

Abstract
Background
Inflammation and thrombosis co-exist in several disorders. Although it is recognized that leukocytes may induce a pro-coagulant state at sites of inflammation, the critical molecular determinants of this process remain largely unknown.
Methods and Results
To examine mechanisms of inflammation-induced thrombosis we developed a murine model of thrombotic glomerulonephritis (TGN), a known cause of acute renal failure in patients. This model, induced by LPS and antibody to the glomerular basement membrane, led to rapid glomerular neutrophil recruitment, thrombotic glomerular lesions with endothelial cell injury and renal dysfunction. In mice immunodepleted of neutrophils, or lacking the leukocyte specific integrin Mac-1, neutrophil recruitment, endothelial injury, glomerular thrombosis and acute renal failure were markedly attenuated despite the robust generation of renal cytokines. Neutrophil elastase (NE) is a likely effector of Mac-1 as its activity was reduced in Mac-1 deficient mice and the phenotype in mice deficient in Mac-1 or NE was similar. Platelets accumulated in glomerular capillaries within 4hrs of TGN before evidence of thrombosis. Platelet immunodepletion prior to TGN markedly exacerbated hematuria (hemorrhage), inflammation and injury, while thrombocytopenic Mac-1 deficient mice remained resistant to disease indicating that initial glomerular platelet deposition protects the vessel wall from neutrophil mediated sequelae. The subsequent thrombosis relied on the interaction of Mac-1 on recruited neutrophils with GpIbα on platelets as antibody mediated disruption of this interaction attenuated TGN without affecting renal neutrophil accumulation.
Conclusions
These observations establish Mac-1 on neutrophils as a critical molecular link between inflammation and thrombosis and suggest it as an attractive target for antithrombotic therapy.
Keywords: Cell adhesion molecules, thrombosis, inflammation, leukocytes, platelets, kidney
Introduction
Coagulation and inflammation are closely related entities in many diseases. There is abundant evidence that these two processes intersect at multiple points which raises the possibility that anti-inflammatory therapeutics may be used to manage thrombotic disorders. This requires a better understanding of the molecular players that link leukocyte activation to the coagulation cascade. Glomerular thrombus formation is often found in severe human glomerulonephritides and is a leading cause of acute renal failure. Thrombotic microangiopathy (TMA), which describes a particular histopathologic lesion as opposed to a single clinical pathologic entity1, 2, occurs in various clinical settings including hemolytic uremic syndrome (HUS), thrombotic thrombocytopenic purpura (TTP), transplant rejection, systemic lupus erythematosus and glomerulonephritis exacerbated by infection. TMA is characterized by endothelial cell swelling and detachment mainly in arterioles and capillaries, inflammatory cell infiltration and intraluminal platelet thrombosis leading to organ damage. Functional manifestations of TMA are clinically classified as HUS/TTP3. Microvascular endothelial cell injury is still considered the most likely inciting factor in TMA4. This may be triggered by bacterial derived endotoxins/toxins, viruses (HIV), immune complexes (ICs) and drugs such as chemotherapeutic agents that likely activate neutrophils5-7. However, the pathogenic role of neutrophils and molecular mechanisms underlying their potential contribution to the observed procoagulant state in TMA and other thrombotic disorders remains largely unknown.
Here, we developed a mouse model of thrombotic glomerulonephritis (TGN) induced by the sequential injections of anti-glomerular basement membrane (GBM) antibody and LPS that was characterized by renal endothelial cell damage, occlusive microvascular thrombosis, renal failure and hematuria. These parameters are prominent in patients with HUS/TTP3, 8. The use of genetically modified mice, cell immunodepletion approaches and functional blocking antibodies in this model allowed us to examine the molecular basis of inflammation-induced thrombosis in vivo, and explore the pathogenesis of this clinically important disease. We demonstrate that neutrophils play a primary role in disease pathogenesis. We provide evidence that Mac-1, a member of the leukocyte specific CD18 integrin family promotes neutrophil recruitment, endothelial injury, glomerular thrombosis and acute renal failure by regulating the release and/or activity of the serine proteinase Neutrophil Elastase (NE). We show that platelets initially preserve the integrity of the glomerular microvasculature following Mac-1 mediated neutrophil recruitment and injury while subsequent neutrophil engagement of platelet GpIbα via Mac-1 potently induces thrombosis.
Materials And Methods
Mice
Gene deleted mice backcrossed to C57Bl/6 are denoted as B6 with a F# designating the number of generations the animals were backcrossed to C57Bl/6. Mac-1 deficient (Mac-1-/-) mice9 are B6F9 and were bred and maintained in the Viral Antigen Free facility at the Longwood Medical Research Center (LMRC) animal housing facility at Harvard Medical School. NE-deficient mice (B6F10)10 were bred in the VAF facility at Harvard School of Public Health animal housing facility and maintained in the LMRC facility. Age-matched wild-type C57Bl/6 mice (Jackson Laboratory) were used for all the aforementioned C57Bl/6 gene deleted animals. Experimental procedures were approved by the Animal Care and Use Committee of Harvard Medical School, Boston MA.
Generation of thrombotic glomerulonephritis
Rabbit antibody against mice GBM (anti-GBM serum) was prepared by immunizing rabbits with mouse GBM (Covance Research Products Inc). Control rabbit serum was purchased from Sigma. Anti-GBM serum was incubated at 56°C for 30 min to inactivate complement and filter-sterilized. Male C57Bl/6, were injected twice via tail vein with 300 μl anti-GBM serum or normal rabbit serum at 1 hr intervals. 50 μg lipopolysaccharide (LPS) from phenolic extracts of Salmonella typhimurium (CalBiochem, La Jolla, CA, USA) in sterile PBS was injected into mice via the tail vein 2 hrs after the second anti-GBM serum injection. Peripheral blood was collected from the retroorbital plexus of anesthetized mice into glass tubes to obtain serum and in EDTA-containing vials to collect whole blood. The animals were euthanized by CO2 inhalation, and both kidneys were harvested for histologic analysis at the indicated times following LPS injection. All experimental procedures were performed in 6- to 8-week-old male mice, as animals greater than 10 weeks of age were more resistant to disease induction.
