

Abstract
Growing evidence indicates that the antiapoptotic protein survivin is a major factor of drug and radiation resistance in cancer cells. However, application of this finding to therapeutic drug combination is largely unexplored. In this study, breast cancer cells were used for treatment with anticancer compounds alone or in combination. We report that T138067, a better drug against multiple drug resistance (MDR) tumor cells than taxol (Shan et al., PNAS 96:5686–91,1999), induces survivin expression and consequently decreases its effectiveness on the induction of cancer cell death. Treatment of breast cancer cells with T138067 induced survivin expression in these cells while showing no effect on Bcl-2, indicating its specificity. Upregulation of survivin by T138067 was concomitant with an increased drug resistance and associated with an increased phosphorylation of Akt and Erk1/2 MAPK, and a decreased phosphorylation of p38 MAPK without affecting the phosphorylation of ErbB2. Therefore, it is possible that inhibition of T138067-induced survivin expression by alternative approaches may sensitize cells to T138067-induced cell death. We found that treatment of breast cancer cells with SN38, the active metabolite of irinotecan, inhibits survivin expression. Intriguingly, inhibition of survivin expression by SN38 was more effective at a low concentration than at the high concentration, which makes SN38 a good survivin modulator. Furthermore, in contrast with the decreased phosphorylation of p38 MAPK after T138067 treatment, inhibition of survivin expression by SN38 was associated with an increased phosphorylation of the p38 MAPK, suggesting opposing signals converging to survivin. Consistent with these observations, T138067 in combination with SN38 strongly induced cell death in comparison with each drug alone. Similarly, sequential combination of resveratrol, a component of red grapes that inhibits survivin expression, with T138067 also provoked massive breast cancer cell death compared with T138067 alone. Together, these results highlight a new concept that unique signaling cross talk converged to survivin may be considered for rational drug combination in the clinic.
Keywords: Survivin, T138067, SN38, irinotecan, resveratrol, breast cancer cells
Introduction
Survivin, a unique member of the inhibitor of apoptosis (IAP) protein family, is highly expressed in cancer while it is undetectable in most normal adult tissue [1–3]. Many studies reported in literature support that survivin is a major factor of drug and radiation resistance and a unique therapeutic target. Inhibition of its expression and function in cancer cells appears to be an effective approach for cancer treatment by inducing apoptosis [3–6]. Previously, we demonstrated that induction of survivin by taxol decreases its effectiveness to induce breast cancer cell death and that inhibition of taxol-induced survivin expression by siRNA strikingly provokes apoptosis [7], suggesting that survivin expression in cancer cells is a drug resistant factor to taxol [8] or other anticancer drugs [9]. Vascular endothelial growth factor (VEGF)-mediated survivin induction plays a pivotal role in preserving the microtubule network of tumor associated with vascular endothelial cells (ECs) and may act to shield tumor ECs from the apoptotic effects of chemotherapy [10]. Down-regulation of survivin expression in adriamycin-resistant HL-60/ADR cells reverses multidrug resistance [11]. Transcriptional downregulation of survivin expression by the GC-rich DNA-selective anticancer agent hedamycin is at least partially involved in hedamycin-mediated apoptotic cell killing [12]. In contrast, transcriptional induction of survivin expression by AT-rich DNA-selective ligand Hoechst33342 appears to be partially involved in drug resistance [13]. This is consistent with the ineffectiveness of Hoechst33342 to induce cell death. More recently, it was shown that estrogen-activated estrogen receptor α (ERα) interacts with p53 at its DNA binding site in the survivin promoter and de-represses p53-mediated inhibition of survivin expression and transcription [14]. Importantly, RNAi-mediated knockdown of ERα resulted in downregulation of survivin expression and sensitized staurosporine-induced apoptotic cell death, which was blocked by exogenous expression of survivin [14]. This study further indicates that modulation of survivin expression is likely an important mechanism for regulation of apoptosis and drug sensitivity in a more general term.
Additionally, many natural dietary components, such as resveratrol [3, 15–17], silibinin [15, 18, 19], Sulindac [15, 20, 21], selenium [22, 23], retinoid [24, 25] and vitamin D (e.g. calcitriol) [26] that have cancer-preventive and therapeutic effects, downregulate the expression of survivin in various types of cancer cells. This is consistent with the role of survivin in oncogenesis [27], and cancer progression, metastasis and relapse [28–32]. However, application of these findings for survivin to therapeutic drug combination is largely unexplored. Here, we report that T138067, a novel compound showing better antitumor activity against multiple drug resistance (MDR) tumor cells than taxol [33], induces survivin expression and thus, decreases its effectiveness on the induction of cancer cell death. However, sequential combination of a compound that inhibits survivin expression with T138067 provoked increased breast cancer cell death compared with each drug alone. The differential modulation of survivin expression by T138067 and survivin inhibitors appears to be attributed to opposing signaling modulation initiated by the compounds used. We propose a new concept that unique signaling cross-talk converged to the modulation of survivin expression may be applied to rational drug combination in the clinic as effective therapeutic regimens.
Materials and methods
Cell culture and reagents
MCF-7, ZR-75 and MDA-MB-231 breast cancer cell lines were maintained in DMEM, supplemented with 10% fetal bovine serum (Mediatech Cellgro, Herndon, VA) and penicillin (100 units/ml)/ streptomycin (0.1 μg/ml) (Invitrogen, Grand Island, NY) in a humidified incubator with 5% CO2 at 37°C. Cells were routinely subcultured every 3 – 4 days. T138067 (2-fluoro-1-methoxy-4-pentafluoro-phenyl-sulfonamido-benzene) was provided by Tularik Inc and SN-38 was from the Rustum laboratory (Roswell Park Cancer Institute). Resveratrol, LY294002 and PD98059 were purchased from Cayman Chemical through VWR (Rochester, NY). Antibodies for survivin (FL-142), Akt, and Erk1/2 (p44/42 MAPK) were purchased from Santa Cruz (Santa Cruz, CA). SB203580 and PD169316 were purchased from CalBiochem (La Jolla, CA). Antibodies for phospho-Akt (Ser473), phospho-p44/42 MAPK (Thr202/Tyr203), phospho-HER2/ErbB2 (Tyr877), HER2/ErbB2, phospho-p38 (Thr180/Tyr182) and p38 MAPK were purchased from Cell Signaling (Beverly, MA). Phosphatase inhibitor cocktail 1, monoclonal anti-actin antibody and goat peroxidase-conjugated anti-rabbit IgG antibody were purchased from Sigma (St. Louis, MO). U0126 was purchased from Promega. Lipofectamine™ 2000 reagents were purchased from Invitrogen (Carlsbad, CA).
