

Abstract
Alpha-1 antitrypsin (A1AT or AAT) is a serine protease inhibitor (PI) which, when present at low levels, can cause chronic obstructive pulmonary disease (COPD) and liver disease in both children and adults. Several mutations within the SERPINA1 gene have been found to cause this deficiency. The most common variants are PI*Z and PI*S, each caused by a single nucleotide polymorphism (SNP). We describe a real time polymerase chain reaction (PCR) assay for the rapid genotyping of these polymorphisms. DNA was extracted from fourteen EDTA-anticoagulated whole blood samples using the Qiagen EZ1 blood extraction kit. SNP genotyping was performed using primer/probe sets purchased from Applied Biosystems. These were evaluated for performance and assay conditions on the Applied Biosystems 7500 FAST System. The genotypes of these samples were compared with their phenotype results from isoelectric focusing assays, which were performed by an independent reference laboratory. In addition, twenty samples that were previously genotyped at another laboratory were obtained for accuracy studies. Thirty-four samples were tested; five genotypes were represented and the assay was able to discriminate these successfully. Only one genotype could not be correlated with its phenotype result, as the phenotype was reported as an “unidentified allele”. All other genotyping results were concordant with previously determined genotypes and phenotypes. We describe a rapid real time PCR assay that is suitable for clinical use in genotyping AAT alleles and which can be used as the initial step in A1AT testing algorithms.
Keywords: Real-time PCR, alpha-1 antitrypsin, molecular diagnostics, serine protease inhibitor, chronic obstructive pulmonary disease (COPD), single nucleotide polymorphism (SNP), genotyping
Introduction
Alpha-1-antitrypsin (AAT) deficiency is an inherited disorder involving mutations in the SERPINA1 (SERine Proteinase INhibitor A1) gene and is characterized by low levels of circulating AAT protein, resulting in an increased risk for development of pulmonary emphysema and liver disease [1, 2]. AAT is a serine protease inhibitor (PI) that functions to protect the lungs from neutrophil elastase, an enzyme released from neutrophils triggered in response to pathogens that enter the lungs [3]. AAT is constitutively produced in the liver and its expression is up-regulated during inflammation and infection. Mutations in the SERPINA1 gene can result in both early onset chronic obstructive pulmonary disease (COPD), caused by neutrophil elastase attacking the lung, and liver cirrhosis, caused by mutated AAT protein that accumulates in the rough endoplasmic reticulum of hepatocytes [4, 5].
The SERPINA1 gene, referred to as the PI locus, is a highly pleomorphic gene located at 14q31-32.3 [6–8]. It is 12.2 kb and contains seven exons and six introns. Of the exons, four are coding and three are noncoding [9]. While AAT displays a significant amount of heterogeneity, many of the described genetic variants do not affect expression or function of this protein [9, 10]. Alleles that result in deficient amounts of protein in the circulation, degraded protein with no function (null), or dysfunctional protein with lower activity levels have been identified [10]. The most common alleles in the general population are PI*M, PI*S and PI*Z. The M, S, and Z nomenclature derives from the migration velocity of each of these variants in starch-gel electrophoresis; M migrates at a medium velocity; S, is slow, and Z is very slow [11]. The M allele (and two common subvariants, M1 and M2) is considered the wild-type, or “normal variant”, and is associated with normal levels of AAT in the plasma; it is the most common in the general population [1, 12]. The most common variant associated with clinical disease is PI*Z allele which results from a G to an A transition at nucleotide position 11940 in exon 5 and leads to a glutamic acid to lysine substitution at amino acid 366 (E366K; rs28929474). The PI*S allele results from an A to T transversion at nucleotide position 9628 in exon 3 and leads to a glutamic acid to valine substitution at amino acid 288 (E288V; rs17580) [13, 14]. Older literature, which uses an amino acid numbering convention based on the mature, processed protein, refers to these mutations as E342K and E264V, respectively. A genotype of ZZ or SZ can cause clinical AAT deficiency, but the homozygous Z genotype typically results in the most severe clinical phenotype. This is due to the fact that 85% of the resultant PI*Z protein accumulates in the endoplasmic reticulum of the hepatocytes (Table 1) [15].
