frequencies by source cDNA libraries tended to be correlated with numbers of raw ESTs. Numbers of singletons within contigs by the source cDNA library tended to increase in proportion to the number of collected raw ESTs, whereas the ratios of singletons were distributed around 35–55% of the contigs in all cases without a significant pattern (Table 1).
Figure 2.
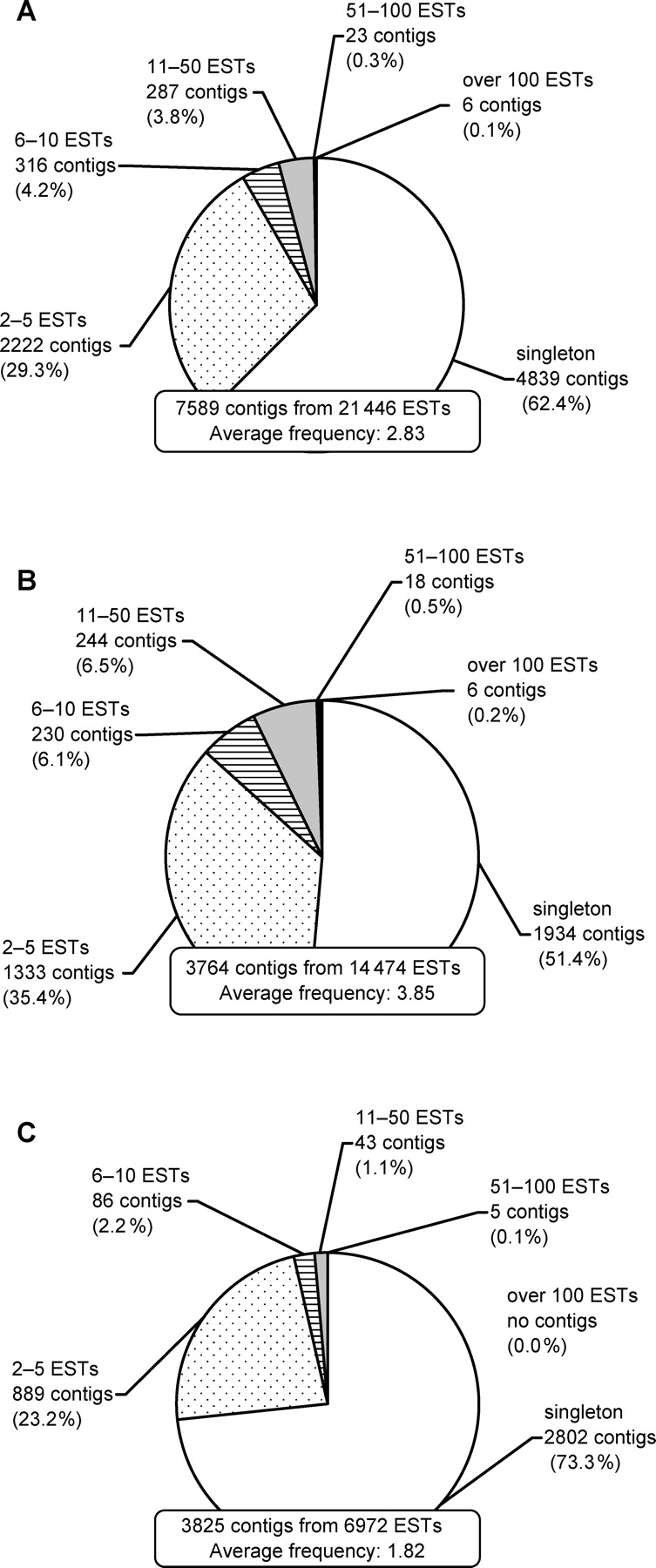
Frequency distribution of the contigs. Contigs were generated by assembling raw ESTs, and they no longer overlapped with each other. The general tendency of frequency, i.e. redundancy of raw ESTs within a contig was analyzed. (A) Total EST contigs. (B) EST contigs of known sequence. (C) EST contigs with no significant similarity.
Contigs consisting of raw ESTs obtained from only a specific cDNA library appeared to be unique to the library, and these sequences were not obtained from other libraries. These contigs were designated ‘unique contigs’ in this report. The singletons fall into the unique contigs. The ratios of unique contigs by source library were distributed around 40–60% in all cases with no significant pattern, and singletons accounted for over roughly 80% of the unique contigs in most cases (Table 1). The number of unique contigs was approximately correlated with the number of raw ESTs. The total number of unique contigs was 5491 and accounted for 72.4% of all contigs (Table 1). This remarkably large ratio of unique contigs, including singletons, showed that independently expressed sequences were collected efficiently by our strategy, i.e. the use of diverse (as many as nine) cDNA libraries for EST sources.