Immunodepletion of neutrophils and platelets, and anti-M2 antibody treatment
For immunodepletion of neutrophils, a rat anti-mouse mAb against the neutrophil maturation antigen, Gr-1 (BD Biosciences-Pharmingen) was used. WT mice were injected intraperitoneally twice with 100 μg of anti-Gr-1 antibody in 200 μl PBS at 24 hrs prior to and after disease induction. A rat IgG2b (R&D systems, Minneapolis, MN, USA.) was used as an isotype control. The platelet-depleting anti-GPIbα antibody and isotype rat IgG control were purchased from Emphret (Wurzburg, Germany). WT or Mac-1 KO mice were injected intravenously with 40 μg anti-GPIbα or rat IgG in PBS 12 hrs prior to disease induction. Wild-type mice were injected i.v. with 100μg of affinity purified peptide-specific polyclonal antibody (termed anti-M2) to the Mac-1 binding site for GPIbα 11 or nonimmune rabbit IgG antibody 1 h prior to the first injection of anti-GBM.
Peripheral blood cell count and differentials
Blood collected in EDTA was used in MBC biochemistry (Tokyo, Japan) to generate a complete blood count with cellular differential.
Functional assessment of renal injury
Serum values for creatinine (Cr), blood urea nitrogen (BUN), lactate dehydrogenase (LDH) were obtained with Hitachi 7700 Automatic Analyzer at MBC (Tokyo, Japan). Hematuria grading was conducted by dip stick analysis using Uro-paper (Eiken Chemical Co., Tokyo, Japan). For collection of serum, blood sampled from the retroorbital plexus of mice was collected in glass tubes, kept at 4°C for 30 min and centrifuged.
Histological and immunohistochemical analysis of renal tissue
4-μm coronal sections of paraffin-embedded kidneys were stained with Periodic-Acid Schiff (PAS) or Masson-Trichrome reagent for analysis of glomerular and interstitial injury. Prevalence of glomerular capillary thrombosis (%) = total number of glomeruli with thrombus in at least one glomerular capillary/total number of glomeruli12. Fibrin deposition was detected by phosphotungstic acid-hematoxilin (PTAH) staining of paraffin renal sections.
Paraffin-embedded kidney sections were stained with rat monoclonal antibody to CD34 (GeneTex, Inc., San Antonio, Texas, USA). The staining intensity within the capillary walls in glomeruli or the interstitium were scored on a scale of 0–3 (0 = none; 1 = weak; 2 = moderate; 3 = strong) by an observer blinded to the identity of the sample. Platelets were stained with goat polyclonal antibody to integrin αIIb (Santa Cruz Inc., Santa Cruz, CA, USA).
Chloroacetate esterase reaction for neutrophil enumeration
Paraffin sections from kidneys were de-paraffinized and incubated in freshly prepared chloroacetate solution containing 0.0125% Naphtol AS-D (Sigma) and 0.0625% Fast Blue BB salt (Sigma) in phosphate buffer (pH 7.3) for 1.5 hr in the dark. The number of esterase positive neutrophils in at least 50 glomeruli per section were calculated and reported as the total number of neutrophils per glomerulus cross-section.
Multiple cytokine and chemokine analysis
Homogenates from mice kidney at indicated times were subjected to the Bio-Plex suspension array system (Bio-Rad laboratories, Hercules, CA, USA) to measure the concentration of 32 cytokines: GM-CSF, G-CSF, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12p40, IL-12p70, IL-13, IL-15 IL-17, IL-18, IFN-γ, TNF-α, MCP-1, RANTES, MIP-1α, MIP-1β, Eotaxin, KC, FGFbasic, LIF, M-CSF, MIG, MIP-2, PDGF-BB, and VEGF.
Analysis of E-selectin
Intrarenal E-selectin levels in kidney homogenates were measured by an E-selectin assay kit (R&D; Minneapolis, MN, USA). Protein concentrations were determined by the Lowry assay with the Bio-Rad DC protein assay dye reagent.
Measurement of granulocyte elastase digests of plasma fibrinogen and fibrin
To obtain plasma, blood sampled from the retroorbital plexus was collected into plastic tubes containing sodium citrate and centrifuged. Fibrin degradation products specifically degraded by granulocyte-elastase (e-XDP) were measured by the “E-XDP TEST” (Mitsubishi Kagaku Iatron Co., Tokyo. Japan) which consists of Tris buffer (R1) and latex reagent (R2) containing IF-123 coated latex beads. IF-123 specifically recognizes e-XDP13. The latex agglutination assay was performed using an automatic analyzer LPIA-NV7 (Mitsubishi Kagaku Iatron Co., Tokyo. Japan). The test sample in Tris-saline buffer was mixed in a reaction cuvette at 37°C. Latex reagent was added to this mixture and the absorbance at 800 nm was measured every 15 seconds for 7 minutes. The V-value, latex agglutination velocity, was defined as delta absorbance at 800 nm per minute. The rate of agglutination was expressed in terms delta A800/min as a function of the concentration of e-XDP.
Statistical analysis
Results are expressed as means ± standard error of the mean (SEM) for data resulting from in vivo analyses of mice. In all cases, an unpaired t test was used to compare two groups. P < 0.05 was considered statistically significant.
Statement of Responsibility
The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written.
Results
Development of a murine model of thrombotic glomerulonephritis
TGN was induced by the injection of LPS and anti-GBM serum (nephrotoxic serum, NTS), which reproducibly produced severe glomerular thrombosis within 72 hrs (Figure 1A). Glomerular neutrophil accumulation was prominent and occurred as early as 4 hrs after disease induction while inflammatory cell infiltration into the tubulointerstitial area and fibrosis was minimal (data not shown). Glomerular crescent formation and sclerosis were absent although tubular damage was observed, as characterized by tubular dilation and casts. This was associated with rapidly progressive renal failure as evidenced by a significant elevation of serum creatinine and blood urea nitrogen (BUN) (Figure 1B). Replacement of NTS with normal rabbit serum failed to induce TGN suggesting that a combination of anti-GBM serum and LPS is required for disease induction (Figure 1A-B). The observed histopathological changes and renal dysfunction were also dependent on LPS, as anti-GBM antibody alone resulted only in acute neutrophil accumulation and mild proteinuira14(data not shown). Thus LPS and in situ glomerular antibody deposits together trigger TGN.
Figure 1. Development of a murine thrombotic glomerulonephritis (TGN) model.

(A) Kidneys were harvested from mice injected with control rabbit serum with LPS or two intravenous injections of anti-GBM serum (αGBM) at 1 hr intervals followed by LPS. Representative Masson-trichrome stained sections of kidneys harvested 96hr after induction of TGN are shown. Only mice given αGBM antibody plus LPS exhibited massive glomerular thrombosis, tubular dilatation and casts (top and middle panels), and plugging of large vessels (bottom panel). Bar: 100 μm (top, bottom), 30 μm (middle). (B) Serum creatinine (sCr) and blood urea nitrogen (BUN) levels were evaluated in WT mice before (Untreated) and 96 hrs following injection with control (Con) or αGBM serum plus LPS. *P<0.05 compared with Con. (C) Mice treated with control IgG (Con) or neutrophil immunodepleting anti-Gr-1 (αGr-1) were subjected to TGN. Shown are representative PAS- and PTAH- stained sections that demonstrate a partial reduction in glomerular thrombus formation (PAS), and fibrin deposition (PTAH) in αGr-1 treated mice. Scoring of PAS positive glomeruli is presented. Bar: 30 μm (D) Neutrophil immunodepletion partially attenuated serum Cr (sCr) and BUN, biochemical markers for renal function, and LDH, a general marker for organ damage. Data represent the mean ± SEM. *P<0.05 compared with Con.