Table 1.
Genetic characteristics of AAT
|
Currently, diagnostic laboratory tests for AAT deficiency include measurement of the circulating AAT protein level, by immunoassay, AAT protein phenotyping by isoelectric focusing (IEF), genotyping or sequencing of SERPINA1, and evaluation of the AAT protein function, aimed at detecting dysfunctional alleles that produce proper protein amounts. Normal AAT levels in an adult are >104 mg/dL, but patients with AAT deficiency have levels between 15–75% of normal depending on their genotype [16]. Individuals with pulmonary symptoms and with AAT levels less than 60 mg/dl are typically eligible for replacement therapy with recombinant AAT protein [17].
This study describes the detection of the most common variants (M, Z, and S) of the SERPINA1 gene using a real time PCR assay and Taqman probes in patient samples that were previously tested by phenotyping and/or genotyping.
Materials and methods
Specimens
DNA was extracted from fourteen EDTA-anticoagulated whole blood samples using the Qiagen EZ1 automated system. Concurrently, the plasma from these samples was collected and sent to a reference laboratory for IEF phenotyping. In addition, DNA from twenty blinded samples that were previously genotyped at another institution (Mayo Clinic, Rochester, MN) was used for test validation.
Genotyping
All samples were genotyped using an allelic discrimination assay on the Applied Bio-systems 7500 FAST Real Time PCR instrument with Taqman® probes. For the PI*S allele, a TaqMan® Pre-Designed SNP Genotyping Assay was purchased from Applied Biosystems (ABI) (assay ID: C_594695_20); for the PI*Z allele, a TaqMan® Custom SNP Genotyping Assay was purchased. For the custom assay, primers and probes based on the PI*S target sequence were designed using the Custom TaqMan® Assay Design Tool (Applied Biosystems) (Table 2). Both the pre-designed and the custom assays included primers and fluorescently labeled (FAM and VIC) MGB™ probes for detection of both the wild-type and variant alleles.
Table 2.
Primers and Probes for the ABI 7500 AAT Pi*Z assay
| Forward: | TCCAGGCCGTGCATAAGG |
| Reverse: | GCCCCAGCAGCTTCAG |
| WT Probe (VIC): | CCATCGACGAGAAAG |
| MUT Probe (FAM): | CATCGACAAGAAAG |
All samples were genotyped using the Allelic Discrimination assay on the Applied Bio-systems 7500 FAST Real-Time PCR System. Cycling conditions were as follows: 95°C for 10 min; 50 cycles of 92°C for 15 sec and 60°C for 90 sec. Fluorescence was detected during the 60°C annealing step of each PCR cycle.
Results
Using the allelic discrimination analysis method on the AB 7500 FAST, we were able to differentiate wild type, heterozygous and homozygous alleles based on specific probe hybridizations (Figure 1). An initial evaluation of the primer/probe sets identified for genotyping included dilution of known mutant DNAs. As expected, the CT values corresponded to the amount of starting DNA and showed that the assay was able to detect a genotype even with very low concentrations of starting DNA (Table 3).
Figure 1.
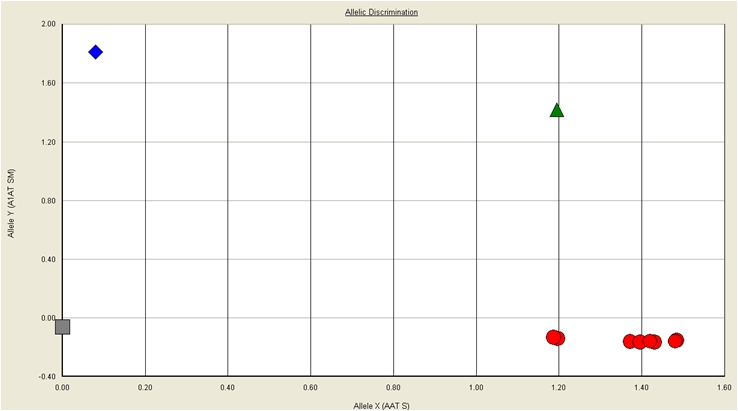
Allelic discrimination plot for real time PCR results detecting the homozygous mutant PI*Z, heterozygous PI*Z and homozygous non-S/non-Z alleles.