Treatment and western blot
Cells were treated with T138067, SN-38 or resveratrol alone or in combination in medium containing 10% serum in all experiments in the presence or absence of various kinase inhibitors. Cells were then harvested for analysis. For Western blots, cells were washed with phosphate-buffered saline (PBS) and lysed at 4°C for 30 minutes in PBS containing 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 10 μg/ml phenylmethyl sulfonyl fluoride, and 20 μM leupeptin with or without the phosphatase inhibitor cocktail 1. Cell extracts were cleared by centrifugation at 15,000 g for 20 minutes at 4°. Fifty μg total proteins from each sample were heated at 95°C for 5 minutes after mixing with equal volume of 2 × SDS loading buffer. Samples were separated on 12 – 15% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) gels and electrotransferred to Pure Nitrocellulose Membranes (Bio-Rad, Hercules, CA). The membrane was blocked in 5% skim milk or BSA (for phospho-specific antibodies) in TBS-T buffer (20 mM Tris/HCl (pH 7.5), 0.137 M NaCl, and 0.05% Tween 20) at room temperature for 2–3 hours. The membranes were incubated with different primary antibodies in TBS-T overnight at 4°C in the range of dilutions from 1:500 to 1:1000. After washing with TBS-T, the membrane was incubated in 5% skim milk in TBS-T buffer containing a secondary antibody (1:5000), for 45–60 minutes at room temperature with shaking. Proteins of interest were detected by Chemiluminecence Reagent (Perkin Elmer, Waltham, MA) and visualized by autoradiography after various times (5–120 seconds) of exposure. For normalization of protein loading, the same membranes were stripped with stripping buffer (100 mM 2-mercaptoethanol, 2% sodium dodecyl sulphate, 62.5 mM Tris-HCl pH 6.7) and used for Western blot with a monoclonal antibody against actin at a dilution of 1:1000 by the same procedure.
Trypan blue exclusion staining for determination of cell viability
Cells for counting were collected by trypsinization/centrifugation and resuspended in PBS buffer. A small sample of the cell suspension was diluted in 0.4% (w/v) trypan blue (one sample a time since viable cells absorb trypan blue over time as well). A cover glass was centered over the hemacytometer chambers and one chamber was filled with the cell dilution using a Pasteur pipette. Stained (dead) and unstained (viable) cells were counted in each of the four corner and central squares under an inverted microscope using 100X magnification, respectively. Each cell sample was counted in this way for three times. The percentage of viable cells in each sample was calculated with the formula of “% viability = total viable cell numbers/total cell numbers × 100”.
Small interfering RNA (siRNA) preparation
Human survivin mRNA-specific RNA oligo-nucleotides with 3'-TT overhangs [92GCGC CUGCACCCCGGAGCG110TT (forward chain) and 110CGCTCCGGGGTGCAGGCGC92TT (reverse chain)] were chemically synthesized and purified by HPLC (Invitrogen, San Diego, CA). Equal moles of each RNA oligonucleotide were mixed together to a final concentration of 20 μM in annealing buffer (100 mM KAc, 30 mM HEPES-KOH, 2 mM MgAc2, pH 7.4). After denaturation at 90°C for 1 minute, the survivin siRNA mixture (designated SRi-2) was annealed at 37°C for 60 minutes and stored at -80°C for transfection experiments. A scramble RNA duplex (designated scraSRi) was also prepared as above for a negative control in this study. The scramble sequence [5′CAG UCG CGU UUG CGA CUG GTT (forward chain) and 5′CCA GUC GCA AAC GCG ACU GTT (reverse chain)] was not present in mammalian cells by BLAST search at NCBI. The effectiveness of survivin SRi-2 on survivin inhibition was 75–90% and confirmed in HeLa [34] and MCF-7 cells (Figure 8A).
Figure 8.
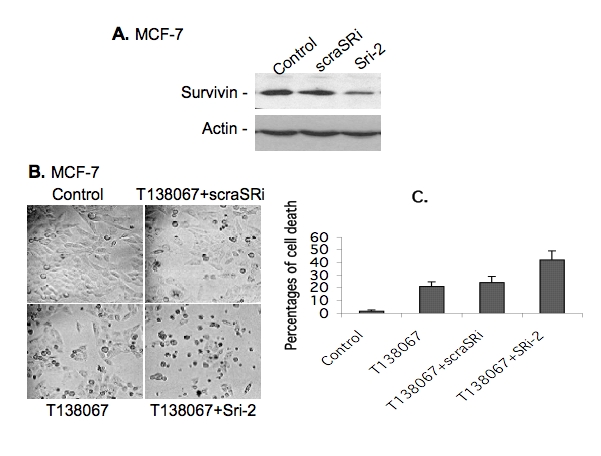
Inhibition of T138067-induced survivin expression by survivin-specific siRNA significantly increased cell death. A. Survivin-specific siRNA (Sri-2) inhibits survivin expression determined by western blots. Control: transfected with transfection reagent only; scraSRi: transfected with scramble RNA; and Sri-2: transfected with Sri-2. B. MCF-7 cells were plated in 12-well plates (105 cells/well) and grown for 24 hours. Cells were sequentially transfected with or without survivin siRNA (SRi-2) or scramble siRNA (scraSRi) for 24 hours and then treated with or without T138067 (100 nM) for 24 hours as shown, followed by photographing. C. Experimental conditions were the same as in A, cell death was analyzed by trypan blue exclusion assays as described in the Methods, data were plotted as a histogram and each bar represents the mean ± SD from three independent assays.
