

Abstract
Protein–protein interactions (PPIs) play crucial roles in protein function for a variety of biological processes. Data from large-scale PPI screening has contributed to understanding the function of a large number of predicted genes from fully sequenced genomes. Here, we report the systematic identification of protein interactions for the unicellular cyanobacterium Synechocystis sp. strain PCC6803. Using a modified high-throughput yeast two-hybrid assay, we screened 1825 genes selected primarily from (i) genes of two-component signal transducers of Synechocystis, (ii) Synechocystis genes whose homologues are conserved in the genome of Arabidopsis thaliana, and (iii) genes of unknown function on the Synechocystis chromosome. A total of 3236 independent two-hybrid interactions involving 1920 proteins (52% of the total protein coding genes) were identified and each interaction was evaluated using an interaction generality (IG) measure, as well as the general features of interacting partners. The interaction data obtained in this study should provide new insights and novel strategies for functional analyses of genes in Synechocystis, and, additionally, genes in other cyanobacteria and plant genes of cyanobacterial origin.
Key words: Synechocystis sp. strain PCC6803, protein–protein interactions, yeast two-hybrid system
1. Introduction
The unicellular cyanobacterium Synechocystis sp. strain PCC6803 was the first phototrophic organism to be fully sequenced.1 The availability of the complete genome sequence of Synechocystis has drastically changed the strategy for studying genetic systems in cyanobacteria.2 Gene identification and functional assignment have been accelerated utilizing the genome sequence. The genome sequence has also allowed systematic analyses of gene regulation and function on the genomic level. Microarray and proteome analyses have made it possible to monitor the expression of a substantial proportion of genes at both transcriptional and translational levels.3,4 Genetic analyses, such as targeted disruption or random tagging analysis, have suggested many functional links between gene products.2 Two genome databases, CyanoBase and CyanoMutants, have been established and act as centralized information resources.5,6 Based on these material and information resources, Synechocystis provides an ideal model system for genetic studies of photosynthetic organisms.
Although a large quantity of data has been accumulated through functional analyses of the genome, many of these analyses were driven by the predicted functions of the annotated genes. Therefore, a limited amount of information is available for unannotated genes. One of the effective approaches to obtain information regarding the function of these uncharacterized proteins is an analysis of protein–protein interactions (PPIs). Since nearly half of the predicted gene products of Synechocystis remain unannotated, a systematic approach for analysis of PPIs is needed.
We conducted a large-scale analysis of Synechocystis PPIs using a yeast two-hybrid (YTH) system. YTH analysis is one of the well-established methods to detect binary protein interactions and is feasible for large-scale analysis. Large-scale YTH screens have been conducted in a wide range of organisms and have provided several important biological and bioinformatics platforms for the study of protein networks in different organisms.7–13 These analyses have also successfully placed functionally uncharacterized proteins in their biological context.
In this study, we applied the YTH approach to three major target gene groups. For the initial screening, we selected genes of two-component signal transducers in order to evaluate our YTH screening system, as well as to elucidate the two-component signal transduction pathways in Synechocystis. The two-component signal transduction system generally consists of a histidine kinase (Hik) and a response regulator (Rre), and signal transduction is achieved by phosphorylation as a result of the interaction between cognate pairs of Hiks and Rres. Since the interactions of Hiks and Rres are binary, these two-component signal transducers are suitable targets for evaluation using the YTH screening system. On the Synechocystis chromosome, 44 putative genes for Hiks and 42 genes for Rres are predicted.1,14,15 In contrast to the Escherichia coli and Bacillus subtilis genomes, in which most of the genes for cognate pairs of Hiks and Rres are located close to each other, many of the genes for Hiks and Rres in Synechocystis are distributed randomly in the chromosome. Among 44 genes for Hiks on the Synechocystis chromosome, 14 are located in the vicinity of genes for potential cognate Rres, whereas other 30 are not located near any genes for Rres.15 Therefore, identification of PPIs between two-component signal transducers will provide valuable information to elucidate the cognate pairs of Hiks and Rres and signal transduction pathways in Synechocystis.
For the second screening group, we selected Synechocystis genes whose homologues are conserved in the A. thaliana genome with the aim to obtain interaction information that is applicable to plant genes of cyanobacterial origin as well as Synechocystis genes. Chloroplasts are descendants of free-living cyanobacteria that became endsymbionts. In the course of the evolutionary processes that transformed the cyanobacterial symbiont into an organelle, chloroplasts have donated many genes to nuclear chromosomes.16 Martin et al.17 reported that approximately 1700 of 9369 A. thaliana genes that were investigated are of cyanobacterial origin and these encompass all functional categories. PPI information on Synechocystis genes conserved in plants would provide useful insights for functional analysis of plant genes of cyanobacterial origin, since Synechocystis has been used as a model for analysis of these plant genes.
For the third screening group, we selected genes of unknown function in order to obtain interaction data that could be used to predict function. Since approximately 60% of the Synechocystis genes of unknown function have putative orthologues in at least one of the cyanobacteria whose genome sequence is available, the data obtained from a screen of Synechocystis genes will be applicable to other cyanobacteria.