The deduced amino acid sequences of EST contigs were compared with the non-redundant public protein database using BLASTX. However, sequences derived from genome sequencing of Aspergilli were not considered as targets for similarity searches because this would mask the distribution of sequences similar to the query in the database.
The results of the analysis indicated that 2953 (38.9%) and 793 (10.5%) contigs showed significant similarity to annotated sequences and to hypothetical proteins, respectively. The remaining 3843 (50.6%) contigs had no significant similarity to sequences deposited in the public databases (Tables 1 and 2). The latter would be novel sequences, correspond to non-coding RNA gene, or untranslated regions of mRNA of protein coding gene. The ratios of the contigs, by the source cDNA library, with no significant similarity to sequences in the public databases, were distributed around 35–45%, except for the LG library (Table 1). The extremely high value in the LG library (61.1%) may be because events occurring during the germination stage of A. oryzae are more poorly understood at the molecular level than those at other mycelial stages or under culture conditions.
Table 2.
Results of similarity search against the public non-redundant protein database
| Similarity | Number of contigs | (%) |
|---|---|---|
| Function-predicted genes | 2953 | (38.9) |
| Hypothetical proteina | 793 | (10.5) |
| No significant similarity | 3843 | (50.6) |
| Total contigs | 7589 | (100.0) |
aSimilarity to deduced amino acid sequences with no definitive functions.
A marked tendency was seen between the results of the similarity search and frequency of raw EST in contigs, i.e. redundancy of the contigs. Fig. 2B and C indicate that the frequencies of raw EST in contigs of function-unknown sequences (showing no significant similarity with sequences in the databases) were lower than those of contigs similar to known sequences. The average frequencies of total contigs, that of contigs similar to known sequences and that of contigs of function-unknown sequences, were 2.83, 3.85, and 1.82, respectively (Fig. 2A–C). That is, the lower frequency group tended to contain a higher ratio contigs homologous to function-unknown sequences than the higher frequency group (data not shown). Low-frequency genes have fewer opportunities to be noticed for research than high-frequency genes, and hence their functions may tend to remain unknown.
3.3. Frequency analysis of the EST contigs
No cDNA libraries used in this study were experimentally normalized during library preparation, and therefore they were expected to retain the populations of the original mRNAs almost entirely. Thus, the frequency information of ESTs from the contigs is likely to reflect the populations of the cDNA library. Consequently, gene transcription profiles would be roughly predictable by comparative analysis of frequency information from the library or type of culture used. However, simple comparative analysis of the raw frequency value would not reach plausible frequency information because numbers of the collected ESTs ranged widely in the cDNA libraries examined (Table 1). Accordingly, frequency information of raw ESTs among all contigs was numerically normalized by the source cDNA library to frequency value per 1000 ESTs, which we termed the ‘normalized frequency value,’ and used for subsequent frequency analysis.
Supplementary Table S1 shows a ranking of the most frequent contigs from the total EST contig list by normalized frequency value. Here, a number of basic metabolic pathway genes and so-called housekeeping genes, such as those encoding ribosomal proteins, translation factors, or histones, were placed in the upper part of the total frequency ranking. Many of these contigs were genes showing nonspecific expression among various cultures (Supplementary Table S1). However, some library-specific or culture type-specific contigs with extremely high normalized frequency values were also included in this list (Supplementary Tables S1–S12). The average normalized frequency values within culture type such as the LC and the SC were compared and most frequent LC- and SC-specific contigs were extracted (Supplementary Tables S2 and S3). In terms of the environment for A. oryzae, the most major difference in culture types would be the physical phase of the medium: i.e. liquid phase or solid phase. The differences between the liquid and solid-state media range over many aspects, e.g. water activity, diffusion of nutrients and gases, continuity of medium distribution, physical barriers for hyphal extension invading the substrates, etc.10,18,19 To adapt to these environmental differences, A. oryzae would change its gene expression profile between LC and SC.10,22 Further detailed analyses of the frequency by cultivation conditions, i.e. LC and SC, may provide some insights into the critical factors required for differential transcriptional regulation. Such studies may also allow discovery of valuable (regulatable and/or strong) promoters, and facilitate industrial utilization.23 Moreover, functional analyses of such genes with biased expression profiles may reveal mechanisms for generating differences in mycelial characteristics among cultivation conditions. Such analyses may also reveal advantages of SC as efficient enzyme-producing systems.
Although many of the contigs in Supplementary Table S2 or S3 were not obtained solely from a particular LC or SC library, some contigs were library-specific, such as AoEST2714 (Supplementary Tables S2 and S8: similar to conidiation-specific protein 10 of Neurospora crassa), AoEST2823 (Supplementary Tables S3 and S11: similar to lactone hydrolase of Rhodococcus ruber), AoEST2980 (Supplementary Tables S2 and S4: similar to hexose transporter of Aspergillus parasiticus), AoEST2991 (Supplementary Tables S3 and S12: encoding aspergillopepsin O), AoEST3150 (Supplementary Tables S1, S2, and S8: no significant similarity to sequences in databases). Their appearance in these tables was due to their extremely high normalized frequency values in each source library.