Table 3.
DNA dilution series
| PI*Z (CT) | PI*S (CT) | |
|---|---|---|
| 5 ng/μl | 31.15 | 26.78 |
| 10 ng/μl | 29.2 | 25.63 |
| 15 ng/μl | 29.03 | 26.44 |
| 20 ng/μl | 28.81 | 24.34 |
| 50 ng/μl | 27.7 | 23.99 |
| 100 ng/ul | 27.12 | 24.12 |
Of the 14 patient samples collected at our medical center, 11 were homozygous non-S, non-Z genotype, 2 were heterozygous (non-S, non-Z)Z, and 1 was heterozygous (non-S, non-Z)S. No homozygous mutant (Z or S) or heterozygous mutant (SZ) genotypes were detected. The genotypes of the extracted DNA samples from the Mayo Clinic were as follows: 5 homozygous non-S, non-Z, 12 heterozygous (6 (non-S, non-Z)Z and 6 (non-S, non-Z)S), 2 presumably compound heterozygous, and 1 homozygous mutant (ZZ). Homozygosity for the S allele was not detected in any of the DNA samples.
The patient sample results of the genotyping assay were compared to the results obtained through IEF from an outside laboratory. Of the 14 patient sample IEF results, 13 matched the real-time PCR results (Table 4). The discordant sample was determined to be an unknown, clinically insignificant variant by phenotyping; our assay determined the genotype was heterozygous (non-S, non-Z)Z. Genotyping results for all 20 Mayo Clinic samples were found to be concordant with the independently determined genotypes. All control samples gave expected results.
Table 4.
IEF comparison to genotyping results
| Lab # | Phenotyping Result | Protein Result | PCR Result |
|---|---|---|---|
| 8 | MZ | 199 mg/dL | (Non-S, non-Z)Z |
| 10 | MM | 139 mg/dL | Non-S, non-Z |
| 11 | MM | 136 mg/dL | Non-S, non-Z |
| 14 | MM | 189 mg/dL | Non-S, non-Z |
| 16 | MM | 112 mg/dL | Non-S, non-Z |
| 17 | MM | 168 mg/dL | Non-S, non-Z |
| 18 | MM | 123 mg/dL | Non-S, non-Z |
| 19 | MM | 175 mg/dL | Non-S, non-Z |
| 20 | MM | 183 mg/dL | Non-S, non-Z |
| 21 | MM | 179 mg/dL | Non-S, non-Z |
| 24 | MM | 148 mg/dL | Non-S, non-Z |
| 25 | MM | 145 mg/dL | Non-S, non-Z |
| 26 | MS | 131 mg/dL | (Non-S, non-Z)S |
| 27 | M/? | 81 mg/dL | (Non-S, non-Z)Z |
Discussion
In this study, we evaluated a real time PCR method for genotyping AAT in patients being screened to confirm or rule-out a diagnosis of AAT deficiency. Genotyping of the SERPINA1 gene is important in the algorithms used to diagnose AAT deficiency, and can reduce the need for costly IEF or sequencing assays [18]. Determination of serum AAT concentration is not sufficient as a diagnostic test for AAT deficiency because AAT is an acute phase reactant, and as such may be elevated under circumstances of inflammation, leading to incorrect measurement of baseline concentrations in whole blood. Levels typically remain well below the normal range in AAT deficient individuals. In addition, specific genetic variations, independent of the serum concentration, can be critical to the patient's risk for developing COPD and liver disease.
Currently, clinical testing for AAT deficiency involves four methods that are commonly used to diagnose AAT deficiency: (1) measurement of the serum or plasma protein level, (2) AAT protein phenotyping from serum or plasma, (3) AAT genotyping, and (4) evaluation of AAT protein function. The measurement of circulating AAT levels is performed in many clinical laboratories as an initial screening test. The limitations associated with this type of testing include the test's inability to detect individuals who are heterozygous for a deficiency allele and those individuals who often have levels at or near the normal cutoff value. Phenotpying and/or genotyping are recommended when the AAT protein level is below the normal range.
