

Abstract
The Medaka is an excellent genetic system for studies of vertebrate development and disease and environmental and evolutionary biology studies. To facilitate the mapping of markers or the cloning of affected genes in Medaka mutants identified by forward-genetic screens, we have established a panel of whole-genome radiation hybrids (RHs) and RH maps for three Medaka chromosomes. RH mapping is useful, since markers to be mapped need not be polymorphic and one can establish the order of markers that are difficult to resolve by genetic mapping owing to low genetic recombination rates. RHs were generated by fusing the irradiated donor, OLF-136 Medaka cell line, with the host B78 mouse melanoma cells. Of 290 initial RH clones, we selected 93 on the basis of high retention of fragments of the Medaka genome to establish a panel that allows genotyping in the 96-well format. RH maps for linkage groups 12, 17, and 22 were generated using 159 markers. The average retention for the three chromosomes was 19% and the average break point frequency was ∼33 kb/cR. We estimate the potential resolution of the RH panel to be ∼186 kb, which is high enough for integrating RH data with bacterial artificial chromosome clones. Thus, this first RH panel will be a useful tool for mapping mutated genes in Medaka.
Key words: Medaka, radiation hybrid mapping, genetic mapping, BAC
1. Introduction
Radiation hybrid (RH) mapping is an efficient way for ordering markers in correlation with physical distance on the chromosome and establish links with genetic maps and bacterial artificial chromosome (BAC) contigs.1,2 An RH panel consists of cell hybrids that randomly retain fragments of chromosomes from the donor cells. These hybrids are produced by fusion of irradiated donor cell of the species of interest with a recipient cell line, usually of rodent origin and have the following features: (i) Markers are scored by PCR analysis for the presence or the absence of DNA from the hybrids. Therefore, markers to be mapped need not to be polymorphic in RH mapping, which facilitates the production of dense maps of the genome. Markers with similar patterns of retention in the collection of hybrids are placed close to each other on the map. (ii) RH mapping depends on random physical breakage of chromosomes by irradiation and thereby reflects physical distance, whereas genetic mapping relies on meiotic recombination rate. Markers close to the centromere, which are difficult to be resolved by genetic mapping, can be ordered in RH mapping. (iii) Furthermore, the resolution of the RH panel can be adjusted by the dose of radiation to achieve the resolution required for linking the RH map with a genetic map and/or with BAC contigs.1,2 Thus, RHs of human,3–5 dog,6 rat,7 mouse,8,9 and zebrafish10,11 have played a key role in localizing markers and anchoring BAC contigs on the chromosomes.
The Medaka, Oryzias latipes, a small freshwater fish,12 has proven to be an excellent genetic system for developmental, environmental, and evolutionary biology studies. A genetic linkage map of Medaka that consists of 24 linkage groups (LG) corresponding to the haploid chromosome number of the organism was established.13 Recently, genomic resource of Medaka including EST,14 genetic map,13,15 and genomic DNA sequence,16 whole-genome shotgun data on UT browser (http://medaka.utgenome.org/), has been developed. Furthermore, a large-scale systematic mutagenesis screen was performed in Medaka to explore gene functions in developmental processes.17 To facilitate the identification of affected genes in Medaka mutants, we established a whole-genome Medaka RH panel consisting of 93 clones. As the first step toward the development of a Medaka RH map, we constructed RH maps for three chromosomes LG12, 17, and 22.
2. Generation of Medaka RHs
Two-hundred and ninety RH clones were produced by fusing donor cells from the Medaka fin fibroblast cell line OLF-13618 (obtained from Riken Cell Bank, RCB 0184), derived from HB32 South Strain with B78 mouse melanoma cells, as described by Hukriede et al.11 For this purpose, a subline of OLF-136 with the highest percentage of euploid cells was selected by karyotyping and used to generate RHs. As the mouse melanoma B78 recipient cell line is not deficient in any enzyme that would allow selection of hybrid cells, we generated OLF-136 clones that randomly integrated the aminoglycoside phosphotransferase gene that confers resistance to G418 into the chromosomes. More than 500 independent G418 OLF-136 clones were pooled and 3 × 107 cells from this pool were subjected to X-ray irradiation at a dose of 5000 rad (50 Gy). The irradiated cells were mixed with an equal number of B78 cells and fused in the presence of polyethylene glycol. After 3 weeks, G418-resistant colonies were picked and expanded for DNA extraction or frozen to maintain stocks.