Frequency rankings of particular culture-specific contigs are shown in Supplementary Tables S4–S12. Parameters of each culture employed in this study, even if excluding the LC- or SC-specific features, differed with other culture conditions in some physical, chemical, and/or biological respects, e.g. nutrient contents, temperature, growth phase, and/or other environmental factors. Consequently, lists of library-specific or -biased frequent contigs may reflect the gene expression profiles due to some individual features of each set of culture conditions (Supplementary Tables S4–S12).
Approximately 30–70% of the contigs with frequency biased to a particular library, except the LR library, in which the ratio was greater than 90%, showed similarity to known sequences in the databases. Laboratory cultivation of A. oryzae is generally done under LR culture conditions, and genes that show good expression under such conditions, such as genes that participate in glycolytic pathways, have tended to be targeted for investigation. Consequently, the ratio of genes with unknown function in LR may be lower than under other culture conditions. The approach to functional and expressional analyses on such genes with expression profiles biased to a particular type of culture (Supplementary Tables S2 and S3) or set of culture conditions (Supplementary Tables S4–S12) may be helpful for understanding the behavior of A. oryzae under these conditions.
The major genes for secreted enzymes necessary for the sake brewing process appeared frequently in several rankings: alpha-amylase, which is a starch-liquefying enzyme (AoEST2370, AoEST3004, AoEST3185; Supplementary Tables S1, S6, and S9); glucoamylase, which is a diastatic enzyme (AoEST03182; Supplementary Tables S1 and S3); and Aspergillopepsin O, which is an acid protease and contributes to liquefaction of rice materials (AoEST2991; Supplementary Tables S3 and S12). The ESTs of the latter two enzyme genes were obtained SC- or SR-specifically (Supplementary Tables S3 and S12). This is consistent with the results of previous studies, and the reported superiority of SC in the sake brewing process.9,12,24 In addition to contigs obtained under a particular set of culture conditions (Supplementary Tables S4–S12), it is interesting that a considerable number of function-unknown genes appeared in the upper parts of Supplementary Tables S1–S3. This suggests that some major but unnoticed features expressed constitutively in common to LC or SC remain in functional, physiological, and/or structural aspects of A. oryzae.
Supplementary Tables S1–S12 contain ORF ID which was registered in the A. oryzae genome database and correspond to each contigs. However, some of them and some contigs not shown in these tables did not correspond to any ORF in the database although there were corresponding genomic nucleotide sequences. They should correspond to non-coding RNA genes, untranslated region of transcripts, or could fail to be predicted as ORFs. In addition, nucleotide sequence corresponding to AoEST2089 (Supplementary Table S9: no similarity to any sequence in databases) and some others not shown in Supplementary Tables S1–S12 were not found in the genome database. There are several sequence gaps in A. oryzae genome information,14 and they are perhaps mapped to one of those regions, respectively.
Next, using all the EST frequency data converted to normalized frequency values, the culture conditions employed for collection of ESTs in this study were classified by a hierarchical clustering method, and the results are illustrated as a dendrogram in Fig. 3. In this figure, LS and SW were clustered as the most similar groups, and SR was next clustered with them, and followed by LM. A previous study suggested a similarity between SC and starved-LC. SC-specific genes were identified by cDNA subtraction, and some were also transcribed in stationary phase, but not in the growth phase in LC.10 Closeness on the dendrogram between LS and these SCs, SW and SR, may be due to a common environmental factor under these culture conditions: i.e. insufficiency of nutrients. The carbon source is deficient in LS, and available nitrogen and carbon sources tend to be deficient in SC, especially in the early stages because of the time lag in hydrolysis of substrates by lytic enzymes and limited diffusion of the resultant nutrients.10,18
Figure 3.

Dendrogram of culture conditions used for collecting ESTs. Culture conditions were classified based on the normalized frequency values of contigs by hierarchical clustering using the nearest neighbor-joining method.
Some ESTs unusually redundant (over 30.0) in a particular culture condition (AoEST3150 and AoEST3179 in LG, and AoEST3164, AoEST3186, and AoEST3189 in SS) (Supplementary Tables S1–S3, S8, and S11) affected neighbor-joining distance of the clusters that contained the above contigs (Fig. 2). In fact, the cluster, LG, which positioned off from the other clusters except SS, moved inside the tree consisting of those clusters when analyzed excluding the two contigs discussed earlier (data not shown). The proteins encoded on those highly redundant contigs may play important roles specifically in the two culture conditions, however, their functions are currently unpredictable based on the sequence similarity to the genes of known functions.