Protein phenotyping is performed by isoelectric focusing gel electrophoresis to detect the isoform patterns of the AAT protein. The limitations of this approach are its inability to identify PI*Null alleles due to the absence of circulating protein and that this procedure is usually performed in specialized laboratories. Genotyping assays are designed for the molecular identification of the most common abnormal AAT variants (PI*S and PI*Z) and thus can miss one of the more than 30 rare genetic variants that lead to reduced protein levels, absent protein levels, or normal levels of a dysfunctional protein. It is for this reason we used the terms “non-S” and “non-Z” for our results; the genotyping assay cannot determine if a variation in the gene sequence exists outside the area being tested.
In order to optimize testing for AAT deficiency, a combination of genotyping of SERPINA1 and measurement of serum AAT protein concentration provides a very informative and less expensive method for determining disease status in a patient. Patients with an abnormally low serum AAT protein concentration, yet normal genotyping results, may be subsequently submitted for IEF or sequencing studies, since this genotyping assay only genotypes the PI*S and the PI*Z mutants. Phenotyping can identify many more alleles than genotyping yet is not capable of detecting null alleles. The genetic status of any given patient becomes critical when deciding on screening mechanisms for other family members who may be at risk for developing disease or being a carrier of a variant.
All patients in this study had normal levels of serum AAT protein with the exception of one patient whose level was approximately 75% of normal. Interestingly, it was this same sample that was found to have an indeterminate phenotype. Our genotyping assay was able to resolve this result and determine that the patient was heterozygous (non-S, non-Z)Z. This result highlights the utility of this assay, however, further studies need to be conducted with larger patient sample sizes to validate these findings.
AAT deficiency continues to be underdiagnosed, especially in many patients with COPD where approximately 1% of these patients have AAT deficiency [17]. The underdiagnosis of AAT has been in part attributed to the perceived risks associated with testing for a genetic condition and informed consent for such testing [19]. Testing for AAT deficiency has been recommended for all patients with COPD, asthma with irreversible airflow obstruction, unexplained liver disease, or necrotizing panniculitis [20].
Conclusion
AAT deficiency is often unrecognized and may lead to COPD and severe liver disease. AAT deficiency can be readily diagnosed by measurement of the serum or plasma protein level, which should be confirmed by assessing the genotype or protein phenotype when AAT levels are below the normal range. The method for genotyping the SERPINA1 gene described in this study provides a significant cost advantage compared to IEF for labs that must send samples out for analysis. The combined use of AAT serum level and genotyping is sufficient to characterize the majority of normal, carrier, and affect cases [18]. Some cases, however, will require IEF reflex testing, because this assay has a very short turn around time; it can be performed to determine if IEF is necessary.
References
- 1.Kohnlein T, Welte T. Alpha-1 antitrypsin deficiency: pathogenesis, clinical presentation, diagnosis, and treatment. American Journal of Medicine. 2008;121:3–9. doi: 10.1016/j.amjmed.2007.07.025. [DOI] [PubMed] [Google Scholar]
- 2.Crystal RG. Alpha 1-antitrypsin deficiency, emphysema, and liver disease. Genetic basis and strategies for therapy. Journal of Clinical Investigation. 1990;85:1343–1352. doi: 10.1172/JCI114578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee WL, Downey GP. Leukocyte elastase: physiological functions and role in acute lung injury. American Journal of Respiratory & Critical Care Medicine. 2001;164:896–904. doi: 10.1164/ajrccm.164.5.2103040. [DOI] [PubMed] [Google Scholar]