Transfection of siRNA and cell viability analysis
One day prior to transfection, 3 × 105 cells per well were seeded in 6-well plates containing culture medium without antibiotics. Cells at 40–60% confluence were transfected with the scramble (scraSRi) and survivin (SRi-2) siRNAs as follows. Serum-free DMEM (100 μl) containing 3 μg siRNAs were mixed with 100 μl serum-free DMEM containing 9 μl Lipofectamine™ 2000 reagents and held at room temperature. After the medium in a 6-well plate was replaced by serum-free DMEM (800 μl/well), the siRNA-Lipofectamine™2000 mixture prepared above was added to each well within 20–45 minutes after the mixture was prepared. Two to four hours later, 330 μl DMEM containing 30% fetal bovine serum was added into each well. Cells were treated with and without T138067 24 hours post transfection. Survivin expression was analyzed by Western blots and cell viability was analyzed microscopically 24 hours after T138067 treatment by trypan blue exclusion as described above.
DNA fragmentation assay
DNA fragmentation assay was performed using Cell Death Detection ELISAPlus assay kit (Roche) following the protocol recommended by the manufacturer. Briefly, MCF-7 and ZR-75 breast cancer cells (5000 cells per well) were seeded in triplicates in 96-well plates. Cell numbers per well at the beginning were decided based on the cell doubling rate that compatible with time point setting. Cells were then treated with and without T138067 and SN38, alone and in combination for 24 and 48 hours, respectively. Plates were centrifuged at 200g for 5–10 min and then, the medium in each well was removed. Cell pellets were then lysed with 200 μl lysis buffers per well (supplied in the kit). Cell lysates were centrifuged at 200g for 10 min after incubation for 30 min at room temperature. Twenty μl of supernatant from each well were dispensed into streptavidin-coated well-removable 96-well plates followed by addition of 80 μl of immuno-reagents. The immuno-reagent consisted of a mixture of biotin-labeled anti-histone antibody and HRP-conjugated anti-DNA antibody, which are major constituents of the nucleosomes. After a 2-hour incubation at room temperature with gentle shaking, unbound components were removed by washing with 250μl of 1X incubation buffer for 3 times. One hundred μl of HRP substrate (2,2′-azino-di-(3-ethylbenzthiazoline sulfonate (6)) diammonium salt, ABTS) was added to each well, and the plate was placed on a shaker at 250 rpm for color development. Measurements were made at 405 nm against an ABTS solution as a blank control (reference wavelength ∼490 nm) using an Ultra Microplate Reader (Bio-Tek Instruments). The absorbance value at 405nm represented the quantities of the DNA fragments of the apoptotic cells induced by treatment.
Results
Differential modulation of the expression of survivin by anticancer compounds
T138067 was shown to disrupt microtubule polymerization in cancer cells by covalently modifying β-tubulin [33]. Therefore, T138067 was shown more efficacious against MDR (multidrug resistance) gene-overexpressed tumor cells in both cell culture and mouse xenograft models than taxol and vinblastine [33]. However, we found that T138067 induces the expression of survivin in breast cancer cell lines in a time (Figure 1A) and concentration (Figure 1B, C) dependent manner, while it shows no effect on the expression of Bcl-2 (Figure 1A), suggesting its specificity. On the other hand, we found that SN-38, the active metabolite of pro-drug irinotecan, inhibits survivin expression in breast cancer cell lines (Figure 2). Intriguingly, a low concentration of SN-38 appears to more effectively downregulate survivin expression than its high concentration (Figure 2 B-D). Therefore, this compound can act as a survivin inhibitor for drug combinational breast cancer treatment.
Figure 1.

T138067 induces the expression of survivin. A. Time-dependent upregulation of survivin but not Bcl2 by T138067 in MCF-7 breast cancer cells. B. Concentration-dependent induction of survivin expression by T138067 in MCF-7 cells. C. Concentration-dependent induction of survivin by T138067 in ZR-75 breast cancer cells. Cells were treated with T138067 at the time and concentration as shown. Cells were lysed and analyzed for the expression of survivin and Bcl-2 by western blots. β–Actin was determined as an internal control for equal protein loading.
Figure 2.

SN-38 inhibits the expression of survivin. A. Time-dependent inhibition of survivin expression by SN38 in MCF-7 cells. Cells were treated with SN-38 (0.3 μM) for 2 hours and then without drug for additional hours as indicated. Cells were then lysed and survivin expression was determined by western blots. B. A low concentration of SN-38 shows a better inhibitory effect on survivin expression in comparison with a relative high concentration of SN-38. C and D. Different breast cancer cell lines were treated with SN-38 for 16 hours with different concentration of SN-38 as indicated, following by western blots. β–Actin was used in all experiments as internal controls (A –D).
Differential modulation of Akt and MAPK pathways by anticancer compounds
Our previous studies revealed that activation of Akt and Erk1/2 by taxol is involved in induction of survivin expression [7], while activation of p38 by vitamin D compounds inhibits survivin expression [26]. Therefore, we tested whether the modulation of survivin expression initiated by T138067 and SN-38 is associated with the modulation of these signaling molecules. Western blots indicated that treatment of MCF-7 cells with T138067 rapidly increases the phos-phorylation of Akt and Erk1/2 (Figure 3A and 3B), decreases the phosphorylation of p38 MAPK, and showed no effects on the phosphorylation of ErbB-2 (Figure 3C). In contrast, SN-38 treatment increases p38 MAPK phosphorylation, while this treatment showed no effect on the phosphorylation of ErbB-2 (Figure 4), indicating the specificity of these events.
Figure 3.