3.4. Functional classification by KOG major categories
Contig sequences were compared with the eukaryotic orthologous groups (KOGs), which consist of several eukaryotic ORF sequence sets, by BLASTX (Fig. 4A and B), and classified into KOG categories of similar sequence.16 Of the 7589 contigs, 2964 contigs showed similarity with the KOG sequence set. A portion of these contigs belonged to multiple categories. If the categorical redundancy was not removed, the total number of contigs, 2964, corresponded to 3284 categorized contigs. Meanwhile, 4625 contigs did not hit to any of KOG sequence. In the following discussion, we expediently considered the group of these 4625 contigs as one category, ‘No hit to KOGs’ (Fig. 4A and B), and accordingly, total number of categorized contigs was 7909. Of these contigs, 2914 (36.8%) were classified into the functional categories, including ‘Poorly characterized,’ of the corresponding counterpart in the KOGs, whereas the remaining 370 (4.7%) had no KOG categories (Fig. 4A, numerical values not shown). They were also classified into the major KOG categories by culture type category, such as SC and LC (Fig. 4A), or by source library (Fig. 4B). The ratios of KOG categories from LC, SC, or each library showed similar patterns to total contigs, except for LG (Fig. 4A and B). Similarly, the results of BLASTX search of the non-redundant database also showed an abundance of function-unknown sequences within A. oryzae EST sequences, especially in the case of contigs containing raw ESTs derived from the LG library (Fig. 2A, Tables 1 and 2).
Figure 4.
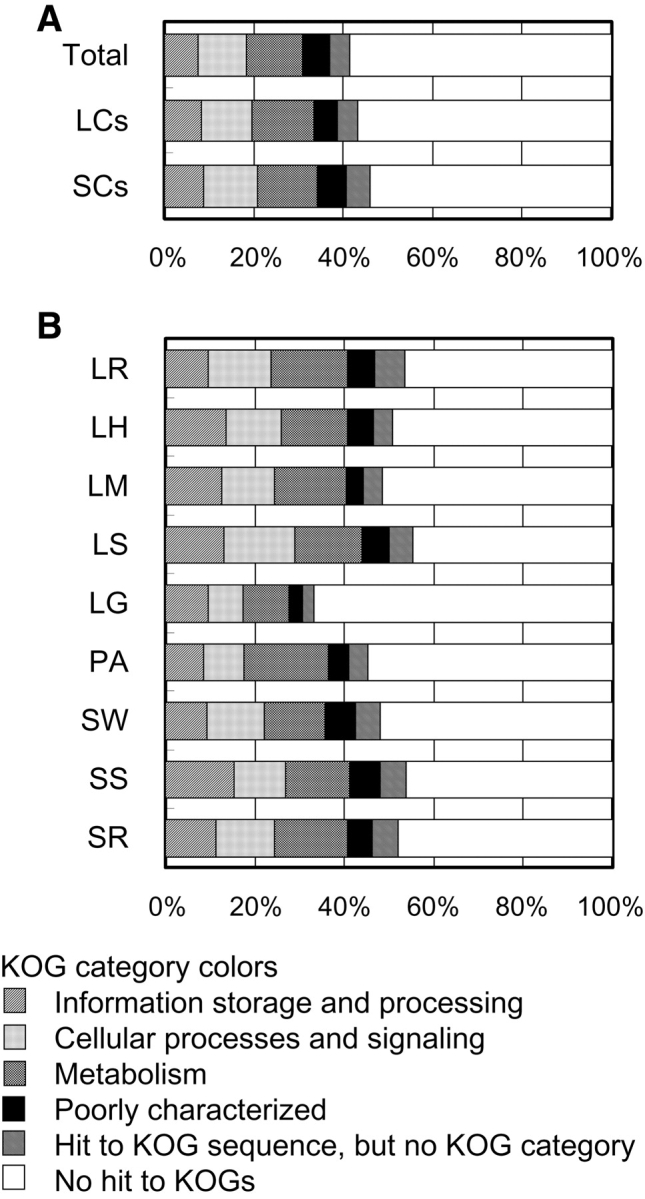
Functional classification of contigs to major KOG categories. Contigs were compared with the KOG sequence set using BLASTX, and then classified into the major KOG major categories of most similar sequence (E-value < 10E-9). Categorical redundancy of contigs was not removed when they belonged to multiple KOG categories. (A) Total contigs, contigs from LCs, and contigs from SCs (B) contigs from each library.
It is noteworthy that a large number of contigs, as many as 4625 (58.5% of 7909, 60.9% of 7589) had no significant counterpart in the KOGs (Fig. 4A, numerical values not shown). The contigs without similarity to KOGs may correspond to some genes with functional similarity to genes of KOGs, but no structural similarity. However, it is more likely that large segments of these sequences are involved in biological functions or characteristics of filamentous fungi lacking in the model eukaryote genomes: e.g. filamentous growth, hyphal differentiation, asexual development, high-capacity protein secretion, various enzymes themselves, and certain secondary metabolism pathways. Therefore, we inferred that these sequences, in preference to other sequences, may be effective for the study of characteristics unique to filamentous fungi.