2. Materials and methods
2.1. Construction of bait clones
The yeast genetic techniques and media used in this study have been described in previous reports.8,18
For the GAL4 DNA-binding domain-fusion (bait) vector, we constructed pAS2-1-Asc I by inserting an Asc I site between the Nco I and Eco RI sites of pAS2-1 (Clontech, Mountain View, CA, USA). Specific primer sets were designed for the target region of each Synechocystis gene, and an Asc I site was added to the 5′ end of each gene-specific forward primer. Target gene fragments were obtained by PCR amplification from cosmid clones or genomic DNA using the gene specific primer sets. To minimize potential misincorporation during PCR, the high-fidelity pfu DNA polymerase (Stratagene, La Jolla, CA, USA) was used. The resulting fragments were subjected to Asc I digestion and ligated into pAS2-1-Asc I predigested with Asc I and Sma I. The target genes cloned into pAS2-1-AscI (bait clone) were transformed into the yeast strains CG1945 (MATa, ura3-52, his3-200, ade2-101, lys2-801, trp1-901, leu2-3, 112, gal4-542, gal80-538, cyhr2, LYS2::GAL1UAS-GAL1TATA-HIS3, URA3::GAL417-mers(x3)-CYC1TATA-lacZ) or AH109 (MATa, trp1-901, leu2-3, 112, ura3-52, his3-200, gal4Δ, gal80Δ, LYS2::GAL1UAS-GAL1TATA-HIS3, MEL1, GAL2UAS-GAL2TATA-ADE2, URA3::MEL1UAS-MEL1TATA-lacZ) (Clontech, Mountain View, CA, USA).
2.2. Library construction
The genomic DNA of Synechocystis sp. strain PCC6803 was subjected to sonication followed by size-fractionation by agarose gel electrophoresis. The 0.5−2.0 kb fraction was cloned into the Sma I digested activation domain-fusion (prey) vector, pACT2 (Clontech, Mountain View, CA, USA), and transformed into E. coli (XLI-Blue; Stratagene, La Jolla, CA, USA). The library DNA isolated from 106 independent clones was then introduced into the yeast strain Y187 (MATα, ura3-52, his3-200, ade2-101, trp1-901, leu2-3, 112, gal4Δ, met−, gal80Δ, URA3::GAL1UAS-GAL1TATA-lacZ, MEL1) using standard lithium acetate-mediated procedures.19 Three million independent yeast colonies were collected, pooled, and stored at −80°C.
2.3. Two-hybrid screening procedure
Prior to screening, the self-activity of each bait clone was tested by mating with the Y187 strain harboring an empty pACT2 vector and then plating on SD/-His/-Leu/-Trp/ medium supplemented with 2.5, 5, or 10 mM 3-amino 1,2,4-triazole (3-AT). Each bait clone was then mated with the prey library containing approximately 1.6 × 107 clones and plated on SD/-His/-Leu/-Trp/ agar medium supplemented with the optimal concentration of 3-AT based on the results of the self-activity test. After 7 days of growth at 30°C, independent positive colonies were collected in 96-well plates and cultured for an additional 3 days at 30°C. Aliquots of cultured positive clones were used for β-galactosidase assays and DNA sequencing of the prey inserts, and the cultures were stored at −80°C. The collected positive clones were treated with a zymolyase solution [2.5 mg/mL Zymolyase-100T (Seikagaku America Inc., East Falmouth, MA, USA), 1.2 M sorbitol, 0.1 M Na phosphate, pH 7.4] for 30 min at 30°C, and then used as templates for direct amplification of the prey clone inserts using the following primers: T0017 (5′-TACCACTACAATGGATGATG-3′) and T0018 (5′- GGGGTTTTTCAGTATCTACG-3′). Amplified inserts were directly sequenced using T0017 (5′) and T0018 (3′) to obtain sequence tags. The sequence tags were subsequently subjected to a BLAST search against the sequence database in CyanoBase (http://www.kazusa.or.jp/cyanobase/) using a semi-automated data processing system. A web-based interface was created to display the search results and to support the submission of interaction data into a interaction database. Sequence tags corresponding to antisense or intergenic regions were discarded. Sequence tags corresponding to the sense strand of an ORF were selected as PPI candidates. The PPI candidates were selected not only from in-frame prey clones (phase 2 position) but also out-of-frame fusion positions for the following reasons. PPIs supported by both in-frame and out-of-frame fusion prey clones have occasionally been found, and translational frameshifting has been reported and biochemically confirmed in previous YTH analyses.18,20 Similarly, the prey sequence tags corresponding to the up-stream region of a gene (within 200 nucleotides) were selected as PPI candidates that may be translated from an upstream start site.
2.4. Paralogue search and calculation of IG
Information on genes paralogous to Synechocystis genes was obtained from the CYORF database [http://cyano.genome.ad.jp/]. The lower threshold of acceptability was set at a Smith–Waterman score of 200.
Calculation of IG values for each interaction was conducted using the methods described in the previous report.21
3. Results and discussion
3.1. Construction of a high throughput YTH system and selection of Synechocystis genes for screening
One of the labor intensive steps of YTH analysis is designing the bait clone construction to establish directional and in-frame cloning. To overcome this point, we introduced an Asc I site into the multiple cloning site of a bait vector and used a simplified procedure for bait clone construction, as described in the Materials and Methods. This procedure can be applied to almost all genes in the Synechocystis genome, since there are only five Asc I sites in coding regions.1
To increase the efficiency of screening, we used a yeast mating-based screen, as described in the Material and Methods, and used a GAL4 activation domain fusion library of random genomic fragments of Synechocystis sp strain PCC6803. By using the random genomic fragment library as prey clones, information on PPI pairs and interacting domains of the prey proteins can be obtained simultaneously. The selected positive clones were analyzed using a 96-well plate format to increase throughput. For data processing of the large number of sequence tags from positive clones, we developed a semi-automatic system of two programs for vector sequence trimming and blast search against the Synechocystis sequence database. We also created an interface to display the search results and to support submission of interaction data to an interaction database.