- 4.Perlmutter DH. Pathogenesis of chronic liver injury and hepatocellular carcinoma in alpha-1-antitrypsin deficiency. Pediatric Research. 2006;60:233–238. doi: 10.1203/01.pdr.0000228350.61496.90. [DOI] [PubMed] [Google Scholar]
- 5.Law RHP, Zhang Q, McGowan S, Buckle AM, Silverman GA, Wong W, Rosado CJ, Langendorf CG, Pike RN, Bird PI, Whisstock JC. An overview of the serpin superfamily. Genome Biology. 2006;7:216. doi: 10.1186/gb-2006-7-5-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Darlington GJ, Astrin KH, Muirhead SP, Desnick RJ, Smith M. Assignment of human alpha 1-antitrypsin to chromosome 14 by somatic cell hybrid analysis. Proceedings of the National Academy of Sciences of the United States of America. 1982;79:870–873. doi: 10.1073/pnas.79.3.870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox DW, Markovic VD, Teshima IE. Genes for immunoglobulin heavy chains and for alpha 1-antitrypsin are localized to specific regions of chromosome 14q. Nature. 1982;297:428–430. doi: 10.1038/297428a0. [DOI] [PubMed] [Google Scholar]
- 8.Luisetti M, Seersholm N. Alpha1-antitrypsin deficiency. 1: epidemiology of alpha1-antitrypsin deficiency. Thorax. 2004;59:164–169. doi: 10.1136/thorax.2003.006494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wood AM, Stockley RA. Alpha one antitrypsin deficiency: from gene to treatment. Respiration. 2007;74:481–492. doi: 10.1159/000105536. [DOI] [PubMed] [Google Scholar]
- 10.Fregonese L, Stolk J, Frants RR, Veldhuisen B. Alpha-1 antitrypsin Null mutations and severity of emphysema. Respiratory Medicine. 2008;102:876–884. doi: 10.1016/j.rmed.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Fagerhol MK, Laurell CB. The Pi systeminherited variants of serum alpha 1-antitrypsin. Progress in Medical Genetics. 1970;7:96–111. [PubMed] [Google Scholar]
- 12.Lisowska-Myjak B. AAT as a diagnostic tool. Clinica Chimica Acta. 2005;352:1–13. doi: 10.1016/j.cccn.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 13.Long GL, Chandra T, Woo SL, Davie EW, Kurachi K. Complete sequence of the cDNA for human alpha 1-antitrypsin and the gene for the S variant. Biochemistry. 1984;23:4828–4837. doi: 10.1021/bi00316a003. [DOI] [PubMed] [Google Scholar]
- 14.Curiel DT, Chytil A, Courtney M, Crystal RG. Serum alpha 1-antitrypsin deficiency associated with the common S-type (Glu264—-Val) mutation results from intracellular degradation of alpha 1-antitrypsin prior to secretion. Journal of Biological Chemistry. 1989;264:10477–10486. [PubMed] [Google Scholar]
- 15.Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature. 1992;357:605–607. doi: 10.1038/357605a0. [DOI] [PubMed] [Google Scholar]
- 16.Sandford AJ, Weir TD, Spinelli JJ, Pare PD. Z and S mutations of the alpha1-antitrypsin gene and the risk of chronic obstructive pulmonary disease. American Journal of Respiratory Cell & Molecular Biology. 1999;20:287–291. doi: 10.1165/ajrcmb.20.2.3177. [DOI] [PubMed] [Google Scholar]
- 17.Silverman EK, Sandhaus RA. Clinical practice. Alpha1-antitrypsin deficiency. New England Journal of Medicine. 2009;360:2749–2757. doi: 10.1056/NEJMcp0900449. [DOI] [PubMed] [Google Scholar]
- 18.Snyder MR, Katzmann JA, Butz ML, Wiley C, Yang P, Dawson DB, Halling KC, Highsmith WE, Thibodeau SN. Diagnosis of alpha-1-antitrypsin deficiency: An algorithm of quantification, genotyping, and phenotyping. Clinical Chemistry. 2006;52:2236–2242. doi: 10.1373/clinchem.2006.072991. [DOI] [PubMed] [Google Scholar]
- 19.Apse KA, Biesecker BB, Giardiello FM, Fuller BP, Bernhardt BA. Perceptions of genetic discrimination among at-risk relatives of colorectal cancer patients. Genetics in Medicine. 2004;6:510–516. doi: 10.1097/01.gim.0000144013.96456.6c. [DOI] [PubMed] [Google Scholar]
- 20.WHO Alpha 1-antitrypsin deficiency: memorandum from a WHO meeting. Bulletin of the World Health Organization. 1997;75:397–415. [PMC free article] [PubMed] [Google Scholar]