3.5. Absence of sequences derived from aflatoxins-biosynthetic genes in the EST collection
Certain species that belong to the genus Aspergillus, such as A. parasiticus and A. flavus, closely related species of A. oryzae, produce aflatoxins (AF)—highly toxic products of secondary metabolism, which have carcinogenicity and acute toxicity. Approximately, 25 genes involved in AF biosynthesis are located in a 70-kb genomic region as a cluster.25 A member of this cluster, the gene aflR, encodes a transcription factor that positively regulates expression of the AF biosynthetic genes, including aflR itself, by a feedforward mechanism, and therefore has a crucial role in AF biosynthesis in these species. In aflatoxinogenic A. flavus, the AF biosynthetic genes must be functional under AF-producing culture conditions. Indeed, it has been reported that an A. flavus EST collection obtained under these culture conditions contained 21 of the total of 25 AF biosynthetic genes in the cluster region.5
In contrast, none of the known strains of A. oryzae produce AF under any conditions. Certain A. oryzae strains either entirely (group III) or partially (group II) lack the cluster region in their genomes, and therefore cannot produce AF.26,27 However, another type of A. oryzae (group I), including the RIB40 strain used in this study, contains the entire AF biosynthetic homologous gene cluster region.26,27 Although the long history of use in food industries has demonstrated the safety of A. oryzae, including the group I strains, the reasons for the safety of group I strains have not been clarified at the molecular level. Sequences of the cluster region in the RIB 40 genome27 were compared with all EST contig sequences. No aflR-like sequence was found among the contigs, although AF-producing culture conditions of aflatoxinogenic species, such as SC, were included in sources of the ESTs. This was consistent with a previous study in which no aflR-like transcripts of A. oryzae were detected under AF-producing conditions.28 Moreover, no other sequences in the AF biosynthetic homologous gene cluster were found in the EST contigs, except for only two singletons: aflJ (also named aflS)-like AoEST3534 from SR and aflT-like AoEST4046 from LR (both not shown in Supplementary Tables S1–S12).25,27 It was reported that aflJ is involved in regulation of the pathway by mechanisms that are as yet unclear,25 and aflT may encode a major facilitator superfamily transporter with no significant roles in AF secretion.29
If the AF pathway were activated in RIB40, a series of ESTs from the pathway genes, including the aflR-like gene, would have been obtained, as in the case of A. flavus.5 In A. oryzae RIB40, none of the AF biosynthetic homologs seemed to be transcriptionally activated by lack of transcription of the aflR-like gene, even under conditions that would result in AF production in the related species. These observations provide circumstantial evidence for the safety of A. oryzae as an industrial microorganism. However, the reason for the inactive aflR-like gene remains to be determined.
3.6. Application of the EST resource for further studies
This study provided not only numerous ESTs from A. oryzae, but also a vast collection of cDNA clones. Consequently, cDNA clones of novel genes can be obtained rapidly in many cases through in silico screening of the EST database instead of using conventional wet screening methods. Several studies have already cloned genes using the EST information.30–45 This study also provided the foundations for post-genomic approaches in A. oryzae.22,46 The clone collection is available for preparation of probes for transcriptome analyses. The sequence information deduced at the amino acid level can be exploited for proteome analyses. In addition, ESTs generally supply useful information for ORF prediction in whole-genome sequencing of eukaryotes. Indeed, the EST database markedly contributed to the A. oryzae whole-genome analysis project.14
The EST data is available on our website (http://www.nrib.go.jp/ken/EST/db/index.html or http://www.aist.go.jp/RIODB/ffdb/EST-DB.html). Nucleotide sequences of the EST contigs have been deposited in the GenBank/EMBL/DDBJ databanks (accession numbers AB223171–AB230863, including some vacant numbers). Genome sequence information of each EST contigs is referrable on the A. oryzae genome database (http://www.bio.nite.go.jp/dogan/Top). This study was performed as a joint project by a consortium of research organizations from industry and academy.
Acknowledgements
The authors thank Kazutoshi Sakamoto for helpful suggestions. This work was partly supported by private companies: Amano Enzyme Inc., Gekkeikan Sake Co. Ltd; Kikkoman Corp.; Higashimaru Shoyu Co. Ltd; Higeta Shoyu Co. Ltd; Ozeki Co. Ltd; Yamasa Shoyu Co. Ltd; Japan Federation of Miso Manufacturers Cooperatives; and Tanekoji-Kumiai (The Association of Koji Seed Companies that produce A. oryzae conidiophores).