Using this high-throughput YTH system, we explored large-scale PPI analysis in the photoautotorophic cyanobacterium Synechocystis sp. PCC6803. For bait clones of YTH screening, we selected the following groups of Synechocystis genes: (i) genes of putative two-component signal transducers, (ii) genes whose homologues are conserved in the A. thaliana genome, and (iii) genes of unknown function.
For two-component signal transducers, 44 predicted genes for Hiks and 42 predicted genes for Rres were selected.14,15 Selection of Synechocystis genes with homologues in the A. thaliana genome was conducted as follows. First, all A. thaliana genes were compared with those of Synechocystis, E. coli, yeast, and Caenorhabditis elegans, and the A. thaliana genes with highest degree of sequence similarity to those of Synechocystis were identified. Then by assigning Synechocystis genes corresponding to the identified A. thaliana genes, a total of 657 genes were selected as candidate Synechocystis genes conserved in the A. thaliana genome. The selected genes accounted for 10–50% of each functional category in Synechocystis. In the Synechocystis chromosome, 1641 genes (49%) are still categorized as hypothetical or of unknown function. Among the genes of unknown function, 190 candidate genes were selected because homologues were present in the A. thaliana genome. In addition, 413 genes encoding proteins with multiple transmembrane domains were excluded, because of the difficulty in detecting PPI of transmembrane proteins using the YTH system. Thus, a total of 1038 genes of unknown function were selected as the third screening group. For approximately 70% of these selected function unknown genes, putative orthologues are present in at least one of the completely sequenced cyanobacteria. Altogether a total of 1825 genes, including 44 genes selected for other purposes, were selected as target genes in this study (Fig. 1, Table 1, Supplementary Table 1).
Figure 1.
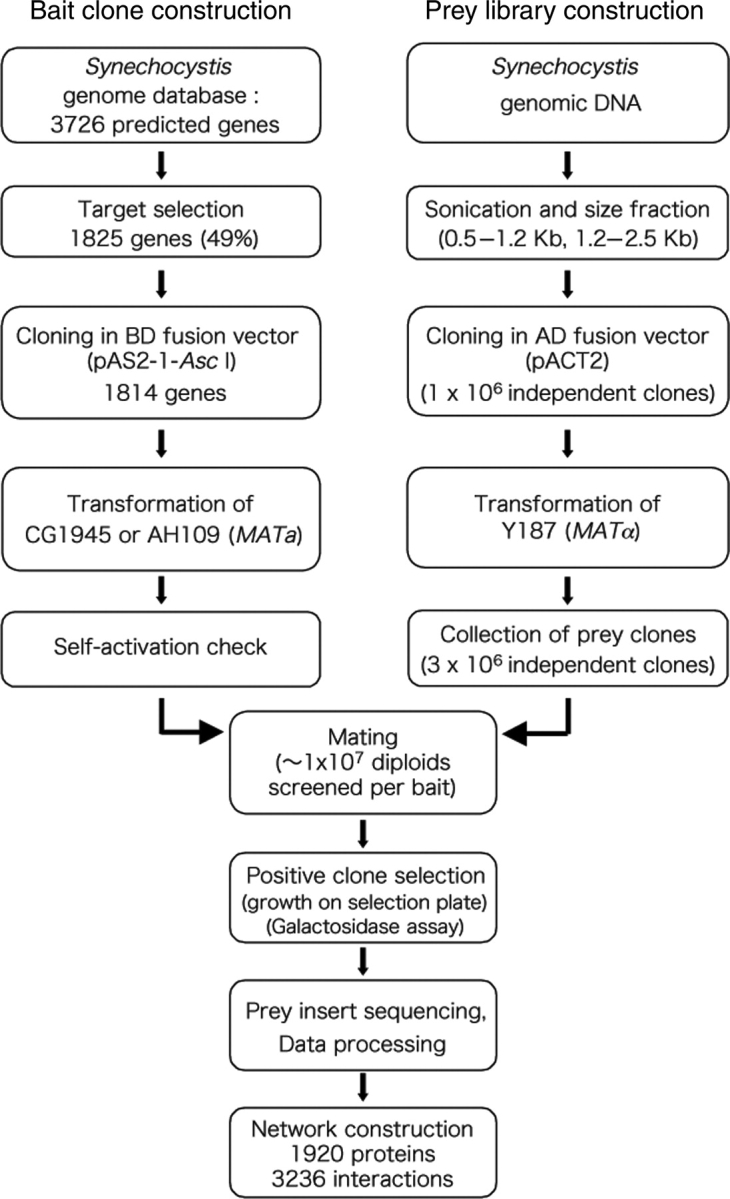
Flow chart of the sequential steps used for identification of PPIs of Synechocystis. The details are given in the Materials and Methods. BD, GAL4 DNA-binding domain; AD, GAL4 activation domain.
Table 1.
Synechocystis genes used for YTH analysis as baits
| Description | Number | Screened | Positive | No positive | Self-active |
|---|---|---|---|---|---|
| Genes of two component signal transducer | 86 | 86 | 53 | 20 | 13 |
| Genes conserved in the A. thaliana genome | 657 | 655 | 380 | 177 | 98 |
| Genes of unknown function | 1038 | 1029 | 571 | 207 | 251 |
| Others | 44 | 44 | 33 | 8 | 3 |
| Total | 1825 | 1814 | 1037 | 412 | 365 |
3.2. Classification and assessment of the protein interaction data
Of the 1825 target genes, bait clones were successfully constructed for 1814 (Fig. 1, Table 1, Supplementary Table 1). The YTH screen detected 3236 independent PPIs from 57% (1037) of the tested bait clones. The PPI network that was obtained contains 1920 Synechocystis proteins (Fig. 1, Supplementary Table 2).