To examine the contribution of neutrophils to TGN, neutrophils were immunodepleted with anti-Gr-1 monoclonal antibody 24 hr prior to disease induction (13.3±6.0 versus 548.8±196.4 neutrophils/μl for Gr-1 mAb (n=4) and control (n=4) groups respectively, 24 hr after treatment). Neutrophil depleted mice exhibited a marked reduction in glomerular thrombosis that correlated with significantly reduced indices of renal failure. These data suggest a primary role for neutrophils in the initiation of TGN (Figure 1C-D).
Mac-1 is essential for the development of TGN
Mac-1-/- and WT cohorts were subjected to TGN. Mac-1-/- mice had minimal glomerular thrombosis, and significantly reduced fibrin deposition (Figure 2A). The integrity of the glomerular microvasculature, a primary site of injury in microangiopathic disorders in humans was evaluated by examining CD34, a widely used marker of capillary endothelial cells that is transcriptionally downregulated in inflammatory settings15, 16. Immunohistochemically, TGN led to a significant reduction of CD34 in the glomerular capillaries of wild-type mice, which is the primary site of injury in this model, while its expression in the interstitium was similar to untreated mice. In contrast, CD34 remained intact in Mac-1-/- mice subjected to TGN (Figure 2B). In addition, the cytokine inducible endothelial specific adhesion molecule E-selectin was elevated in the renal tissue of wild-type mice indicating endothelial activation, while this was much less pronounced in Mac-1-/- animals (Figure 2C). Together these data indicate a prominent role for Mac-1 in endothelial activation and damage following TGN. Indices of renal dysfunction were significantly less pronounced in Mac-1-/- versus wild-type animals. Hematuria, a marker of hemorrhage was milder in Mac-1-/- mice compared to WT counterparts (Figure 2D) and Mac-1-/- mice were resistant to TGN-induced renal failure as evidenced by a significant attenuation in the elevation of serum creatinine, BUN and LDH (Figure 2E). The protection from TGN in Mac-1-/- mice was associated with a marked reduction in glomerular neutrophil accumulation at both 4 and 24 hrs after disease induction (Figure 2F). To assess whether changes in renal cytokine levels contribute to the observed protection in Mac-1-/- mice, multiplex cytokine analysis of renal tissue 4 hrs after induction of TGN was conducted. Among 32 cytokines/chemokines measured, 16 were induced and these were comparable in wild-type and Mac-1-/- mice (Table 1).
Figure 2. Mac-1 deficient mice are protected from developing TGN.


TGN was induced in WT and Mac-1-/- mice and analyzed 96 hrs after induction of disease. (A) Analysis of PAS and PTAH stained kidney sections and scoring of PAS+ deposits revealed a significant reduction in severity of TGN in Mac-1-/- mice compared to wild-type. Bar: 30 μm. (B) Immunohistochemical analysis of CD34 on renal tissue of WT untreated, and WT and Mac-1-/- mice subjected to TGN. A reduction in glomerular CD34, but not interstitial CD34, was observed in WT/TGN mice compared to untreated WT, while Mac-1-/-/TGN mice retained glomerular CD34. Bar: 50 μm. (C) E-selectin in renal tissue of indicated animals was measured and showed a decrease in Mac-1-/ mice compared to wild-type mice subjected to TGN. (D) Hematuria, and (E) serum creatinine (sCr), BUN and LDH levels were significantly reduced in Mac-1-/- mice subjected to TGN compared to wild-type. (F) At indicated times after induction of TGN, kidneys were harvested and the number of neutrophils (PMNs) per glomerular cross-section were determined. A representative kidney section from WT and Mac-1-/- mice 4 hrs after induction of nephritis is shown. Neutrophils (stained blue, arrow) were recruited only into the glomerulus with no observable interstitial infiltration. Data represent the mean ± SEM. *P<0.05 compared to wild-type subjected to TGN, #P<0.05 compared to untreated wild-type mice. Bar: 30 μm
Table 1. Intrarenal cytokine profile in wild-type and Mac-1 deficient mice at 4h after induction of TGN.
TGN was induced in WT or Mac-1-/- mice and kidneys were harvested 4 hrs after induction. Cytokine concentrations in pg/mg of total protein in kidney homogenates were measured by the Bio-Plex System. Values are mean±SEM; n=4 per group.
| WT control | WT TGN | Mac-1-/- control | Mac-1-/- TGN | |
|---|---|---|---|---|
| IL-1α | 22.2 ± 6.0 | 47.3 ± 4.7* | 17.4 ± 1.6 | 48.0 ± 2.6† |
| IL-1β | 151.4 ± 15.8 | 208.5 ± 13.9* | 102.8 ± 9.7 | 229.1 ± 13.1† |
| IL-6 | 22.6 ± 4.7 | 181.2 ± 46.9* | 15.6 ± 1.2 | 253.7 ± 35.3† |
| IL-12 (p40) | 7.3 ± 1.0 | 124.9 ± 26.8* | 6.1 ± 0.8 | 95.3 ± 15.3† |
| G-CSF | 1.1 ± 0.3 | 310.1 ± 43.8* | 0.6 ± 0.1 | 369.1 ± 58.4† |
| KC | 4.0 ± 1.1 | 1282.1 ± 293.5* | 3.1 ± 0.2 | 1682.4 ± 491.2† |
| RANTES | 64.6 ± 10.9 | 550.9 ± 85.7* | 52.5 ± 4.9 | 546.5 ± 26.5† |
| MCP-1 | 42.0 ± 6.7 | 411.7 ± 85.7* | 35.1 ± 2.9 | 488.5 ± 120.9† |
| MIP-1β | 26.3 ± 5.1 | 56.4 ± 5.4* | 18.1 ± 2.3 | 56.2 ± 3.2† |
| IL-15 | 181.2 ± 13.8 | 340.4 ± 53.8* | 88.9 ± 25.0 | 290.5 ± 51.1† |
| FGF basic | 326.2 ± 43.9 | 2329.5 ± 794.5* | 730.1 ± 288.8 | 2131.5 ± 458.4† |
| LIF | 7.6 ± 0.8 | 58.5 ± 7.2* | 3.85 ± 0.58 | 64.9 ± 10.0† |
| M-CSF | 19.7 ± 1.3 | 88.1 ± 6.5* | 20.8 ± 0.3 | 95.2 ± 12.6† |
| MIG | 2628.4 ± 209.6 | 63889.8 ± 13826.4* | 2777.3 ± 901.6 | 62931.0 ± 4478.1† |
| MIP-2 | 6.4 ± 0.6 | 265.1 ± 41.1* | 4.2 ± 0.7 | 342.7 ± 77.3† |
| PDGF-BB | 1300.8 ± 149.9 | 7742.9 ± 1491.0* | 3808.4 ± 728.0 | 8805.2 ± 1370.8† |
p<0.05 compared to WT control
p<0.05 compared to Mac-1-/- control
Hematologic abnormalities associated with TGN were less severe in Mac-1-/- mice. Mac-1 deficiency led to a partial attenuation of the observed thrombocytopenia in wild-type mice that likely reflects less platelet consumption. Mild anemia was present in both wild-type and Mac-1-/- mice suggesting that it is not a consequence or predictor of glomerular thrombosis. Circulating WBC counts decreased within hours after induction of TGN in both wild-type and Mac-1-/- mice but the recovery at day 4 was greater in Mac-1-/- mice compared to wild-type counterparts possibly as a compensation for reduced efficiency of renal neutrophil accumulation (Supplemental data, S1).