Supplementary Data
Supplementary data are available online at www.dnaresearch.oxfordjournals.org.
References
- 1.Zhu H., Nowrousian M., Kupfer D., et al. Analysis of expressed sequence tags from two starvation, time-of-day-specific libraries of Neurospora crassa reveals novel clock-controlled genes. Genetics. 2001;157:1057–1065. doi: 10.1093/genetics/157.3.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim S., Ahn I. P., Lee Y. H. Analysis of genes expressed during rice-Magnaporthe grisea interactions. Mol. Plant Microbe. Interact. 2001;14:1340–1346. doi: 10.1094/MPMI.2001.14.11.1340. [DOI] [PubMed] [Google Scholar]
- 3.Trail F., Xu J. R., San Miguel P., Halgren R. G., Kistler H. C. Analysis of expressed sequence tags from Gibberella zeae (anamorph Fusarium graminearum) Fungal Genet. Biol. 2003;38:187–197. doi: 10.1016/s1087-1845(02)00529-7. [DOI] [PubMed] [Google Scholar]
- 4.Nugent K. G., Choffe K., Saville B. J. Gene expression during Ustilago maydis diploid filamentous growth: EST library creation and analyses. Fungal Genet. Biol. 2004;41:349–360. doi: 10.1016/j.fgb.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 5.Yu J., Whitelaw C. A., Nierman W. C., Bhatnagar D., Cleveland T. E. Aspergillus flavus expressed sequence tags for identification of genes with putative roles in aflatoxin contamination of crops. FEMS Microbiol. Lett. 2004;237:333–340. doi: 10.1016/j.femsle.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 6.Tailor M. J., Richardson T. Application of microbial enzymes in food systems and in biotechnology. Adv. Appl. Microbiol. 1979;25:7–35. doi: 10.1016/s0065-2164(08)70144-8. [DOI] [PubMed] [Google Scholar]
- 7.Barbesgaard P., Heldt-Hansen H. P., Diderichsen B. On the safety of Aspergillus oryzae: a review. Appl. Microbiol. Biotechnol. 1992;36:569–572. doi: 10.1007/BF00183230. [DOI] [PubMed] [Google Scholar]
- 8.Machida M. Progress of Aspergillus oryzae genomics. Adv. Appl. Microbiol. 2002;51:81–106. doi: 10.1016/s0065-2164(02)51002-9. [DOI] [PubMed] [Google Scholar]
- 9.Hata Y., Ishida H., Ichikawa E., Kawato A., Suginami K., Imayasu S. Nucleotide sequence of an alternative glucoamylase-encoding gene (glaB) expressed in solid-state culture of Aspergillus oryzae. Gene. 1998;207:127–134. doi: 10.1016/s0378-1119(97)00612-4. [DOI] [PubMed] [Google Scholar]
- 10.Akao T., Gomi K., Goto K., Okazaki N., Akita O. Subtractive cloning of cDNA from Aspergillus oryzae differentially regulated between solid-state culture and liquid (submerged) culture. Curr. Genet. 2002;41:275–281. doi: 10.1007/s00294-002-0314-y. [DOI] [PubMed] [Google Scholar]
- 11.Kitamoto K. Molecular biology of the koji molds. Adv. Appl. Microbiol. 2002;51:129–153. doi: 10.1016/s0065-2164(02)51004-2. [DOI] [PubMed] [Google Scholar]
- 12.Sakaguchi K., Takagi M., Horiuchi H., Gomi K. Fungal enzymes used in oriental food and beverage industries. In: Kinghorn J. R., Turner G, editors. Glasgow: Applied molecular genetics of filamentous fungi, Blackie Academic and Professional; 1992. pp. 54–99. [Google Scholar]
- 13.Gomi K., Iimura Y., Hara S. Integrative transformation of Aspergillus oryzae with a plasmid containing the Aspergillus nidulans argB gene. Agric. Biol. Chem. 1987;51:2549–2555. [Google Scholar]
- 14.Machida M., Asai K., Sano M., et al. Genome sequencing and analysis of Aspergillus oryzae. Nature. 2005;438:1157–1161. doi: 10.1038/nature04300. [DOI] [PubMed] [Google Scholar]
- 15.Cathala G., Savouret J.-F., Mendez B., et al. A method for isolation of intact, translationally active ribonucleic acid. DNA. 1983;2:329–335. doi: 10.1089/dna.1983.2.329. [DOI] [PubMed] [Google Scholar]
- 16.Tatusov R. L., Fedorova N. D., Jackson J. D., et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41–54. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sambrook J., Fritsch E. F., Maniatis T. 2nd edn. New York: Cold Spring Harbor Laboratory Press, Cold Spring Harbor; 1989. Molecular cloning: A Laboratory Manual. [Google Scholar]