3. Establishment of the Medaka RH panel
To carry out the genotyping of the RH panel in a 96-well microtiter plate format, we selected 93 RH clones by the following procedures: (i) Among the 290 RHs, we first selected 136 RHs that gave clear bands in PCR genotyping, using 932 STS (sequence-tagged site) markers randomly selected from the 24 LGs. (ii) Among these 136 RHs, 93 RHs were selected on the basis of their high retention of Medaka chromosomes fragments. For independent estimates of retention,19 among those markers that gave no more than one typing error in triplicate genotyping assays, only one or two markers with the largest distance on the genetic map were chosen from a single LG. The retention frequency of the selected 93 RH clones based on 26 STS markers from 15 LGs was 16%.
The RHs were genotyped by PCR amplification, followed by gel electrophoresis, and the results of genotyping for a marker, designated as an RH vector, were documented as described.19 Sequences of STS markers were obtained from the MBASE (http://mbase.bioweb.ne.jp/). PCR reactions were set up in a 384-well format, using the Biomeck 2000 robotic system (Beckman, USA). Since the donor Medaka chromosomal fragments are retained at different molarities among the RH cell lines, the intensity of amplified bands may vary among them.3 Therefore, genotyping of the RHs was carried out three times to minimize discordance. Discordance between the multiple runs of a marker was kept below 7% of the total number of RHs. The markers that showed higher discordance were eliminated.
4. Generation of RH maps for three Medaka chromosomes
To generate RH maps for the three chromosomes (Fig. 1), we took the following steps. The numbers of markers relevant to each step are listed in Table 1.
Figure 1.
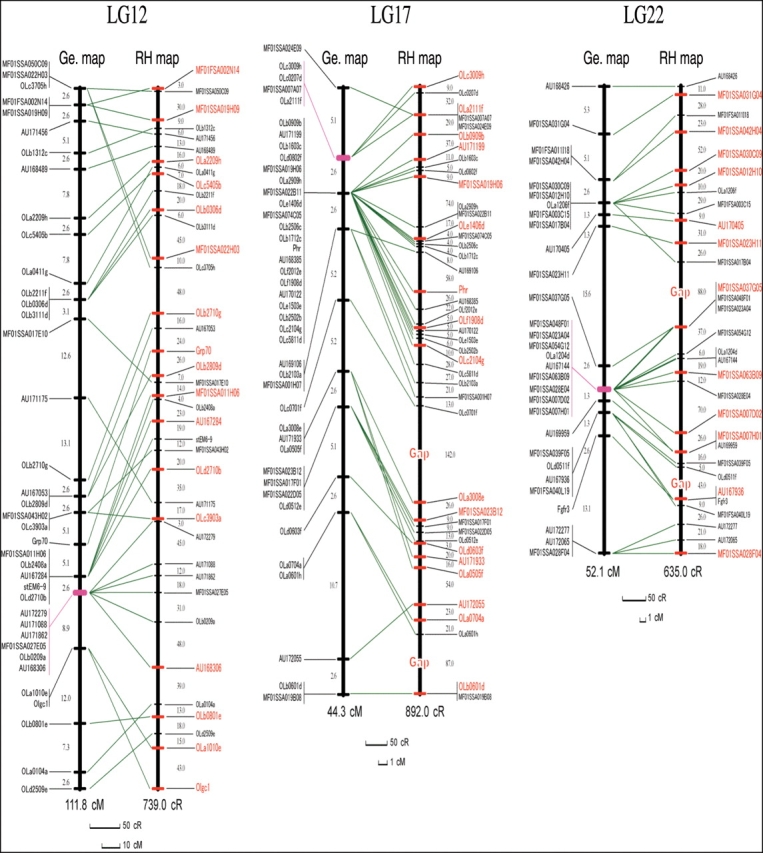
Comparison of the RH and genetic maps for the three Medaka chromosomes. Positional relationships of the markers on the RH (left) and genetic (right) maps are indicated by green lines. The distances between adjacent markers are shown in centiRay in the RH maps and centiMorgan in the genetic maps. In the RH maps, there are two gaps indicated as ‘Gap’ in LG17 and 22. The markers used to build the framework map are shown in red and those used for later placement are in black. On the genetic maps, centromeric regions are indicated in purple.