Upregulation of survivin by T138067 is associated with the modulation of the phosphorylation of Akt and MAPK molecules. A. T138067 increases Akt phosphorylation. B. T138067 increases Erk1/2 phosphorylation. C. T138067 decreases p38 MAPK phosphorylation but shows no effect on the phosphorylation of ErbB2. MCF-7 cells were treated with T138067 over the time indicated. Cells were then lysed and analyzed for phosphorylation of Akt, Erk1/2, p38 MAPK and ErbB-2 by western blots with the phospho-Akt-specific, phospho-Erk1/2-specific, phospho-p38-specific and phospho-ErbB2-specific antibodies. Total Akt and Erk1/2 and p38 proteins were monitored as internal controls.
Figure 4.
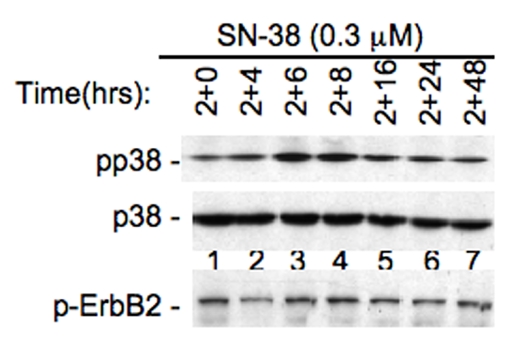
Inhibition of survivin by SN-38 is associated with increased p38 MAPK phosphorylation. MCF-7 cells were treated with SN-38 for 2 hrs and then without drug over the time indicated. Cells were then lysed and analyzed for the phosphorylation of p38 and ErbB2 by western blots using phospho-p38-specific or phospho-ErbB2-specific antibodies. Total p38 protein was monitored as an internal control.
Activation of Akt, Erk1/2 and p38 is involved in anticancer compounds-mediated modulation of survivin expression
To study whether the increased phos-phorylation of Akt and Erk1/2 by T138068 is required for T138067-induced survivin expression, MCF-7 breast cancer cells were treated for 24 hours with T138067 in the presence and absence of LY294002 (an inhibitor of the PI3K, which is a direct kinase upstream of Akt). Cells were then lysed and analyzed for survivin expression using western blots. As shown (Figure 5A), in the presence of LY294002, the effect of T138067 on survivin induction was abrogated. Similar results were obtained by inhibition of MEK (an immediate kinase upstream of Erk1/2) using PD98059 and U0126 (Figure 5B). These results indicate that both PI3K/Akt and MEK/Erk1/2 pathways were activated by T138067 and their activation was involved in T138067-induced survivin expression.
Figure 5.
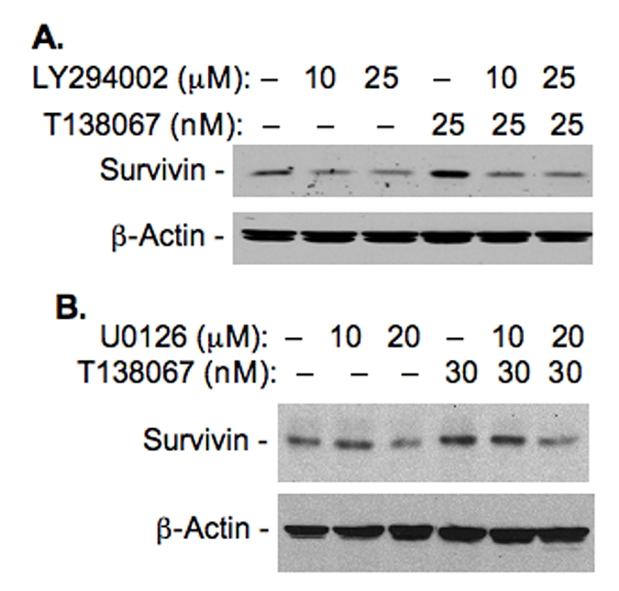
Inhibition of T138067-induced activation of Akt and Erk1/2 decreases T138067-mediated induction of survivin expression. A. MCF-7 breast cancer cells were treated with T138067 in the presence and absence of PI3K inhibitor LY294002. Cells were lysed 24 hours after treatment and analyzed for survivin expression by western blots as shown. B. MCF-7 cells were treated with T138067 in the presence and absence of MEK inhibitors, PD98059 and U0126 as shown. Cells were lysed 24 hours after treatment and analyzed for survivin expression by western blots. β-Action was used as internal controls for protein loading for both A and B.
To determine whether the increased phosphor-rylation of p38 MAKP induced by SN-38 is involved in SN38-mediated downregulation of survivin expression, MCF-7 cells were treated with SN-38 for 24 hours in the presence and absence of PD169316 and SB203580 (p38 MAPK-specific inhibitors). Cells were then lysed and analyzed for survivin expression using western blots. As shown (Figure 6), in the presence of p38 inhibitors the inhibition of survivin expression by SN-38 was alleviated, suggesting that activation of p38 by SN-38 treatment is at least in part involved in SN38-mediated inhibition of survivin expression.
Figure 6.
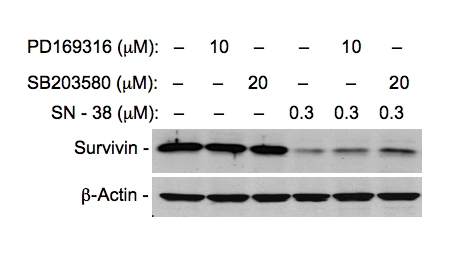
Inhibition of SN38-induced activation of p38 MAPK alleviates SN38-mediated inhibition of survivin expression. MCF-7 breast cancer cells were treated with SN-38 in the presence and absence of p38 kinase inhibitors as shown. Cells were lysed 24 hours after treatment and analyzed for survivin expression by western blots as shown. β-Action was used as an internal control for equal protein loading.