One of the major concerns of large-scale interaction analysis is its reliability. Generally, PPI data sets obtained from YTH analysis, as well as other comprehensive analyses, contain many false positives. Therefore, the detected interactions need to be evaluated. In the case of YTH analysis, promiscuous interaction and self-activation by bait clones are two of the major causes of false positives.20–23 In a large-scale YTH analysis in yeast, Ito et al.8 classified the reliability of the PPI data by the number of interaction sequence tags (ISTs), the pair of tagged sequences, obtained from positive clones. They designated PPI data with more than three IST hits as the core data, and used this data set for construction of an interaction map and data comparison.8 Similar to this method, we classified all detected interactions into four categories (Category A–D), as an indicator of data reliability, based on how many positive prey clones supported the interaction. Category A and Category B comprised interactions supported by multiple positive prey clones with different (A) or identical (B) inserts. Category C comprised interactions supported by a single positive clone. PPI data supported by prey clones which were identified to have interactions with more than 18 different bait clones, i.e. 1% of tested bait clones, were considered to be promiscuous interactions and grouped in Category D. Most of the PPIs classified in Category D are likely to be false positives. Although there was no clear common feature among the 15 proteins classified as promiscuous prey, four of them had multiple trans-membrane domains and two contained a protein domain previously shown to interact promiscuously (Supplementary Table 3).24,25 The proportion of genes in each category is shown in Table 2.
Table 2.
Classification of interaction category
| Description | Total interactions | Proportion (%) |
|---|---|---|
| Category A (supported by multiple positive clones with different inserts ) | 795 | 25 |
| Category B (supported by multiple positive clones with same inserts) | 269 | 8 |
| Category C (supported by a single positive clone) | 1768 | 55 |
| Category D (interaction of putative promiscuous prey clones) | 404 | 12 |
To assess the validity of our categorizations, we evaluated all the detected interactions using an IG measure, a method for computationally assessing the reliability of PPI.21 Interactions with lower IG values are more likely to be reliable than interactions with higher IG values. The IG values for all detected interactions ranged from 1 to 17 and the average IG value was 2.4. When we extracted interactions having an IG value <3, approximately 90% of interactions from Categories A, B, and C were included, whereas only 44% of of interactions from Category D were included (Supplementary Fig. 1). The average IG values of Categories A (2.00 ± 0.16), B (1.87 ± 0.08), and C (2.01 ± 0.33) were significantly lower (P < 0.01) than that of Category D (5.42 ± 0.08) when fifty independent interactions were selected randomly from each category and compared. This result suggests that the interaction Categories A–D appropriately indicate the reliability of the PPI data.
To minimize false positives caused by self-activation, we used multiple reporter genes driven by different GAL4-responsive promoters and carefully determined the level of self-activation of each bait clone (see Materials and Methods). As shown in Supplementary Table 4, a total of 363 bait clones displayed strong self-activation as they could not be suppressed by leaky HIS3 reporter gene expression even in the presence of 10 mM 3-AT and these clones were eliminated from the screen. To examine the protein domains responsible for self-activation, we examined the protein domains of the genes that showed self-activation. Bacterial regulatory protein domains, such as the response regulator receiver (IPR001789) and TPR-1 (IPR001440), were frequently identified among the annotated proteins (Supplementary Table 4). In addition, several domains of unknown function, such as DUF29 (IPR002636) and UPF0150 (IPR005357), were occasionally observed among the proteins of unknown function that showed self-activation.
Another feature of large-scale YTH analyses is the high frequency of false negatives or missed interactions. The precise proportion of false-negative interactions can be assessed by comparison with published data. However, this approach is difficult to apply to Synechocystis PPI analysis because of the limited data available regarding protein interactions in Synechocystis. As an alternative, we estimated the proportion of false-negatives based on the interactions of two-component signal transducers whose interaction can be expected by the location of the corresponding genes in the genome. Among 14 Hiks located in the vicinity of genes for potentially cognate Rres in the Synechocystis genome, seven pairs were detected in our screening, indicating that about 50% of the interactions may not have been detected in our analysis. Considering that the two-component signal transducers were extensively analyzed during this study, the false-negative rate among other bait clones could be higher.
3.3. General features of protein interactions
A global view of the protein interaction network is illustrated in Fig. 2A using Cytoscape 2.2 (http://www.cytoscape.org/).26 In Fig. 2A, protein disks and interaction lines are color-coded according to functional categories and interaction categories, respectively. The 1920 proteins involved in this interaction network includes proteins from all functional categories in Synechocystis (Fig. 2B).
Figure 2.

Global view of the PPIs of Synechocystis. (A) All detected PPIs. Proteins (circles) are color-coded according to their functional category, as assigned in Kaneko et al.1 Interactions (lines) are color-coded by interaction category (A–D), which is based on the frequency of detection of identical pairs, as described in the text. (B) Number of identified interactions in each function category. The white bar indicates the total number of genes from each functional category assigned in the Synechocystis genome and the black bar indicates number of genes shown to have interactions. The red bar indicates the number of genes used as bait in the screen. The percentages represent the proportion of interacting genes in each function category.