Table 1.
Markers used at the various steps of RH map construction
| Chromosome | Initial markers analyzed | Markers scored after PCR (percentage of initial markers) | Markers on framework maps at LOD = 7 | Markers on final maps at LOD = 5 (percentage of scored markers) |
|---|---|---|---|---|
| LG12 | 50 | 42 (84) | 17 | 38 (90) |
| LG17 | 60 | 51 (85) | 17 (two gaps) | 42 (82) (two gaps) |
| LG22 | 49 | 40 (82) | 12 (two gaps) | 30 (75) (two gaps) |
| Total | 159 | 133 (84) | 110 (83) |
Percentages of initial markers and percentages of scored markers are given in parentheses.
From LG12, 17, and 22 of the genetic maps, markers that gave reliable RH vectors were chosen. A total of 159 STS markers from the genetic map, that is, 50, 60, and 49 markers from LG12, 17, and 22, respectively, were subjected to genotyping on the RH panel. Twenty-six of these markers were excluded because they yielded no PCR products or ambiguous bands, leaving 133 markers (84%) for the following RH map construction. We used the TSP/CONCORD program to analyze the RH vectors, as described in the following section.
We checked whether the 133 markers fall into three groups corresponding to the three genetic LGs by searching linked markers with a threshold of pairwise LOD scores up to 5. This resulted in three LGs in agreement with the genetic map, as well as 17 singletons. The singletons were discarded. The three LGs included 42, 42, and 32 markers (total 116) from LG12, 17, and 22, respectively.
To generate high-confidence framework maps, markers from each LG in the previous step were analyzed with a pairwise LOD score threshold of 7. Three disconnected framework marker sets with two gaps were formed for LG17 and LG22, whereas a single-framework map with no gap was formed for LG12. Those disconnected framework marker sets were oriented and linked by referring to the linkage information of the genetic map to make one framework map for LG17 and 22.
Finally, to compose the final RH maps, we put the rest of the markers on the framework map, using the placement program. The two gaps in the framework maps of LG12 and 22 could not be filled up by this procedure.
The final RH maps of the three LGs and the comparison between the RH and genetic maps are shown in Fig. 1. Remarkably, comparisons of the placement of the markers in the RH maps with those in the genetic maps indicated that the markers with poor resolution in genetic maps in the putative centromeric regions (shown in purple) could be well resolved in the RH maps.
As shown in Table 2, given the estimated physical lengths, 30, 32, and 29 Mb, of LG12, 17, and 22, respectively, the physical distances corresponding to 1 cR5000 were calculated to be 41, 35, and 46 kb for LG12, 17, and 22, respectively. The average marker retention frequencies for LG12, 17, and 22 were 26, 16, and 13%, respectively.
Table 2.
Characteristics of RH maps for three Medaka LGs
| Chromosome | Map length (cR5000) | Average retention (%) | Breakpoint frequency (kb/cR) | Estimated physical length (Mb) |
|---|---|---|---|---|
| LG12 | 739 | 26 | 41 | 30 |
| LG17 | 915 | 16 | 35 | 32 |
| LG22 | 635 | 13 | 46 | 29 |
| Average | 19 | 41 |
The three RH maps have a high confidence since the framework linkage maps were obtained with an LOD score > 7 and each marker was linked at least to one other marker with an LOD score > 5 (corresponding to a likelihood of >100 000:1) in the final RH maps (Table 1 and Fig. 1). A total of 110 markers, 83% of the 133 markers that gave the RH vectors, could be placed on the final RH maps (Table 1). The lower success rate (75%) for LG22 could be due to its lower retention (13%; Tables 1 and 2). The average resolution of the three RH maps is 186 kb, which is high enough to place BAC clones (150–200 kb) on the RH maps. The average retention frequency of the 110 markers mapped to the three LGs was 19% (Table 2). This is comparable with whole-genome RH panels of other species whose overall retention ranges between 16 and 30%.9–11,20,21
Recently, the Medaka genome sequence has become accessible: the complete sequence of LG22 by BAC-based sequencing approach15 and the assembled sequence data from the whole-genome shotgun approach (UT genome browser). The alignment of the RH and genetic maps with the complete sequence of LG22 showed a high correlation of the RH map with the physical length of the genome (Fig. 2i). The poorly resolved region in the male genetic map of LG22 (shown by the red line at 33 cM, nine out of 30 markers) was resolved into 170 cR, which corresponds to 11.4 Mb. This region spans nearly one-third of the LG22 genome sequence. Similar results were obtained for LG12 and 17 assembled sequences (Fig. 2ii and iii). Markers that were not found in the genome sequences (those plotted at 0 Mb), 2, 3, and 1 on LG22, 12, and 17, respectively, were successfully mapped on the RH maps. These results further corroborate the usefulness of RH maps for establishing the complete sequence of the Medaka genome.