- 18.Mitchell D. A., Berovic M., Krieger N. Biochemical engineering aspects of solid state bioprocessing. Adv. Biochem. Eng. 2000;68:61–138. doi: 10.1007/3-540-45564-7_3. [DOI] [PubMed] [Google Scholar]
- 19.Ishida H., Hata Y., Kojima Y., et al. Regulation of the glucoamylase-encoding gene (glaB), expressed in solid-state culture (koji) of Aspergillus oryzae. J. Ferment. Bioeng. 1998;86,:301–307. [Google Scholar]
- 20.Christensen T., Woeldike H., Boel E., et al. High level expression of recombinant genes in Aspergillus oryzae. Bio/Technology. 1988;6:1419–1421. [Google Scholar]
- 21.Gouka R. J., Punt P. J., van den Hondel C. A. M. J. J. Efficient production of secreted protein by Aspergillus: progress, limitation and prospects. Appl. Microbiol. Biotechnol. 1997;47:1–11. doi: 10.1007/s002530050880. [DOI] [PubMed] [Google Scholar]
- 22.Maeda H., Sano M., Maruyama Y., et al. Transcriptional analysis of genes for energy catabolism and hydrolytic enzymes in the filamentous fungus Aspergillus oryzae using cDNA microarrays and expressed sequence tags. Appl. Microbiol. Biotechnol. 2004;65:74–83. doi: 10.1007/s00253-004-1608-4. [DOI] [PubMed] [Google Scholar]
- 23.Ishida H., Hata Y., Kawato A., Abe Y., Kashiwagi Y. Isolation of a novel promoter for efficient protein production in Aspergillus oryzae. Biosci. Biotechnol. Biochem. 2004;68:1849–1857. doi: 10.1271/bbb.68.1849. [DOI] [PubMed] [Google Scholar]
- 24.Kitano H., Kataoka K., Furukawa K., Hara S. Specific expression and temperature-dependent expression of the acid protease-encoding gene (pepA) in Aspergillus oryzae in solid state culture (rice-koji) J. Biosci. Bioeng. 2002;93:563–567. doi: 10.1016/s1389-1723(02)80238-9. [DOI] [PubMed] [Google Scholar]
- 25.Yu J., Chang P. K., Ehrlich K. C., et al. Clustered pathway genes in aflatoxin biosynthesis. Appl. Environ. Microbiol. 2004;70:1253–1262. doi: 10.1128/AEM.70.3.1253-1262.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kusumoto K., Nogata Y., Ohta H. Directed deletions in the aflatoxin biosynthesis gene homolog cluster of Aspergillus oryzae. Curr. Genet. 2000;37:104–111. doi: 10.1007/s002940050016. [DOI] [PubMed] [Google Scholar]
- 27.Tominaga M., Lee Y. H., Hayashi R., et al. Molecular analysis of a homologous aflatoxin biosynthetic gene cluster in Aspergillus oryzae RIB strains. Appl. Environ. Microbiol. 2006;72:484–490. doi: 10.1128/AEM.72.1.484-490.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kusumoto K., Yabe K., Nogata Y., Ohta H. Transcript of a homolog of aflR, a regulatory gene for aflatoxin synthesis in Aspergillus parasiticus, was not detected in Aspergillus oryzae strains. FEMS Microbiol. Lett. 1998;169:303–307. doi: 10.1111/j.1574-6968.1998.tb13333.x. [DOI] [PubMed] [Google Scholar]
- 29.Chang P. K., Yu J., Yu J. H. aflT, a MFS transporter-encoding gene located in the aflatoxin gene cluster, does not have a significant role in aflatoxin secretion. Fungal Genet. Biol. 2004;41:911–920. doi: 10.1016/j.fgb.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 30.Nakajima K., Kunihiro S., Sano M., et al. Comprehensive cloning and expression analysis of glycolytic genes from the filamentous fungus, Aspergillus oryzae. Curr. Genet. 2000;37:322–327. doi: 10.1007/s002940050534. [DOI] [PubMed] [Google Scholar]
- 31.Juvvadi P. R., Arioka M., Nakajima H., Kitamoto K. Cloning and sequence analysis of cnaA gene encoding the catalytic subunit of calcineurin from A. oryzae. FEMS Microbiol. Lett. 2001;204:169–174. doi: 10.1111/j.1574-6968.2001.tb10881.x. [DOI] [PubMed] [Google Scholar]
- 32.Chigira Y., Abe K., Gomi K., Nakajima T. chsZ, a gene for a novel class of chitin synthase from Aspergillus oryzae. Curr. Genet. 2002;41:261–267. doi: 10.1007/s00294-002-0305-z. [DOI] [PubMed] [Google Scholar]