Prokaryotic genes are generally organized into operons in which a gene cluster is transcribed as a polycistronic mRNA. Several previous works have demonstrated that genes encoded in the same operon are likely to have related functions.27,28 In the PPI data obtained here, we found 44 interacting protein pairs that are encoded by genes mapped to adjacent loci in the Synechocystis genome (Supplementary Table 5). It is noteworthy that 31 of these 44 PPIs were classified in Category A and the frequency of Category A PPIs occurring between proteins encoded by adjacent genes is significantly higher than that of the entire network (Table 2). This result supports the validity of our evaluation of the protein interaction data by interaction category. These interactions contained six interacting pairs between annotated proteins and proteins of unknown function mapping to adjacent loci. Two of these interactions were between extracytoplasmic function (ECF) sigma factors and their immediate downstream gene products, SigI (Sll0687)–Sll0688 and SigG (Slr1545)–Slr1546. ECF sigma factor is usually transcribed by one or more negative regulators (anti-sigma factors) which bind and inhibit the cognate sigma factor.29 Therefore, these PPI data suggest that the hypothetical proteins Sll0688 and Slr1546 may function as anti-sigma factors.
We detected 110 self-interacting proteins (Supplementary Table 6). Of these, 34 were proteins with functional annotations, and some are known to execute their function as homo-multimers or multi-protein complexes containing homo-dimers, such as the 10 kD chaperonin (Slr2075) and nitrogen regulatory protein P-II (Ssl0707). However, more than half of the self-interacting proteins detected in this screen were categorized as hypothetical or unknown. This information on the self-interaction of uncharacterized proteins will further our understanding of the function of these proteins. Domain level analysis of the uncharacterized self-interacting proteins revealed that five of the proteins have domains that are known to be involved in PPIs, such as ribbon–helix–helix (IPR10985) and the HxlR type helix–turn–helix, (IPR002577). Furthermore, a protein domain of unknown function, DUF820 (IPR008538), was found in nine of the self-interacting proteins and, thus, we hypothesize that this domain is involved in PPIs.
We also identified 15 hetero-dimeric interactions that occurred between two paralogous proteins (Smith–Waterman score >200). Among these interactions, seven were between proteins of unknown function, and these proteins also tended to be self-interacting. For instance, Slr1152, which showed self-interaction, interacted with a paralogous protein Sll1446. Also among the proteins containing the DUF820 domain (IPR008538) described above, the paralogous interactions Sll1250–Sll0296 and Sll1250–Sll1355 were detected in addition to the self-interactions of Sll0296 and Sll1355. These results suggest that a duplication of self-interacting proteins can generate a pair of paralogous proteins that interact with each other. Similar tendencies have been identified in PPI networks of yeast and other eukaryotes.30
3.4. Interactions of two-component signal transducers
A total of 105 bait clones corresponding to 86 genes of two-component signal transducers were constructed, since genes longer than 2.5 kb were divided into two or more constructs. After removal of 23 self-active bait clones, 82 bait clones were used for screening. A total of 195 PPIs were obtained from 54 bait clones. Among these PPI data, 44 interactions were between two component signal transducers, including seven pairs of bidirectional interactions and six self-interactions (Fig. 3). Of these, we successfully identified seven putative cognate pairs of Hiks and Rres predicted by genome location. Among the 10 PPIs detected between these cognate pairs, six were classified in Category A, and three putative cognate pairs were supported by bidirectional PPIs (Fig. 3). Detection of putative cognate pairs of Hiks and Rres as relatively reliable interactions indicates the validity of this screening approach and the specificity of our YTH analysis. Moreover, 21 of the identified PPIs between Hiks and Rres corresponded to 19 Hik and Rre pairs that were localized to different regions of the Synechocystis genome (Fig. 3). Since it is difficult to know the specific functional Hik and Rre partners from their sequence, the interaction pairs obtained by this study provide evidence to reinforce the functional relationships of the putative cognate pairs of Hiks and Rres and enabled us to identify the functional partners of Hiks and Rres that were localized to different regions of the Synechocystis genome.
Figure 3.
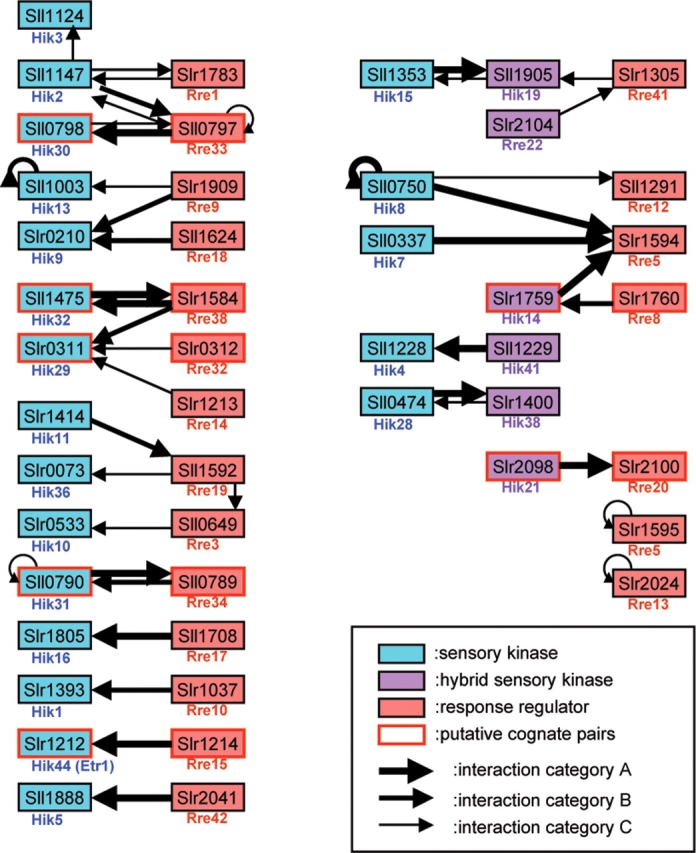
Interaction pairs of two-component signal transducers. Sensory histidine kinases, hybrid sensory kinases, and response regulators are shown by blue, purple, and orange boxes, respectively. Boxes marked with a red line indicate the interaction between Hik and Rre encoded by putative cognate pairs. The arrow in each interaction indicates the direction of bait protein to prey protein and the reliability of each interaction.