Figure 2.
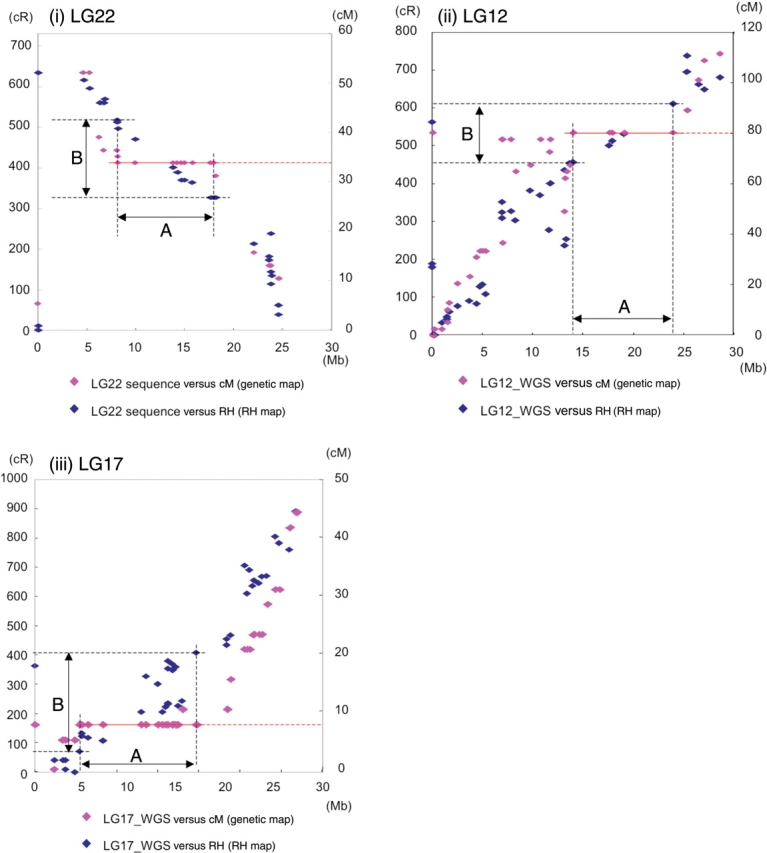
Consistency of the RH maps with the genome sequence. Markers on the LG22, 12, 17 were plotted on the Y axis as genetic map position in centi-Morgan (cM) (pink) and RH map position in cR (blue) and on the X axis as the position in the BAC-based genome sequence of (i) LG22 or assembled the whole genome shotgun sequence reads of (ii) LG12 and (iii) LG17. In the case of LG22, markers poorly resolved on the red line (nine out of 30 markers) at 33.8 cM on the genetic map were resolved into 170 cR (B) on the RH map that corresponds to 11.4 Mb (A), one-third of the physical length of LG22. The poorly resolved regions in LG12 and 17 were resolved into 154 and 338 cR, which correspond to13.0 and 14.2 Mb, one-third and nearly a half of the chromosome. Markers that were not found in the genome sequence (those plotted at 0 Mb), 2, 3, and 1 on LG22, 12, and 17, respectively, were successfully mapped on the RH maps.
Taken together, we produced the first whole-genome RH panel for mapping genes and markers in Medaka and demonstrated that it is suitable to build a genome-wide RH map. The construction of RH maps for the other Medaka LGs is ongoing. We will distribute the Medaka RH panel upon request to the corresponding author and provide assistance in RH mapping efforts.