Together, the data indicate that Mac-1-/- mice exhibit reduced neutrophil accumulation and reduced susceptibility to TMA associated endothelial injury, glomerular pathology, thrombosis, renal failure and thrombocytopenia despite an abundance of renal cytokines and chemokines.
A role for neutrophil elastase in renal damage
Mac-1 engagement can lead to NE release17, 18 suggesting it as a possible effector of Mac-1. Mice deficient in NE subjected to TGN exhibited a reduction in disease indices compared to wild-type cohorts. Glomerular neutrophil recruitment was reduced in NE-deficient mice, which phenocopies the Mac-1-/- mice (Table 2). To determine if NE was downstream of Mac-1 we compared NE-activity in plasma of wild-type and Mac-1-/- mice by quantitating levels of NE-digested fibrinogen degradation products e-XDP 13. A significant reduction in NE-derived fibrinogen products was observed in plasma samples of Mac-1-/- compared to wild-type mice at day 1 after disease induction (Table 3).
Table 2. Neutrophil elastase-deficient mice are protected from TGN.
TGN was induced in NE-deficient mice. Quantitation of glomerular PAS, hematuria, functional markers (BUN, sCr, LDH) and glomerular PMN accumulation in wild-type and NE-/- mice are given. All analyses were done at 96 hrs after TGN except glomerular neutrophil recruitment which was evaluated at 4 hrs after TGN induction. Data represent the mean ± SEM.
| WT | NE-/- | |
|---|---|---|
| PAS (%) | 83.55 ± 4.88 | 60.78 ± 5.65* |
| Hematuria grade | 2.86 ± 0.14 | 1.43 ± 0.30* |
| Cr (mg/dl) | 0.30 ± 0.05 | 0.17 ± 0.02* |
| BUN (mg/dl) | 153.70 ± 28.70 | 78.90 ± 9.80* |
| LDH × 103 (IU/l) | 2.00 ± 0.26 | 1.11 ± 0.12* |
| PMNs/glom | 5.74 ± 1.04 | 0.91 ± 0.15* |
P<0.05 compared with wild-type
Table 3. Quantitation of Elastase-digested fibrin degradation products in plasma samples.
TGN was induced in WT or Mac-1-/- mice and plasma from mice at the indicated times following TGN were examined for NE activity by examining fibrin degradation products reported to be specifically generated by granulocyte-elastase (e-XDP).
| Day | 0 | 1 | 2 | 4 |
|---|---|---|---|---|
|
Wild-type (μg/ml) |
0.06 ± 0.06 (n=4) |
2.26 ± 0.17 (n=12) |
1.69 ± 0.19 (n=5) |
1.97 ± 0.28 (n=4) |
|
Mac-1-/- (μg/ml) |
0.01 ± 0.01 (n=4) |
1.68 ± 0.16 (n=12) |
1.04 ± 0.29 (n=4) |
2.30 ± 0.67 (n=4) |
| p value | 0.40 | 0.02 | 0.09 | 0.66 |
Platelet immunodepletion accelerates TGN in wild-type mice but thromobocytopenic Mac-1 deficient mice remain resistant to disease
It is widely recognized that platelets play important roles in the pathogenesis of thrombotic diseases but there is also convincing evidence that inflammation is a potent trigger of hemorrhage in the absence of platelets19. Here we evaluated platelet accumulation and their role in TGN development. Platelets deposited in glomerular capillaries within 4hrs of TGN induction (Figure 3A) and this was dependent on Mac-1 (Intensity of GPIbα staining, WT/Con: 0.047 ± 0.01; WT/TGN: 0.557 ± 0.032 (median: 0.576); Mac-1-/-/TGN: 0.313 ± 0.073 (median: 0.303), p<0.038 for WT/TGN vs Mac-1-/-/TGN). Next, platelets were immunodepleted with anti-GP1bα antibody 12 hrs prior to disease induction. This regimen maintained low circulating platelet counts up to 24 hrs after TGN induction (0h, <2.0% of control; 24h, <5.0% of control), as previously described20. TGN induction in thrombocytopenic animals accelerated renal injury, resulting in lethality in a proportion of wild-type animals within 72 hrs. Thus, mice were evaluated 48hr after disease induction. A significant increase in histological parameters of glomerular injury (Figure 3B) and glomerular endothelial damage (as assessed by CD34 staining) (Figure 3C) was observed in thrombocytopenic animals. An increase in hemorrhage (hematuria), a rapid elevation of BUN, serum creatinine and LDH, and increased glomerular PMN accumulation were also observed (Figure 3D-F). Platelet immunodepletion 24hrs after induction of TGN did not exacerbate disease and indeed led to a trend of reduced indices of renal failure (Supplemental data S2). The accumulation of platelets within hours of TGN induction, before any evidence of thrombosis, coupled with our data that elimination of platelets before TGN increases glomerular injury suggest that early platelet deposition is cytoprotective.
Figure 3. Immunodepletion of platelets accelerates TGN.