In addition to the 31 interactions between Hiks and Rres, nine interactions within Hiks were detected. Among the interactions within Hiks, five PPIs were obtained between a sensor kinase and hybrid sensory kinases, which contain both transmitter and receiver domains and are involved in multistep phospho-relays.14,31 As a result, candidates for a putative multistep phospho-relay were identified as Hik15 (Sll1353)–Hik19 (Sll1905)–Rre41 (Slr1305).
Interactions between two-component transducers were detected not only as simple one-to-one interactions between Hiks and Rres, but also as interactions between multiple Hiks and a single Rre, and vice versa. These interactions could reflect cross-regulation occurring between multiple two-component signal transducers. The possibility of cross-regulation in two-component signal transduction systems has been proposed previously.32,33 In Synechocystis, the presence of cross-regulation has been demonstrated experimentally. Detailed analysis of Hiks using a series of gene-knockout libraries revealed that Hik33 (Sll0698) regulates several different sets of genes by interacting with deferent Rres in response to low-temperature stress, hyperosmotic stress, and salt stress signals.15,34–36 Although we failed to identify interactions between Hik33 (Sll0698) and Rres, the PPI data obtained for multiple Hiks and Rres will help to elucidate novel cross-regulation in two-component systems in Synechocystis.
3.5. Interactions of proteins encoded by genes conserved in the plant genome
Among 657 genes selected as candidates that are conserved in the A. thaliana genome, a total of 557 bait clones were used for screening (98 self-active clones and two failed constructs were removed). A total of 1281 PPIs were obtained from 380 bait clones. Among these interactions, 30% (389) were PPIs between the products of the selected 657 genes (Supplementary Fig. 2). These PPI data between the products of candidate genes conserved in the A. thaliana genome will be useful in functional analysis of plant genes of endosymbiont cyanobacterial origin. An example of the applicable PPI information is the acetyl-CoA carboxylase (ACCase) complex. ACCase is essential for fatty acid synthesis in both Synechocystis and plants. Many plants have two types of ACCases in two subcellular locations, a eukaryotic homomeric form in the cytoplasm and a prokaryotic heteromeric form in the chloroplast.37 The plant heteromeric form of ACCase is considered to be of cyanobacterial origin, and is composed of four subunits, a biotin carboxyl carrier protein (BCCP), biotin carboxylase (BC), and the α and β subunits of carboxyltransferase (CT). The plant heteromeric form of ACCase easily dissociates into two components, the α and β subunits of CT and a BC–BCCP complex.37 Consistent with the physiological properties of the plant heteromeric ACCase, the PPIs in Synechocystis were found between AccB (Slr0435: encoding BCCP) and AccC (Sll0053: encoding BC), and between AccA (Sll0728: encoding the CT α subunit) and AccD (Sll0336: encoding the CT β subunit). These results demonstrate the applicability of the PPI data to the study of plant genes of cyanobacterial origin. Additionally, the results also indicate the advantage of YTH analysis in detecting basal binary interactions among multi-subunit complex components as well as the limitations in detecting secondary interactions in the multi-subunit complex.
An example of applicable PPI information was also obtained from that of conserved hypothetical proteins. A bidirectional interaction was detected between the hypothetical YCF65 protein (Slr0923) and the 30S ribosomal protein S1 (Slr1356). The A. thaliana protein PSRP-3 encoded by a homologue of the ycf65 gene on the nuclear genome has previously been identified as a component of the 30S subunit of the chloroplast ribosome by comprehensive proteome analysis of the chloroplast ribosome.38 Therefore, the PPI obtained between YCF65 (Slr0923) and the 30S ribosomal protein S1 (Slr1356) provides important experimental evidence revealing the functional conservation between cyanobacteria and plants, and also supports the hypothesis that Psrp-3/ycf65 may be a gene transferred from the organelle genome to the nuclear genome during chloroplast evolution.38
3.6. Interactions of proteins of unknown function
When 1228 genes of unknown function were screened (including 190 genes screened as conserved genes), 2029 PPIs were obtained from 682 bait clones (54%). Among these interactions, 1104 PPIs from 525 bait clones interacted with proteins of known function. PPI data on proteins of known function were also obtained as prey clones during the course of screening. In the entire PPI network obtained in this study, 1022 proteins of unknown function were included (Table 3). Of these, 727 have at least one partner of known function and 104 showed interactions with two or more known proteins of the same functional category (Table 3). For example, Slr0978 interacts with four distinct hybrid type Hiks (Hik22, Hik24, Hik38, and Hik40), which are paralogues with common domains, suggesting that Slr0978 may be involved in multiple two component signal transduction pathways. Sll0269 and Slr1636 interacted with two methyl-accepting chemotaxis proteins, Sll0041 and Slr1044, and Sll0041 and Sll1294, respectively. Thus Sll0269 and/or Slr1636 may play roles in the chemotaxis signaling pathway.
Table 3.