Inhibition of T138067-induced survivin expression by survivin inhibitors strikingly induced breast cancer cell death
The findings that T138067 upregulation and SN-38 downregulation of survivin expression in breast cancer cells raise the possibility that treatment of breast cancer cells by T138067 in combination with a low concentration of SN38 as a survivin-negative modulator might increase T138067 effectiveness to induce breast cancer cell death. To test this possibility, MCF-7 breast cancer cells were treated with T138067 in combination with a low concentration of SN-38. The rational combination treatment strikingly increased T138067 effectiveness inhibiting cell growth and inducing cell death when compared with either drug alone (Figure 7A and 7B). Similar results were obtained with ZR-75 breast cancer cells (Figure 7C). Alternatively, direct inhibition of T138067-induced survivin expression using validated survivin-specific small interfering RNA (siRNA) in cancer cells (Figure 8A) [34] resulted in a similar effect of induction of breast cancer cell death (Figure 8B and C). Importantly, resveratrol, a naturally occurring dietary compound found in grapes, wine, nuts and berries, downregulates survivin expression (Figure 9A), while it shows no inhibitory effect on the expression of Bcl-2, Bcl-XL, Bax and c-IAP1 [35]. Combination treatment of T138067 with resveratrol strikingly inhibited cell growth and induced cell death (Figure 9B). These results are consistent with our new concept that increased survivin expression during therapeutic drug treatment is an essential drug-resistant factor and inhibition of drug-induced survivin expression using a survivin-negative expression modulator strikingly induces cancer cell death.
Figure 7.

Increased induction of cell death by T138067 in the presence of a low concentration of SN-38. A. Cell morphologies were photographed under an inverted phase-contrast microscope. Cells were plated in cell cultural plates and grown for 24 h. Cells were sequentially treated with SN-38 for 16 hours at 30 nM and then treated with or without T138067 (30 nM) for 48 hours. B. Experimental conditions were as in A, cell death was analyzed by trypan blue exclusion assay descried in the method section. Data were plotted in a histogram. Each bar is the means ± SD from three independent assays. C. Experimental conditions and photographing were the same as in A with the exception of using ZR-75 breast cancer cells with lower drug concentrations as indicated.
Figure 9.
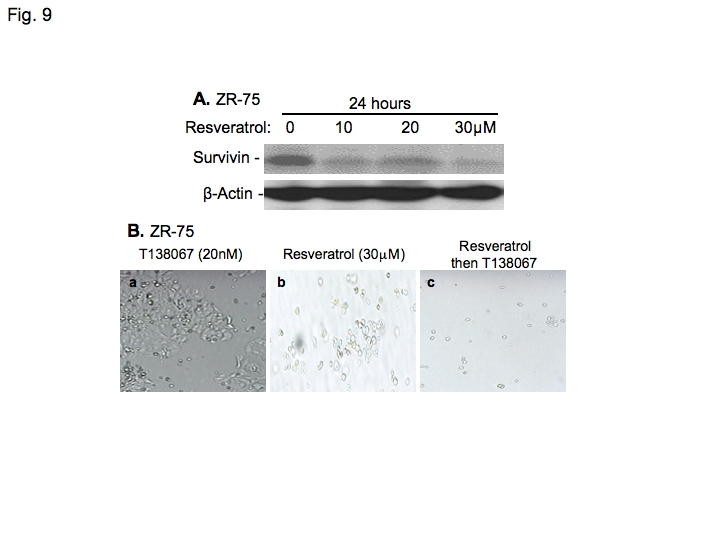
Resveratrol inhibits survivin expression and significantly enhances the effectiveness of T138067 to induce breast cancer cell death. A. ZR-75 breast cancer cells were treated with T138067 at different concentrations for 24 hours as shown. Cells were then lysed and analyzed by western blots to determine survivin expression. β-Action is an internal control for protein loading. B. ZR-75 cells were treated with T138067 or resveratrol alone or in combination. For combination treatment, cells were treated sequentially with resveratrol (30 μM) for 24 hours, followed by T138067 (20 nM) for 24 hours. Pictures were taken at 7 days after Resveratrol treatment.
Discussion
Chemotherapy plays an important role in cancer treatment. However, a challenging problem is that most types of cancer (if not all) can acquire drug resistance during chemotherapy. For example, it was shown that the efficacy of taxol is affected by expression of ATP-dependent efflux pumps (P-glycoprotein) to increase taxol efflux and lower intracellular drug concentrations [36], or by tubulin mutation [37, 38]. In contrast, resistance to cisplatin was shown to be due to reduced drug uptake [39, 40] and resistance to etoposide was attributed to activation of a DNA repair system [41, 42]. However, current studies have shown that upregulation of survivin is involved in tumor cell resistance to anticancer agents [7–9, 13, 43, 44] and ionizing radiation [45, 46] in different type of cancers. In contrast, downregulation of survivin expression contributes to cancer cell drug sensitivity [11, 12, 47]. These observations indicate that survivin expression in cancer cells is a broad drug resistant factor for different classes of chemotherapeutic drugs. Therefore, this led us to investigate whether we could use the modulation of survivin expression by therapeutic drugs as a novel parameter for rational drug combination treatment to overcome drug resistance.