(A) TGN (LPS + αGBM) was induced in wild-type mice and renal tissue was harvested 4hrs later from these mice and untreated wild-type mice (normal) to immunohistochemically assess platelet accumulation using antibody that recognizes the αIIb subunit of αIIbβ3 on platelets. (B-F) WT mice were intravenously injected with anti-GPIbα (40μg/mice) or control IgG (Con) 12 hrs prior to TGN induction. Glomerular PAS and fibrin deposition (PTAH staining) were significantly increased (B) and glomerular CD34 expression was reduced (C) in platelet-depleted mice in comparison with controls. The hematuria grade (D) was greater as were biochemical markers for renal function (BUN, Cr) and general organ damage (LDH) (E). The number of neutrophils (PMNs) per glomerulus (F) was also increased in platelet depleted mice. Data are the mean ± SEM. *P<0.05 compared with control IgG group. Bar: 30 μm
Others have provided compelling evidence that inflammation induces hemorrhage in the background of thrombocytopenia19 but the contribution of inflammatory cells to this process has not been directly evaluated. Thus we examined the role of Mac-1 in thrombocytopenia-induced exacerbation of TGN. Gp1bα antibody treatment prior to TGN immunodepleted platelets in both wild-type and Mac-1-/- mice (platelet counts, WT: <2% and Mac-1-/-: <3% of control 12 hrs after anti-GpIbα injection). Notably, platelet immunodepleted Mac-1-/- animals remained completely resistant to TGN-induced renal failure (Figure 4A). This indicates that Mac-1 mediated vessel injury is modulated by platelets.
Figure 4. Thrombocytopenic Mac-1-deficient mice remain resistant to TGN. Mac-1 interaction with platelet GpIbα promotes thrombosis.

(A) TGN was induced in WT or Mac-1-/- mice treated with Con IgG or anti-GPIbα to immunodeplete platelets. Serum Cr (sCr), BUN, and LDH were significantly elevated only in thrombocytopenic WT mice following induction of TGN. Thrombocytopenic Mac-1-/- mice exhibited no increase in indices of renal failure following induction of TGN. (B-C) TGN was induced in wild-type mice treated with a polyclonal anti-M2 (αM2) or rabbit IgG control (Con). (B) PAS deposition (left panel) and neutrophil accumulation (right panel) were evaluated in renal tissue. A significant reduction in PAS deposition but no decrease in glomerular neutrophil accumulation was observed in anti-M2 versus IgG control animals. (C) Indices of renal failure including sCr, BUN and LDH were reduced in anti-M2 treated animals compared to Control. Data represent the mean + SEM. *P<0.05 compared with wild-type.
Mac-1 interaction with platelet GpIbα promotes thrombosis
Mac-1 binds GpIbα on platelets. This interaction is required for neutrophil adhesion and transmigration at sites of endothelial denudation and platelet deposition following wire-induced vascular injury11. Here, an antibody targeting the GpIbα binding site on CD11b (termed anti-M2)11 was injected in wild-type mice to assess the contribution of Mac-1-GpIbα interaction to the development of TGN. Anti-M2 treatment had no effect on glomerular neutrophil accumulation (Figure 4B). Despite this, a significant reduction in thrombosis was observed in anti-M2 versus IgG isotype control treated wild-type animals (Figure 4B). As anti-M2 did not effect neutrophil recruitment, the data suggests a direct role for Mac-1 interaction with GpIbα on platelets in the initiation of glomerular thrombosis.
Discussion
The major finding of our work is that Mac-1 represents a critical molecular link between inflammation and thrombosis in a model of TGN that recapitulates features of the human disease. Our studies introduce the concept that Mac-1 mediated neutrophil recruitment and neutrophil-platelet interactions are key steps in inflammation-induced thrombosis leading to organ damage in TGN. We demonstrate that Mac-1 supports neutrophil recruitment likely through regulation of NE release and/or activity and stimulates the accumulation of platelets that initially function to preserve vessel integrity in the context of neutrophil mediated vascular damage. However, subsequent interaction of neutrophil Mac-1 with GpIbα on deposited platelets promotes thrombosis, a major aspect of renal injury in this model (Figure 5).
Figure 5. Model of Mac-1 mediated inflammation induced thrombosis in TGN.
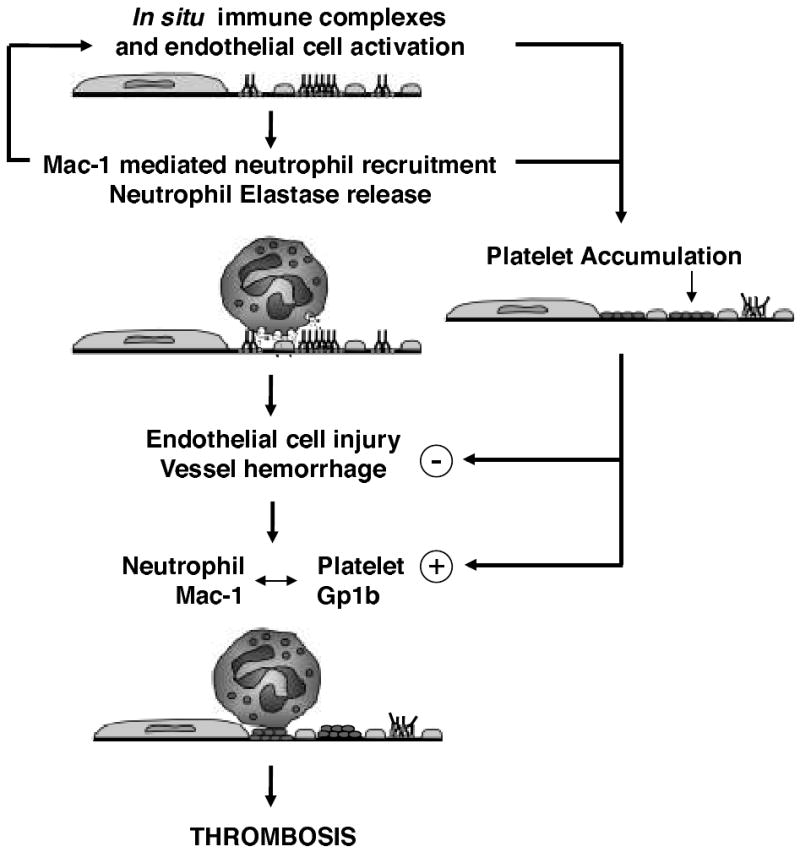
Anti-GBM antibody in combination with LPS results in in situ immune complex formation and endothelial cell activation. This triggers neutrophil recruitment through Mac-1, which further promotes endothelial cell activation. Engagement of Mac-1 results in neutrophil elastase release, which enhances neutrophil influx. Recruited neutrophils promote platelet accumulation. Platelets initially protect the vessel wall from neutrophil mediated sequelae (-). However, subsequent interaction of Mac-1 on recruited neutrophils with GpIbα on platelets triggers thrombosis (+) that is responsible for vessel occlusion and organ damage.