- 33.Kuroki Y., Juvvadi P. R., Arioka M., Nakajima H., Kitamoto K. Cloning and characterization of vmaA, the gene encoding a 69-kDa catalytic subunit of the vacuolar H + -ATPase during alkaline pH mediated growth of Aspergillus oryzae. FEMS Microbiol. Lett. 2002;209:277–282. doi: 10.1111/j.1574-6968.2002.tb11144.x. [DOI] [PubMed] [Google Scholar]
- 34.Kunihiro S., Kawanishi Y., Sano M., et al. A polymerase chain reaction-based method for cloning novel members of a gene family using a combination of degenerate and inhibitory primers. Gene. 2002;289:177–184. doi: 10.1016/s0378-1119(02)00547-4. [DOI] [PubMed] [Google Scholar]
- 35.Sasangka P., Matsumoto A., Tanaka A., et al. Structural features of the glycogen branching enzyme encoding genes from Aspergilli. Microbiol. Res. 2002;157:337–344. doi: 10.1078/0944-5013-00170. [DOI] [PubMed] [Google Scholar]
- 36.Yamada O., Na Nan S., Akao T., et al. dffA gene from Aspergillus oryzae encodes L-ornithine N5-oxygenase and is indispensable for deferriferrichrysin biosynthesis. J. Biosci. Bioengi. 2003;95:82–88. doi: 10.1016/S1389-1723(03)80153-6. [DOI] [PubMed] [Google Scholar]
- 37.Kaya M., Matsumura K., Higashida K., et al. Cloning and enhanced expression of the cytochrome P450nor gene (nicA; CYP55A5) encoding nitric oxide reductase from Aspergillus oryzae. Biosci. Biotechnol. Biochem. 2004;68:2040–2049. doi: 10.1271/bbb.68.2040. [DOI] [PubMed] [Google Scholar]
- 38.Mizutani O., Nojima A., Yamamoto M., et al. Disordered cell integrity signaling by disruption of kexB gene in Aspergillus oryzae. Eukaryotic Cell. 2004;3:1036–1048. doi: 10.1128/EC.3.4.1036-1048.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Obata H., Ishida H., Kawato A., et al. Cloning of a novel tyrosinase-encoding gene (melB) from Aspergillus oryzae and its overexpression in solid-state culture (rice koji) J. Biosci. Bioeng. 2004;97:400–405. doi: 10.1016/S1389-1723(04)70226-1. [DOI] [PubMed] [Google Scholar]
- 40.Matsumura K., Obata H., Hata Y., Kawato A., Abe Y., Akita O. Isolation and characterization of a novel gene encoding α-L-Arabinofuranosidase from Aspergillus oryzae. J. Biosci. Bioeng. 2004;98:77–84. doi: 10.1016/S1389-1723(04)70246-7. [DOI] [PubMed] [Google Scholar]
- 41.Maruyama J., Juvvadi P. R., Ishi K., Kitamoto K. Three-dimensional image analysis of plugging at the septal pore by Woronin body during hypotonic shock inducing hyphal tip bursting in the filamentous fungus Aspergillus oryzae. Biochem. Biophys. Res. Commun. 2005;331:1081–1088. doi: 10.1016/j.bbrc.2005.03.233. [DOI] [PubMed] [Google Scholar]
- 42.Hisada H., Hata Y., Kawato A., Abe Y., Akita O. Cloning and expression analysis of two catalase genes from Aspergillus oryzae. J. Biosci. Bioeng. 2005;99:562–568. doi: 10.1263/jbb.99.562. [DOI] [PubMed] [Google Scholar]
- 43.Takahashi T., Maeda H., Yoneda S., et al. The fungal hydrophobin RolA recruits polyesterase and laterally moves on hydrophobic surfaces. Mol. Microbiol. 2005;57:1780–1798. doi: 10.1111/j.1365-2958.2005.04803.x. [DOI] [PubMed] [Google Scholar]
- 44.Koseki T., Miwa Y., Akao T., Akita O., Hashizume K. An Aspergillus oryzae acetyl xylan esterase: Molecular cloning and characteristics of recombinant enzyme expressed in Pichia pastoris. J. Biotechnol. 2006;121:381–389. doi: 10.1016/j.jbiotec.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 45.Akao T., Yamaguchi M., Yahara A., et al. Cloning and expression of 1,2-α-mannosidase gene (fmanIB) from filamentous fungus Aspergillus oryzae: in vivo visualization of the FmanIBp-GFP fusion protein. Biosci. Biotechnol. Biochem. 2006;70:471–479. doi: 10.1271/bbb.70.471. [DOI] [PubMed] [Google Scholar]
- 46.Oda K., Kakizono D., Yamada O., Iefuji H., Akita O., Iwashita K. Proteomic analysis of extracellular proteins from Aspergillus oryzae grown under submerged and solid-state culture conditions. Appl. Environ. Microbiol. 2006;72:3448–3457. doi: 10.1128/AEM.72.5.3448-3457.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]