In this study, we demonstrated that although the novel anti-tumor compound T138067 was demonstrated to be more efficacious against MDR gene-overexpressed tumor cells than taxol and vinblastine [33], T138067 strongly induced survivin expression as a potential inherent resistant factor to drug treatment. On the other hand, treatment of breast cancer cells with a low concentration of SN-38, the active metabolite of the pro-drug irinotecan, significantly downregulated survivin expression, which indicates that SN-38 may be a good survivin expression-negative modulator. Therefore, we tested our hypothesis that combination of T138067 with a survivin expression-negative modulator will significantly increase T138067 effectiveness to kill breast cancer cells. Our studies indicated that this is indeed the case (Figs. 7–9). We further investigated the molecular mechanism for the differential modulation of survivin expression by T138067 and SN-38 (Figs. 3–6). Our data revealed that T138067 upregulates the phosphorylation of Akt and Erk1/2, while decreases p38 MAPK phosphorylation (Figure 3). In contrast, SN-38 treatment of cancer cells induced p38 phosphorylation/activation (Figure 4). We further demonstrated that activation of PI3K/Akt and MEK/Erk pathways by T138067 is involved in T138067-mediated induction of survivin expression (Figure 5), while activation of p38 MAPK by SN-38 is at least partially involved in SN38-mediated survivin inhibition. These observations support the concept that the significant effect of T138067 and SN-38 combination treatment on massive breast cancer cell death likely comes from opposing drug-induced signals of cell survival vs. cell apoptosis that converges to the different modulation of survivin expression in cancer cells. We previously demonstrated that p38 MAPK is activated by vitamin D3 (VD3) treatment during VD3-mediated down-regulation of survivin in MCF-7E breast cancer cells, and downregulation of survivin is required for VD3-induced breast cancer cell G1 arrest and apoptosis induction [26]. Whereas, taxol-mediated activation of Akt and Erk1/2 is involved in upregulation of survivin expression and increase of taxol resistance [7]. These findings, together with the findings in this report, imply that modulation (induction or inhibition) of survivin expression by manipulating survival versus apoptotic signaling in cancer cells may represent a major mechanism that accounts for treatment-associated antitumor drug resistance or sensitivity, and support our new rational drug combination concept that utilizes the effect of therapeutic drugs on the modulation of survivin expression as a criterion for drug combination. We anticipate that if the concept of rational drug combination treatment based on therapeutic drug-induced modulation of survivin expression, will be demonstrated with more examples, this novel concept may result in significant consequences not only for extending our vision on drug action mechanisms but also for developing novel therapeutic approaches to improve cancer chemotherapy and drug resistance in the clinic. We have demonstrated that activation of both PI3K/Akt and MEK/Erk1/2 pathways are involved in T138067-induced expression of survivin and activation of p38 MAPK is at least partially involved in SN38-mediated inhibition of survivin. On the other hand, given that combination of T138067 with SN-38 or resveratrol significantly induces massive cell death, further studies on the detailed mechanism for the modulation of survivin expression by T138067 and SN-38 as well as whether SN-38 or resveratrol take a similar mechanism to inhibit survivin expression, will be required to further enrich this new area. Additionally, we should point out that combination of two compounds that differentially modulate survivin expression does not always obtain increased drug effectiveness as each compound alone. For example, we found that combination of T138067 with 5-Aza-2′-deoxycytidine that could inhibit survivin expression does not show a significant increase of cancer cell death (data not shown). Thus, the concept reported here could have exceptions.
Finally, we would like to point out that a Phase 2 clinical trial of T138067 in patients with malignant glioma was recently performed by the National Cancer Institute of Canada Clinical Trials Group [48]. Specifically, patients with recurrent anaplastic astrocytoma or glioblastoma multiforme were treated intravenously with weekly 330 mg/m2 of T138067-sodium. Treatment was continued until the patient experienced either unacceptable toxicity or progressive disease. Among the nineteen patients who entered the clinical trial, the result is that 1) no dose-limiting toxicity was seen; 2) pharmacokinetic results were consistent with those from nonglioma patients; and 3) no patients dropped out of the trial because of toxicity. Although three patients had stable disease with a median duration of 2.6 months, no responses were seen in the 15 eligible patients who completed at least one cycle. This result at least suggests that at this dose and schedule, T138067 does not show activity on the malignant glioma in these patients. To our view, this result may imply that application of T138067 as a mono-regimen treatment may not be effective enough. This could be resulted from its inherent induction of survivin expression, which could be eliminated by combinational treatment with a survivin expression-negative modulator such as YM155 [49] or these found in this study. Importantly, given that T138067 appears to be an antitumor compound with low toxicity to cancer patients [48], we would like to emphasize that the lack of activity to glioma may also be resulted from the fact that this compound has difficulty to pass through the blood brain barrier [50]. Thus, this compound is warranted for additional clinical trials on other type of cancer with or without a rational drug combination using a survivin expression-negative modulator.
In summary, we provided examples in this report to demonstrate that modulation of survivin expression by antitumor compounds may be an important drug action mechanism to control drug sensitivity or resistance, and may be used as a novel rational drug combination regimen for drug combinational treatment in the clinic.
Acknowledgments
We thank Dr. Youcef M. Rustum for his constructive comments and suggestions on this work as well as his generosity for providing SN-38 compounds. This work was sponsored by the research grant (BCTR63806) from Susan G. Komen Breast Cancer Foundation and the Grants from NIH/NCI (CA109481, CA133241) to FL.