We developed a model of TMA in mice that has functional outcomes/laboratory features that resembled HUS in humans so that the molecular cross-talk between inflammation and thrombosis, two prominent features of HUS, and the pathogenesis of this disease can be explored. Many aspects of HUS were recapitulated in our model such as endothelial injury, glomerular thrombosis, thrombocytopenia, neutrophilia, renal failure and LDH elevation. However, anemia, a major characteristic of HUS was not significant in this mouse model as was also the case in published rat models21, 22. This may be because the glomerular intravascular hemolysis is not severe enough to manifest as systemic anemia since glomerular thrombosis is localized and less extensive in the rodent models compared to the human disease. Glomerular endothelial cell damage is a predominant feature of TMA in humans. CD34 served as a sensitive read-out of glomerular endothelial injury that also correlated with the extent of leukocyte infiltration and thrombosis. The reduction of CD34 in TGN correlated with an increase in renal levels of E-selectin, a selective marker of endothelial activation. Anti-GBM antibody or LPS alone were not sufficient to reduce glomerular endothelial CD34 indicating a positive correlation of this marker and TMA related pathology. Our model relied on ICs formed in situ. Another murine model recently developed with ICs to a planted antigen also resulted in renal endothelial injury and TMA suggesting a close link between immune complexes and TMA-related endothelial damage23.
Neutrophils were a prerequisite for development of thrombosis in TGN and Mac-1 specifically promoted TGN. Mac-1-/- mice subjected to TGN retained CD34 and had reduced renal E-selectin which indicates a primary role for Mac-1 on neutrophils in endothelial activation and injury in this model. A deficiency in Mac-1 or NE resulted in a significant attenuation of neutrophil influx. Elastase activity was decreased in plasma of Mac-1-/- mice leading us to propose that Mac-1 engagement results in the local release of NE17, 18 that then stimulates the secretion of endothelial chemoattractants24 or the generation of chemoattractant cleavage products 25 that promote neutrophil recruitment. Previous studies suggest that the role of Mac-1 in neutrophil accumulation is context dependent. Mac-1 is not required for neutrophil recruitment in IC-induced nephrotoxic nephritis, the Reverse Arthus reaction (JH and TNM, unpublished data) or complement-induced vasculitis17. It is required for sustaining adhesion in a heterologous model of anti-GBM-induced nephritis that is independent of NE14 and has a co-dominant role with its sister integrin LFA-1 in neutrophil accumulation in some other models26. Platelets accumulated in glomerular capillary within hours of induction of TGN, and this was Mac-1 dependent. It is possible that reactive oxygen species generated by recruited neutrophils trigger the release of von Willebrand factor from endothelial cells, that leads to platelet adherence27.
Platelet immunodepletion prior to TGN increased hematuria and renal injury suggesting that platelets are cytoprotective and prevent excessive inflammation. This is counter indicative to reports in the field28, 29 but in all these cases anti-coagulant therapies were initiated after disease was induced. Consistent with this, platelet immunodepletion after TGN induction also reduced disease indices. What is the cytoprotective mechanism of platelets? Platelets are well-established to preserve vessel integrity. A supportive role for platelets is shown following organ perfusion30, 31 and in sprouting vessels in tumors32. Clinically, patients with a drop in platelet counts as in idiopathic thrombocytopenic purpura (ITP) develop spontaneous bleeding while others with equally low platelet counts do not, suggesting that additional factors dictate the propensity for bleeding33. One explanation is that thrombocytopenia increases susceptibility to inflammation-induced hemorrhage. A recent study showed that although thrombocytopenia alone did not lead to hemorrhage34, acute inflammation-induced in the skin, brain, or lung of thrombocytopenic animals resulted in massive bleeding at the inflammatory site that was independent of platelet adhesion receptors required for platelet plug formation19. We show that thrombocytopenic Mac-1-/- mice remained completely resistant to TGN-induced renal injury indicating a potent function for Mac-1 initiated inflammation in inducing hemorrhage and identifying an important point of intersection between inflammation and coagulation. Platelet derived factors may change the quality of the vessel wall thus making it less vulnerable to inflammation mediated damage35 and/or platelet-neutrophil interactions may promote the transcellular biosynthesis of lipoxins, which are potent stop signals for PMN trafficking36. This is a fruitful area for future investigation.
Thrombosis was consistently reduced in Mac-1-/- mice following induction of TGN. Mac-1 on recruited neutrophils could promote thrombosis through several pathways by virtue of its capacity to bind numerous ligands. It binds factor X, which can be activated by neutrophil serine proteinases, and thus provide an alternative mechanism for thrombin generation 27. It also binds platelet counterreceptors Gp1bα and junctional adhesion molecule-3 directly 37, 38, and indirectly interacts with fibrinogen bound to αIIbβ339. Our data indicate that Mac-1 promotes thrombosis by mediating neutrophil engagement of platelet GpIbα. Blocking the GpIbα binding site on Mac-1 with anti-M2 antibody has been previously shown to limit vascular injury by inhibiting neutrophil recruitment to platelets deposited in the injured vessel wall11. Here, blockade of Mac-1/GpIbα interaction inhibited events downstream of neutrophil recruitment as this parameter was unaffected in mice treated with anti-M2. To our knowledge, this is the first in vivo evidence that Mac-1 interaction with platelets directly promotes thrombosis. Mac-1 binding to GpIbα on platelets may provide a physical proximity that permits transcellular metabolic cooperation and the efficient delivery of proteinases such as elastase, which activate platelets through limited proteolytic cleavage of αIIbβ327. In support of this, the interaction of active Mac-1 on neutrophil microparticles with GpIbα was recently shown to promote platelet activation in vitro 40.
In conclusion, we demonstrate that Mac-1 on neutrophils represents a critical molecular link between inflammation and coagulation leading to organ damage. The protective effect of Mac-1 deficiency was apparent despite the strong induction of a number of proinflammatory cytokines in renal tissue that are known to cause activation of T and B cells, macrophages, endothelial cells, complement, and the coagulation cascade. A likely effector of Mac-1 mediated neutrophil recruitment is the serine proteinase NE that was required for neutrophil recruitment and ensuing vessel damage, and whose activity was significantly diminished in the absence of Mac-1. Inflammation-induced hemorrhage in the context of thrombocytopenia was Mac-1 dependent suggesting that platelets support vessel wall integrity in the context of Mac-1 mediated injury. On the other hand, the interaction of Mac-1 on recruited neutrophils with Gp1bα on platelets is a major pathway for the subsequent development of thrombosis. These data suggest the possibility of targeting Mac-1 as a novel therapeutic modality to preserve vascular integrity and attenuate thrombosis in thrombotic disorders such as TGN.