5. Computation of RH maps
To construct RH maps, the TSP/CONCORD V2.0 software package,22 working with IBM DB2, was used on an IBM RS/6000 AIX workstation. TSP/CONCORD was used in conjunction with CONCORD and QSopt software to calculate RH maps by solving the Traveling Salesman Problem.23 This method has been proven to be rapid and efficient for integrating large data and constructing an RH map.22 We referred to TSP/CONCORD's manual to analyze RH vectors and to compute the marker order of each LG and the distance between markers. In the package, there are five objective functions for the evaluation of an RH vector available: two of them are based on obligate chromosome breaks (OCBs) and three on a maximum likelihood estimate (MLE). Since the OCB objective functions do not provide indications for estimating physical distances between markers and for comparing likelihoods of competing marker orders, only the three MLE objectives, namely, BASE TSP + MLE, Extended TSP + MLE, and Normalize TSP + MLE, were used in our computations. To construct the framework maps, most of the parameters were set to the program default values; three of them are sensitive for restricting the number of markers in computation, they were LIMIT_DISTANCE = 3, LIMIT_ LIKELIHOOD = 3, and UNKNOWN_COUNT = 2.
First, pairwise LOD scores and distances for each pair of markers were computed by using the TSP/CONCORD ‘pairlods_dists’ program. At an LOD score threshold of 5, the program ‘make_groups’ was executed to find LGs for all markers. Singletons were abandoned. For each of those LGs, marker sets were identified at an LOD score threshold of 7 by using again the ‘make_groups’ program. Subsequently, the framework maps of each marker set were computed by using the MLE objective functions in the package. We confirmed the robustness of each framework map by making sure that there are no improving flips of up to eight markers by using the program ‘flips’ and that there are no alternatives within 0.25 LOD units of the best marker order by using the program ‘map_eval’. Subsequently, the markers that were not on the framework maps were placed by performing ‘placement <marker_data > <framework_map >’ iteratively, generating maps for all marker sets. The initial maps of each LG were ordered and oriented relative to each other by referring to the genetic map. Finally, the initial maps of each LG were joined by using the three MLE objective functions of the package to generate three candidate maps. From these three candidates, an optimal one was picked up as the final RH map by comparing the results of the program ‘quality’.
Acknowledgments
This research was generously supported by grants from Japan Science and Technology Agency: a PREST grant to M. F.-S., and ERATO and SORST grants to H.K. This study was also supported in part by the Ground-based Research Program for Space Utilization from the Japan Space Forum to M.F.-S. and H.M. We thank Lucille Joly and Patricia Tellis for technical assistance. F.S. was supported by 21st Century COE program ‘Dynamic of Biological System’.
References
- 1.Cox D. R., Burmeister M., Price E. R., Kim S., Myers R. M. Radiation hybrid mapping: a somatic cell genetic method for constructing high-resolution maps of mammalian chromosomes. Science. 1990;250:245–250. doi: 10.1126/science.2218528. [DOI] [PubMed] [Google Scholar]
- 2.Walter M. A., Spillett D. J., Thomas P., Weissenbach J., Goodfellow P. N. A method for constructing radiation hybrid maps of whole genomes. Nat. Genet. 1994;7:22–28. doi: 10.1038/ng0594-22. [DOI] [PubMed] [Google Scholar]
- 3.Hudson T. J., Stein L. D., Gerety S. S., et al. An STS-based map of the human genome. Science. 1995;270:1945–1954. doi: 10.1126/science.270.5244.1945. [DOI] [PubMed] [Google Scholar]
- 4.Stewart E. A., McKusick K. B., Aggarwal A., et al. An STS-based radiation hybrid map of the human genome. Genome Res. 1997;7:422–433. doi: 10.1101/gr.7.5.422. [DOI] [PubMed] [Google Scholar]
- 5.Gyapay G., Schmitt K., Fizames C., et al. A radiation hybrid map of the human genome. Hum. Mol. Genet. 1996;5:339–346. doi: 10.1093/hmg/5.3.339. [DOI] [PubMed] [Google Scholar]