References
- 1.Altieri DC, Marchisio PC, Marchisio C. Survivin apoptosis: an interloper between cell death and cell proliferation in cancer. Lab Invest. 1999;79:1327–1333. [PubMed] [Google Scholar]
- 2.Li F. Survivin Study: What is the next wave? J Cell Physiol. 2003;197:8–29. doi: 10.1002/jcp.10327. [DOI] [PubMed] [Google Scholar]
- 3.Li F, Ling X. Survivin Study: An update of “What is the next wave?”. J Cell Physiol. 2006;208:476–486. doi: 10.1002/jcp.20634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grossman D, Altieri DC. Drug resistance in melanoma: mechanisms, apoptosis, and new potential therapeutic targets. Cancer Metastasis Rev. 2001;20:3–11. doi: 10.1023/a:1013123532723. [DOI] [PubMed] [Google Scholar]
- 5.Zaffaroni N, Pennati M, Daidone MG. Survivin as a target for new anticancer interventions. J Cell Mol Med. 2005;9:360–372. doi: 10.1111/j.1582-4934.2005.tb00361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Altieri DC. Targeted therapy by disabling crossroad signaling networks: the survivin paradigm. Mol Cancer Ther. 2006;5:478–482. doi: 10.1158/1535-7163.MCT-05-0436. [DOI] [PubMed] [Google Scholar]
- 7.Ling X, Bernacki RJ, Brattain MG, Li F. Induction of survivin expression by taxol (paclitaxel) is an early event which is independent on taxol-mediated G2/M arrest. J Biol Chem. 2004;279:15196–15203. doi: 10.1074/jbc.M310947200. [DOI] [PubMed] [Google Scholar]
- 8.Zhang M, Mukherjee N, Bermudez RS, Latham DE, Delaney MA, Zietman AL, Shipley WU, Chakravarti A. Adenovirus-mediated inhibition of survivin expression sensitizes human prostate cancer cells to paclitaxel in vitro and in vivo. Prostate. 2005;64:293–302. doi: 10.1002/pros.20263. [DOI] [PubMed] [Google Scholar]
- 9.Tirro E, Consoli ML, Massimino M, Manzella L, Frasca F, Sciacca L, Vicari L, Stassi G, Messina L, Messina A, Vigneri P. Altered expression of c-IAP1, survivin, and Smac contributes to chemotherapy resistance in thyroid cancer cells. Cancer Res. 2006;66:4263–4272. doi: 10.1158/0008-5472.CAN-05-3248. [DOI] [PubMed] [Google Scholar]
- 10.Tran J, Master Z, Yu JL, Rak J, Dumont DJ, Kerbel RS. A role for survivin in chemoresistance of endothelial cells mediated by VEGF. Proc Natl Acad Sci USA. 2002;99:4349–4354. doi: 10.1073/pnas.072586399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L, Zhang GM, Feng ZH. Downregulation of survivin expression reversed multidrug resistance in adriamycin-resistant HL-60/ADR cell line. Acta Pharmacol Sin. 2003;24:1235–1240. [PubMed] [Google Scholar]
- 12.Wu J, Ling X, Pan D, Apontes P, Song L, Liang P, Altieri DC, Beerman T, Li F. Molecular mechanism of inhibition of survivin transcription by the GC-rich sequence selective DNA-binding antitumor agent, hedamycin: evidence of survivin downregulation associated with drug sensitivity. J Biol Chem. 2005;280:9745–9751. doi: 10.1074/jbc.M409350200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu J, Apontes P, Song L, Liang P, Yang L, Li F. Molecular mechanism of upregulation of survivin transcription by the AT-rich DNA-binding ligand, Hoechst33342: evidence for survivin involvement in drug resistance. Nucleic Acids Res. 2007;35:2390–2402. doi: 10.1093/nar/gkm149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sayeed A, Konduri SD, Liu W, Bansal S, Li F, Das GM. Estrogen receptor alpha inhibits p53-mediated transcriptional repression: implications for the regulation of apoptosis. Cancer Res. 2007;67:7746–7755. doi: 10.1158/0008-5472.CAN-06-3724. [DOI] [PubMed] [Google Scholar]
- 15.Li F. Role of survivin in cancer chemoprevention. Assay Designs-Simply Science. 2005;1:2–6. [Google Scholar]
- 16.Riles WL, Erickson J, Nayyar S, Atten MJ, Attar BM, Holian O. Resveratrol engages selective apoptotic signals in gastric adenocarcinoma cells. World J Gastroenterol. 2006;12:5628–5634. doi: 10.3748/wjg.v12.i35.5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu Y, Rahlfs S, Mersch-Sundermann V, Becker K. Resveratrol modulates mRNA transcripts of genes related to redox metabolism and cell proliferation in non-smallcell lung carcinoma cells. Biol Chem. 2007;388:207–219. doi: 10.1515/BC.2007.023. [DOI] [PubMed] [Google Scholar]
- 18.Mallikarjuna G, Dhanalakshmi S, Singh RP, Agarwal C, Agarwal R. Silibinin protects against photocarcinogenesis via modulation of cell cycle regulators, mitogen-activated protein kinases, and Akt signaling. Cancer Res. 2004;64:6349–6356. doi: 10.1158/0008-5472.CAN-04-1632. [DOI] [PubMed] [Google Scholar]
- 19.Singh RP, Dhanalakshmi S, Agarwal C, Agarwal R. Silibinin strongly inhibits growth and survival of human endothelial cells via cell cycle arrest and downregulation of survivin, Akt and NF-kappaB: implications for angioprevention and antiangiogenic therapy. Oncogene. 2005;24:1188–1202. doi: 10.1038/sj.onc.1208276. [DOI] [PubMed] [Google Scholar]
- 20.Scheper MA, Sauk JJ, Nikitakis NG. COX-independent antineoplastic effects of sulindac in oral cancer are mediated by survivin down-regulation. Anticancer Res. 2006;26:4103–4113. [PubMed] [Google Scholar]
- 21.Jin HO, Yoon SI, Seo SK, Lee HC, Woo SH, Yoo DH, Lee SJ, Choe TB, An S, Kwon TJ, Kim JI, Park MJ, Hong SI, Park IC, Rhee CH. Synergistic induction of apoptosis by sulindac and arsenic trioxide in human lung cancer A549 cells via reactive oxygen species-dependent down-regulation of survivin. Biochem Pharmacol. 2006;72:1228–1236. doi: 10.1016/j.bcp.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 22.Azrak RG, Frank CL, Ling X, Slocum HK, Li F, Foster BA, Rustum YM. The mechanism of methylselenocysteine and docetaxel synergistic activity in prostate cancer cells. Mol Cancer Ther. 2006;5:2540–2548. doi: 10.1158/1535-7163.MCT-05-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chun JY, Hu Y, Pinder E, Wu J, Li F, Gao AC. Selenium inhibition of survivin expression by preventing Sp1 binding to its promoter. Mol Cancer Ther. 2007;6:2572–2580. doi: 10.1158/1535-7163.MCT-07-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christine Pratt MA, Niu M, White D. Differential regulation of protein expression, growth and apoptosis by natural and synthetic retinoids. J Cell Biochem. 2003;90:692–708. doi: 10.1002/jcb.10682. [DOI] [PubMed] [Google Scholar]