Supplementary Material
Acknowledgments
We are grateful to Bio-Rad for support with the Bio-Plex protein array system.
Funding Source: This work was supported by the Ministry of Education, Culture, Sports, Science and Technology, Japan, 17209034 (TF), 17790547(JH), the Uehara Memorial Foundation (Japan)(JH), Arthritis Foundation (NT), a grant from the Ministry of Health, Labour and Welfare in Japan (KS) and NIH RO1 AR050800, HL065095 and DK077111(TNM).
Footnotes
Disclosure Statement: The authors have no conflicts to disclose
Under inflammatory conditions such as infection, autoimmune diseases, and atherosclerosis, thrombosis is an important clinical manifestation, which can lead to fatal organ damage. Thrombotic microangiopathy (TMA) occurs in various clinical settings including hemolytic uremic syndrome (HUS), thrombotic thrombocytopenic purura (TTP), transplant rejection, systemic lupus erythematosus and glomerulonephritis exacerbated by infection. In this report, we report that Mac-1, a leukocyte-specfic CD18 integrin, links inflammation and thrombosis in TMA. Mac-1 is responsible for vascular endothelial damage, and links neutrophil-platelet interaction through GPIbα on platelets. Blockade of Mac-1/GpIbα interaction prevented inflammation-induced glomerular thrombosis, and preserved renal function. In conclusion, we propose that Mac-1 is a novel therapeutic target that may prevent thrombosis in inflammatory diseases and thus preserve vascular integrity.
References
- 1.Stave GM, Croker BP. Thrombotic microangiopathy in anti-glomerular basement membrane glomerulonephritis. Arch Pathol Lab Med. 1984;108:747–751. [PubMed] [Google Scholar]
- 2.Besbas N, Karpman D, Landau D, Loirat C, Proesmans W, Remuzzi G, Rizzoni G, Taylor CM, Van de Kar N, Zimmerhackl LB. A classification of hemolytic uremic syndrome and thrombotic thrombocytopenic purpura and related disorders. Kidney Int. 2006;70:423–431. doi: 10.1038/sj.ki.5001581. [DOI] [PubMed] [Google Scholar]
- 3.Ruggenenti P, Noris M, Remuzzi G. Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney Int. 2001;60:831–846. doi: 10.1046/j.1523-1755.2001.060003831.x. [DOI] [PubMed] [Google Scholar]
- 4.Forsyth KD, Simpson AC, Fitzpatrick MM, Barratt TM, Levinsky RJ. Neutrophil-mediated endothelial injury in haemolytic uraemic syndrome. Lancet. 1989;2:411–414. doi: 10.1016/s0140-6736(89)90591-6. [DOI] [PubMed] [Google Scholar]
- 5.Fernandez GC, Rubel C, Dran G, Gomez S, Isturiz MA, Palermo MS. Shiga toxin-2 induces neutrophilia and neutrophil activation in a murine model of hemolytic uremic syndrome. Clin Immunol. 2000;95:227–234. doi: 10.1006/clim.2000.4862. [DOI] [PubMed] [Google Scholar]
- 6.Ponticelli C. De novo thrombotic microangiopathy. An underrated complication of renal transplantation. Clin Nephrol. 2007;67:335–340. doi: 10.5414/cnp67335. [DOI] [PubMed] [Google Scholar]
- 7.Mayer SA, Aledort LM. Thrombotic microangiopathy: differential diagnosis, pathophysiology and therapeutic strategies. Mt Sinai J Med. 2005;72:166–175. [PubMed] [Google Scholar]
- 8.Cohen JA, Brecher ME, Bandarenko N. Cellular source of serum lactate dehydrogenase elevation in patients with thrombotic thrombocytopenic purpura. J Clin Apher. 1998;13:16–19. doi: 10.1002/(sici)1098-1101(1998)13:1<16::aid-jca3>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 9.Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Andrian UH, Arnaout MA, Mayadas TN. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 10.Belaaouaj A, McCarthy R, Baumann M, Gao Z, Ley TJ, Abraham SN, Shapiro SD. Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat Med. 1998;4:615–618. doi: 10.1038/nm0598-615. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Sakuma M, Chen Z, Ustinov V, Shi C, Croce K, Zago AC, Lopez J, Andre P, Plow E, Simon DI. Leukocyte engagement of platelet glycoprotein Ibalpha via the integrin Mac-1 is critical for the biological response to vascular injury. Circulation. 2005;112:2993–3000. doi: 10.1161/CIRCULATIONAHA.105.571315. [DOI] [PubMed] [Google Scholar]
- 12.Kondo C, Mizuno M, Nishikawa K, Yuzawa Y, Hotta N, Matsuo S. The role of C5a in the development of thrombotic glomerulonephritis in rats. Clin Exp Immunol. 2001;124:323–329. doi: 10.1046/j.1365-2249.2001.01513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kohno I, Inuzuka K, Itoh Y, Nakahara K, Eguchi Y, Sugo T, Soe G, Sakata Y, Murayama H, Matsuda M. A monoclonal antibody specific to the granulocyte-derived elastase-fragment D species of human fibrinogen and fibrin: its application to the measurement of granulocyte-derived elastase digests in plasma. Blood. 2000;95:1721–1728. [PubMed] [Google Scholar]
- 14.Tang T, Rosenkranz A, Assmann KJ, Goodman MJ, Gutierrez-Ramos JC, Carroll MC, Cotran RS, Mayadas TN. A role for Mac-1 (CDIIb/CD18) in immune complex-stimulated neutrophil function in vivo: Mac-1 deficiency abrogates sustained Fcgamma receptor-dependent neutrophil adhesion and complement-dependent proteinuria in acute glomerulonephritis. J Exp Med. 1997;186:1853–1863. doi: 10.1084/jem.186.11.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Norton J, Sloane JP, Delia D, Greaves MF. Reciprocal expression of CD34 and cell adhesion molecule ELAM-1 on vascular endothelium in acute cutaneous graft-versus-host disease. J Pathol. 1993;170:173–177. doi: 10.1002/path.1711700213. [DOI] [PubMed] [Google Scholar]
- 16.Delia D, Lampugnani MG, Resnati M, Dejana E, Aiello A, Fontanella E, Soligo D, Pierotti MA, Greaves MF. CD34 expression is regulated reciprocally with adhesion molecules in vascular endothelial cells in vitro. Blood. 1993;81:1001–1008. [PubMed] [Google Scholar]