Summary of interaction with protein of unknown function
| Description | |
|---|---|
| Number of assessed baits of function unknown protein | 1228 |
| Number of baits of function unknown protein exhibiting interaction | 682 |
| Number of function unknown protein in whole network | 1022 |
| Number of function unknown protein interact with annotated gene | 727 |
| Number of function unknown protein interact with two or more annotated genes of same function category | 104 |
The obtained PPI information regarding genes of unknown function expands our knowledge of known pathways by identifying novel interacting components of the pathways. For instance, interaction between the nitrogen regulatory protein PII (GlnB; Ssl0707) and a function unknown protein (Sll0985, now designated PamA) indicates the need for further analysis of Sll0985 in the PII signaling pathway. A detailed analysis using a pamA deletion mutant revealed that PamA plays a role in the control of transcript abundance of nitrogen-related and sugar catabolic genes.39 Thus, the PPI information obtained in this study will provide useful starting points for further detailed functional analyses to expand the knowledge of known pathways and to place functionally uncharacterized proteins in their biological context.
We have established a YTH screening system feasible for large-scale analysis of Synechocystis sp. strain PCC6803, and have identified 3236 interactions involving 1920 proteins using 1825 selected genes as bait in this screen. The obtained interaction network contains numbers of proteins encoded by genes conserved in plant genomes, as wells as many genes of unknown function. Thus, these data should provide new insights and novel strategies for functional analysis of genes in Synechocystis, and, additionally, genes in other cyanobacteria and plant genes of cyanobacterial origin. To make the interaction data publicly available, the obtained PPI data have been integrated into the cyanobacterial genome database, CyanoBase (http://www.kazusa.or.jp/cyanobase/). We have provided all the PPI data, as well as the interaction categories so that the user can select a data set based on particular analysis needs.
Although it will be essential to confirm and extend our findings by independent tests, we believe that the obtained PPI data should provide a useful starting point for studying the functions of uncharacterized proteins and for predicting functional pathways in Synechocystis, other cyanobacteria, and plant.
Funding
This work was supported by the Kazusa DNA Research Institute Foundation.
Acknowledgements
We would like to thank S. Sasamoto for excellent technical assistance. We are also grateful to Drs T. Kaneko, S. Okamoto and M. Nakao for their help in data analysis and for valuable discussions.
Supplementary Data: Supplementary data are available online at www.dnaresearch.oxfordjournals.org.
References
- 1.Kaneko T., Sato S., Kotani H., et al. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 1996;3:109–136. doi: 10.1093/dnares/3.3.109. [DOI] [PubMed] [Google Scholar]
- 2.Ikeuchi M., Tabata S. Synechocystis sp. PCC 6803—a useful tool in the study of the genetics of cyanobacteria. Photosynth Res. 2001;70:73–83. doi: 10.1023/A:1013887908680. [DOI] [PubMed] [Google Scholar]
- 3.Hihara Y., Kamei A., Kanehisa M., Kaplan A., Ikeuchi M. DNA microarray analysis of cyanobacterial gene expression during acclimation to high light. Plant Cell. 2001;13:793–806. doi: 10.1105/tpc.13.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sazuka T., Yamaguchi M., Ohara O. Cyano2Dbase updated: linkage of 234 protein spots to corresponding genes through N-terminal microsequencing. Electrophoresis. 1999;20:2160–2171. doi: 10.1002/(SICI)1522-2683(19990801)20:11<2160::AID-ELPS2160>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura Y., Kaneko T., Miyajima N., Tabata S. Extension of CyanoBase. CyanoMutants: repository of mutant information on Synechocystis sp. strain PCC6803. Nucleic Acids Res. 1999;27:66–68. doi: 10.1093/nar/27.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakamura Y., Kaneko T., Tabata S. CyanoBase, the genome database for Synechocystis sp. strain PCC6803: status for the year. Nucleic Acids Res. 2000;28:72. doi: 10.1093/nar/28.1.72. 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uetz P., Giot L., Cagney G., Mansfield T. A., et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 8.Ito T., Chiba T., Ozawa R., Yoshida M., Hattori M., Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl Acad. Sci. USA. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walhout A. J., Sordella R., Lu X., et al. Protein interaction mapping in C. elegans using proteins involved in vulval development. Science. 2000;287:116–122. doi: 10.1126/science.287.5450.116. [DOI] [PubMed] [Google Scholar]
- 10.Rain J. C., Selig L., De Reuse H., et al. The protein–protein interaction map of Helicobacter pylori. Nature. 2001;409:211–215. doi: 10.1038/35051615. [DOI] [PubMed] [Google Scholar]
- 11.Giot L., Bader J. S., Brouwer C., et al. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–1736. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- 12.LaCount D. J., Vignali M., Chettier R., et al. A protein interaction network of the malaria parasite Plasmodium falciparum. Nature. 2005;438:103–107. doi: 10.1038/nature04104. [DOI] [PubMed] [Google Scholar]
- 13.Parrish J. R., Yu J., Liu G., et al. A proteome-wide protein interaction map for Campylobacter jejuni. Genome Biol. 2007;8:R130. doi: 10.1186/gb-2007-8-7-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mizuno T., Kaneko T, Tabata S. Compilation of all genes encoding bacterial two-component signal transducers in the genome of the cyanobacterium, Synechocystis sp. strain PCC 6803. DNA Res. 1996;3:407–414. doi: 10.1093/dnares/3.6.407. [DOI] [PubMed] [Google Scholar]