- 6.Priat C., Hitte C., Vignaux F., et al. A whole-genome radiation hybrid map of the dog genome. Genetics. 1998;54:361–378. doi: 10.1006/geno.1998.5602. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe T. K., Bihoreau M.-T., McCarthy L. C., et al. A radiation hybrid map of the rat genome containing 5,255 markers. Nat. Genet. 1999;22:27–36. doi: 10.1038/8737. [DOI] [PubMed] [Google Scholar]
- 8.McCarthy L. C., Terrett J., Davis M. E., et al. A first-generation whole genome-radiation hybrid map spanning the mouse genome. Genome Res. 1997;7:1153–1161. doi: 10.1101/gr.7.12.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Etten W. J. V., Steen R. G., Nguyen H., et al. Radiation hybrid map of the mouse genome. Nat. Genet. 1999;22:384–387. doi: 10.1038/11962. [DOI] [PubMed] [Google Scholar]
- 10.Geisler R., Rauch G.-J., Baier H., et al. A radiation hybrid map of the zebrafish genome. Nat. Genet. 1999;23:86–89. doi: 10.1038/12692. [DOI] [PubMed] [Google Scholar]
- 11.Hukriede N. A., Joly L., Tsang M., et al. Radiation hybrid mapping of the zebrafish genome. Proc. Natl Acad. Sci. USA. 1999;96:9745–9750. doi: 10.1073/pnas.96.17.9745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shima A., Mitani H. Medaka as a research organism: past, present and future. Mech. Dev. 2004;121:599–604. doi: 10.1016/j.mod.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 13.Naruse K., Fukamachi S., Mitani H., et al. A detailed linkage map of medaka, Oryzias latipes: comparative genomics and genome evolution. Genetics. 2000;154:1773–1784. doi: 10.1093/genetics/154.4.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kimura T., Jindo T., Narita T., et al. Large-scale isolation of ESTs from medaka embryos and its application to medaka developmental genetics. Mech. Dev. 2004;121:915–932. doi: 10.1016/j.mod.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 15.Naruse K., Tanaka M., Mita K., Shima A., Postlethwait J., Mitani H. A medaka gene map: the trace of ancestral vertebrate proto-chromosomes revealed by comparative gene mapping. Genome Res. 2004;14:820–828. doi: 10.1101/gr.2004004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sasaki T., Shimizu A., Ishikawa S. K., et al. The DNA sequence of medaka chromosome LG22. Genomics. 2007;89:124–133. doi: 10.1016/j.ygeno.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 17.Furutani-Seiki M., Sasado T., Morinaga C., et al. A systematic genome-wide screen for mutations affecting organogenesis in Medaka, Oryzias latipes. Mech. Dev. 2004;121:647–658. doi: 10.1016/j.mod.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 18.Etoh H., Suyama I., Hyodo-Taguchi Y., Matsudaira H. Establishment and characteristics of various cell lines from medaka (Teleostei) In: Kuroda Y., Kurstak E., Maramorosch K., editors. Invertebrate and Fish Tissue Culture 1988. Tokyo/Springer-Verlag, Berlin: Japan Scientific Societies Press; 1988. [Google Scholar]
- 19.Jones H. B. Hybrid selection as a method of increasing mapping power for radiation hybrids. Genome Res. 1996;6:761–769. doi: 10.1101/gr.6.8.761. [DOI] [PubMed] [Google Scholar]
- 20.Geisler R. Mapping and cloning in zebrafish. In: Nüsslein-Volhard C., Dahm R., editors. A Practical Approach. Oxford: Oxford University Press; 2002. p. 261. [Google Scholar]
- 21.Birren B., Green E. D., Hieter P. Genome Analysis: A Laboratory Manual: Mapping Genome. Vol. 4. New York: Cold Spring Harbor Laboratory Press; 1999. Genome Analysis Series. [Google Scholar]
- 22.Morisson M., Lemière A., Bosc S., et al. ChickRH6: a chicken whole-genome radiation hybrid panel. Genet. Sel. Evol. 2002;34:521–533. doi: 10.1186/1297-9686-34-4-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agarwala R., Applegate D. L., Maglott D., Schuler G. D., Schaffer A. A. A fast and scalable radiation hybrid map construction and integration strategy. Genome Res. 2000;10:350–364. doi: 10.1101/gr.10.3.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Applegate D., Bixby R., Chvatal V., Cook W. On the solution of traveling salesman problems. Doc. Math. 1998;III:645–656. (J. DMV, Extra Volume) ICM 1998. [Google Scholar]