- 17.Hirahashi J, Mekala D, Van Ziffle J, Xiao L, Saffaripour S, Wagner DD, Shapiro SD, Lowell C, Mayadas TN. Mac-1 signaling via Src-family and Syk kinases results in elastase-dependent thrombohemorrhagic vasculopathy. Immunity. 2006;25:271–283. doi: 10.1016/j.immuni.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 18.Ross GD. Regulation of the adhesion versus cytotoxic functions of the Mac-1/CR3/alphaMbeta2-integrin glycoprotein. Crit Rev Immunol. 2000;20:197–222. [PubMed] [Google Scholar]
- 19.Goerge T, Ho-Tin-Noe B, Carbo C, Benarafa C, Remold-O'Donnell E, Zhao BQ, Cifuni SM, Wagner DD. Inflammation induces hemorrhage in thrombocytopenia. Blood. 2008;111:4958–4964. doi: 10.1182/blood-2007-11-123620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nieswandt B, Bergmeier W, Rackebrandt K, Gessner JE, Zirngibl H. Identification of critical antigen-specific mechanisms in the development of immune thrombocytopenic purpura in mice. Blood. 2000;96:2520–2527. [PubMed] [Google Scholar]
- 21.Tomosugi NI, Cashman SJ, Hay H, Pusey CD, Evans DJ, Shaw A, Rees AJ. Modulation of antibody-mediated glomerular injury in vivo by bacterial lipopolysaccharide, tumor necrosis factor, and IL-1. J Immunol. 1989;142:3083–3090. [PubMed] [Google Scholar]
- 22.Naruse T, Tsuchida A, Ogawa S, Yano S, Maekawa T. Selective glomerular thrombosis in rats induced by combined injections of nephrotoxic antiserum and lipopolysaccharide. J Lab Clin Med. 1985;105:146–156. [PubMed] [Google Scholar]
- 23.Hohenstein B, Braun A, Amann KU, Johnson RJ, Hugo CP. A murine model of site-specific renal microvascular endothelial injury and thrombotic microangiopathy. Nephrol Dial Transplant. 2008;23:1144–1156. doi: 10.1093/ndt/gfm774. [DOI] [PubMed] [Google Scholar]
- 24.Berger SP, Seelen MA, Hiemstra PS, Gerritsma JS, Heemskerk E, van der Woude FJ, Daha MR. Proteinase 3, the major autoantigen of Wegener's granulomatosis, enhances IL-8 production by endothelial cells in vitro. J Am Soc Nephrol. 1996;7:694–701. doi: 10.1681/ASN.V75694. [DOI] [PubMed] [Google Scholar]
- 25.Leavell KJ, Peterson MW, Gross TJ. The role of fibrin degradation products in neutrophil recruitment to the lung. Am J Respir Cell Mol Biol. 1996;14:53–60. doi: 10.1165/ajrcmb.14.1.8534486. [DOI] [PubMed] [Google Scholar]
- 26.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 27.Afshar-Kharghan V, Thiagarajan P. Leukocyte adhesion and thrombosis. Curr Opin Hematol. 2006;13:34–39. doi: 10.1097/01.moh.0000190107.54790.de. [DOI] [PubMed] [Google Scholar]
- 28.Drews RE. Critical issues in hematology: anemia, thrombocytopenia, coagulopathy, and blood product transfusions in critically ill patients. Clin Chest Med. 2003;24:607–622. doi: 10.1016/s0272-5231(03)00100-x. [DOI] [PubMed] [Google Scholar]
- 29.Ikeguchi H, Maruyama S, Morita Y, Fujita Y, Kato T, Natori Y, Akatsu H, Campbell W, Okada N, Okada H, Yuzawa Y, Matsuo S. Effects of human soluble thrombomodulin on experimental glomerulonephritis. Kidney Int. 2002;61:490–501. doi: 10.1046/j.1523-1755.2002.00160.x. [DOI] [PubMed] [Google Scholar]
- 30.Gimbrone MA, Jr, Aster RH, Cotran RS, Corkery J, Jandl JH, Folkman J. Preservation of vascular integrity in organs perfused in vitro with a platelet-rich medium. Nature. 1969;222:33–36. doi: 10.1038/222033a0. [DOI] [PubMed] [Google Scholar]
- 31.Danielli JF. Capillary permeability and oedema in the perfused frog. J Physiol. 1940;98:109–129. doi: 10.1113/jphysiol.1940.sp003837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kisucka J, Butterfield CE, Duda DG, Eichenberger SC, Saffaripour S, Ware J, Ruggeri ZM, Jain RK, Folkman J, Wagner DD. Platelets and platelet adhesion support angiogenesis while preventing excessive hemorrhage. Proc Natl Acad Sci U S A. 2006;103:855–860. doi: 10.1073/pnas.0510412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nieswandt B. How do platelets prevent bleeding? Blood. 2008;111:4835. doi: 10.1182/blood-2008-02-139006. [DOI] [PubMed] [Google Scholar]
- 34.Dominguez V, Govezensky T, Gevorkian G, Larralde C. Low platelet counts alone do not cause bleeding in an experimental immune thrombocytopenic purpura in mice. Haematologica. 2003;88:679–687. [PubMed] [Google Scholar]
- 35.Ho-Tin-Noe B, Goerge T, Cifuni SM, Duerschmied D, Wagner DD. Platelet granule secretion continuously prevents intratumor hemorrhage. Cancer Res. 2008;68:6851–6858. doi: 10.1158/0008-5472.CAN-08-0718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brady HR, Serhan CN. Lipoxins: putative braking signals in host defense, inflammation and hypersensitivity. Curr Opin Nephrol Hypertens. 1996;5:20–27. [PubMed] [Google Scholar]
- 37.Santoso S, Sachs UJ, Kroll H, Linder M, Ruf A, Preissner KT, Chavakis T. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J Exp Med. 2002;196:679–691. doi: 10.1084/jem.20020267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simon DI, Chen Z, Xu H, Li CQ, Dong J, McIntire LV, Ballantyne CM, Zhang L, Furman MI, Berndt MC, Lopez JA. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18) J Exp Med. 2000;192:193–204. doi: 10.1084/jem.192.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weber C, Springer TA. Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to alphaIIbbeta3 and stimulated by platelet-activating factor. J Clin Invest. 1997;100:2085–2093. doi: 10.1172/JCI119742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pluskota E, Woody NM, Szpak D, Ballantyne CM, Soloviev DA, Simon DI, Plow EF. Expression, activation, and function of integrin alphaMbeta2 (Mac-1) on neutrophil-derived microparticles. Blood. 2008;112:2327–2335. doi: 10.1182/blood-2007-12-127183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.