- 15.Murata N., Suzuki I. Exploitation of genomic sequences in a systematic analysis to access how cyanobacteria sense environmental stress. J. Exp. Bot. 2006;57:235–247. doi: 10.1093/jxb/erj005. [DOI] [PubMed] [Google Scholar]
- 16.Rujan T., Martin W. How many genes in Arabidopsis come from cyanobacteria? An estimate from 386 protein phylogenies. Trends Genet. 2000;17:113–120. doi: 10.1016/s0168-9525(00)02209-5. [DOI] [PubMed] [Google Scholar]
- 17.Martin W., Rujan T., Richly E., et al. Evolutionary analysis of Arabidopsis, cyanobacterial, and chloroplast genomes reveals plastid phylogeny and thousands of cyanobacterial genes in the nucleus. Proc. Natl Acad. Sci. USA. 2002;99:12246–12251. doi: 10.1073/pnas.182432999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fromont-Racine M., Rain J.-C., Legrain P. Toward a functional analysis of the yeast genome through exhaustive two-hybrid screens. Nat. Genet. 1997;16:277–282. doi: 10.1038/ng0797-277. [DOI] [PubMed] [Google Scholar]
- 19.Ito H., Fukuda Y., Murata K., Kimura A. Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uetz P., Hughes R. E. Systematic and large-scale two-hybrid screens. Curr. Opin. Microbiol. 2000;3:303–308. doi: 10.1016/s1369-5274(00)00094-1. [DOI] [PubMed] [Google Scholar]
- 21.Saito R., Suzuki H., Hayashizaki Y. Interaction generality, a measurement to assess the reliability of a protein-protein interaction. Nucleic Acids Res. 2002;30:1163–1168. doi: 10.1093/nar/30.5.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fields S. High-throughput two-hybrid analysis. FEBS J. 2005;272:5391–5399. doi: 10.1111/j.1742-4658.2005.04973.x. The promise and the peril. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki H., Fukunishi Y., Kagawa I., et al. Protein–protein interaction panel using mouse full-length cDNAs. Genome Res. 2007;11:1758–1765. doi: 10.1101/gr.180101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ng S. K., Zhang Z., Tan S.H., Lin K. InterDom: a database of putative interacting protein domains for validating predicted protein interactions and complexes. Nucleic Acids Res. 2003;31:251–254. doi: 10.1093/nar/gkg079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arifuzzaman M., Maeda M., Itoh A., et al. Large-scale identification of protein-protein interaction of Escherichia coli K-12. Genome Res. 16:686–691. doi: 10.1101/gr.4527806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shannon P., Markiel A., Ozier O., et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Snel B., Bork P., Huynen M. A. The identification of functional modules from the genomic association of genes. Proc. Natl Acad. Sci. 2002;99:5890–5895. doi: 10.1073/pnas.092632599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Overbeek R., Fonstein M., D'Souza M., Pusch G. D., Maltsev N. The use of gene clusters to infer functional coupling. Proc. Natl Acad. Sci. 1999;96:2896–2901. doi: 10.1073/pnas.96.6.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Helmann J. D. The extracytoplasmic function (ECF) sigma factors. Adv. Microb. Physiol. 2002;46:47–110. doi: 10.1016/s0065-2911(02)46002-x. [DOI] [PubMed] [Google Scholar]
- 30.Ispolatov I., Yuryev A., Mazo I., Maslov S. Binding properties and evolution of homodimers in protein–protein interaction networks. Nucleic Acids Res. 2005;33:3629–3635. doi: 10.1093/nar/gki678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alex L. A., Simon M. I. Protein histidine kinases and signal transduction in prokaryotes and eukaryotes. Trends Genet. 1994;10:133–138. doi: 10.1016/0168-9525(94)90215-1. [DOI] [PubMed] [Google Scholar]
- 32.Parkinson J. S., Kofoid E. C. Communication modules in bacterial signaling proteins. Annu. Rev. Genet. 1992;26:71–112. doi: 10.1146/annurev.ge.26.120192.000443. [DOI] [PubMed] [Google Scholar]
- 33.Wanner B.L. Is cross regulation by phosphorylation of two-component response regulator proteins important in bacteria. J. Bacteriol. 1992;174:2053–2058. doi: 10.1128/jb.174.7.2053-2058.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki I., Los D. A., Kanesaki Y., Mikami K., Murata N. The pathway for perception and transduction of low-temperature signals in Synechocystis. EMBO J. 2000;19:1327–1334. doi: 10.1093/emboj/19.6.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paithoonrangsarid K., Shoumskaya M. A., Kanesaki Y., et al. Five histidine kinases perceive osmotic stress and regulate distinct sets of genes in Synechocystis. J. Biol. Chem. 2004;279:53078–53086. doi: 10.1074/jbc.M410162200. [DOI] [PubMed] [Google Scholar]
- 36.Shoumskaya M. A., Paithoonrangsarid K., Kanesaki Y., et al. Identical Hik-Rre systems are involved in perception and transduction of salt signals and hyperosmotic signals but regulate the expression of individual genes to different extents in Synechocystis. J. Biol. Chem. 2005;280:21531–21538. doi: 10.1074/jbc.M412174200. [DOI] [PubMed] [Google Scholar]
- 37.Sasaki Y., Nagano Y. Plant acetyl-CoA carboxylase: Structure, biosynthesis, regulation, and gene manipulation for plant breeding. Biosci. Biotechnol. Biochem. 2004;68:1175–1184. doi: 10.1271/bbb.68.1175. [DOI] [PubMed] [Google Scholar]
- 38.Yamaguchi K., Subramanian A. R. Proteomic identification of all plastid-specific ribosomal proteins in higher plant chloroplast 30S ribosomal subunit. Eur. J. Biochem. 2003;270:190–205. doi: 10.1046/j.1432-1033.2003.03359.x. [DOI] [PubMed] [Google Scholar]
- 39.Osanai T., Sato S., Tabata S., Tanaka K. Identification of PamA as a PII-binding membrane protein important in nitrogen-related and sugar-catabolic gene expression in Synechocystis sp. PCC 6803. J. Biol. Chem. 2005;280:34684–34690. doi: 10.1074/jbc.M507489200. [DOI] [PubMed] [Google Scholar]