- 25.Pratt MA, Niu MY, Renart LI. Regulation of survivin by retinoic acid and its role in paclitaxel-mediated cytotoxicity in MCF-7 breast cancer cells. Apoptosis. 2006;11:589–605. doi: 10.1007/s10495-006-4603-7. [DOI] [PubMed] [Google Scholar]
- 26.Li F, Ling X, Huang H, Brattain L, Apontes P, Wu J, Binderup L, Brattain MG. Differential regulation of survivin expression and apoptosis by vitamin D(3) compounds in two isogenic MCF-7 breast cancer cell sublines. Oncogene. 2005;24:1385–1395. doi: 10.1038/sj.onc.1208330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li F. Role of survivin and its splice variants in tumorigenesis. Br J Cancer. 2005;92:212–216. doi: 10.1038/sj.bjc.6602340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyachi K, Sasaki K, Onodera S, Taguchi T, Nagamachi M, Kaneko H, Sunagawa M. Correlation between survivin mRNA expression and lymph node metastasis in gastric cancer. Gastric Cancer. 2003;6:217–224. doi: 10.1007/s10120-003-0255-2. [DOI] [PubMed] [Google Scholar]
- 29.Wobser M, Keikavoussi P, Kunzmann V, Weininger M, Andersen MH, Becker JC. Complete remission of liver metastasis of pancreatic cancer under vaccination with a HLA-A2 restricted peptide derived from the universal tumor antigen survivin. Cancer Immunol Immunother. 2006;55:1294–1298. doi: 10.1007/s00262-005-0102-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marioni G, Bertolin A, Giacomelli L, Marchese-Ragona R, Savastano M, Calgaro N, Marino F, De Filippis C, Staffieri A. Expression of the apoptosis inhibitor protein Survivin in primary laryngeal carcinoma and cervical lymph node metastasis. Anticancer Res. 2006;26:3813–3817. [PubMed] [Google Scholar]
- 31.Thomas J, Liu T, Cotter MA, Florell SR, Robinette K, Hanks AN, Grossman D. Melanocyte Expression of Survivin Promotes Development and Metastasis of UV-Induced Melanoma in HGF-Transgenic Mice. Cancer Res. 2007;67:5172–5178. doi: 10.1158/0008-5472.CAN-06-3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ouhtit A, Matrougui K, Bengrine A, Koochekpour S, Zerfaoui M, Yousief Z. Survivin is not only a death encounter but also a survival protein for invading tumor cells. Front Biosci. 2007;12:1260–1270. doi: 10.2741/2144. [DOI] [PubMed] [Google Scholar]
- 33.Shan B, Medina JC, Santha E, Frankmoelle WP, Chou TC, Learned RM, Narbut MR, Stott D, Wu P, Jaen JC, Rosen T, Timmermans PB, Beckmann H. Selective, covalent modification of beta-tubulin residue Cys-239 by T138067, an antitumor agent with in vivo efficacy against multidrug-resistant tumors. Proc Natl Acad Sci USA. 1999;96:5686–5691. doi: 10.1073/pnas.96.10.5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ling X, Li F. Silencing of antiapoptotic survivin gene by multiple approaches of RNA interference technology. BioTechniques. 2004;36:450–454. 456–460. doi: 10.2144/04363RR01. [DOI] [PubMed] [Google Scholar]
- 35.Hayashibara T, Yamada Y, Nakayama S, Harasawa H, Tsuruda K, Sugahara K, Miyanishi T, Kamihira S, Tomonaga M, Maita T. Resveratrol Induces Downregulation in Survivin Expression and Apoptosis in HTLV-1-Infected Cell Lines: A Prospective Agent for Adult T Cell Leukemia Chemotherapy. Nutr Cancer. 2002;44:193–201. doi: 10.1207/S15327914NC4402_12. [DOI] [PubMed] [Google Scholar]
- 36.Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 37.Schibler MJ, Cabral F. Taxol-dependent mutants of Chinese hamster ovary cells with alterations in alpha-and beta-tubulin. J Cell Biol. 1986;102:1522–1531. doi: 10.1083/jcb.102.4.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giannakakou P, Sackett DL, Kang YK, Zhan Z, Buters JT, Fojo T, Poruchynsky MS. Paclitaxel-resistant human ovarian cancer cells have mutant beta-tubulins that exhibit impaired paclitaxel-driven polymerization. J Biol Chem. 1997;272:17118–17125. doi: 10.1074/jbc.272.27.17118. [DOI] [PubMed] [Google Scholar]
- 39.Shen D, Pastan I, Gottesman MM. Crossresistance to methotrexate and metals in human cisplatin-resistant cell lines results from a pleiotropic defect in accumulation of these compounds associated with reduced plasma membrane binding proteins. Cancer Res. 1998;58:268–275. [PubMed] [Google Scholar]
- 40.Shen DW, Goldenberg S, Pastan I, Gottesman MM. Decreased accumulation of [14C]carboplatin in human cisplatin-resistant cells results from reduced energy-dependent uptake. J Cell Physiol. 2000;183:108–116. doi: 10.1002/(SICI)1097-4652(200004)183:1<108::AID-JCP13>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 41.Levenson VV, Davidovich IA, Roninson IB. Pleiotropic resistance to DNA-interactive drugs is associated with increased expression of genes involved in DNA replication, repair, and stress response. Cancer Res. 2000;60:5027–5030. [PubMed] [Google Scholar]
- 42.Jacob S, Aguado M, Fallik D, Praz F. The role of the DNA mismatch repair system in the cytotoxicity of the topoisomerase inhibitors camptothecin and etoposide to human colorectal cancer cells. Cancer Res. 2001;61:6555–6562. [PubMed] [Google Scholar]
- 43.Ikeguchi M, Liu J, Kaibara N. Expression of survivin mRNA and protein in gastric cancer cell line (MKN-45) during cisplatin treatment. Apoptosis. 2002;7:23–29. doi: 10.1023/a:1013556727182. [DOI] [PubMed] [Google Scholar]
- 44.Zaffaroni N, Pennati M, Colella G, Perego P, Supino R, Gatti L, Pilotti S, Zunino F, Daidone MG. Expression of the anti-apoptotic gene survivin correlates with taxol resistance in human ovarian cancer. Cell Mol Life Sci. 2002;59:1406–1412. doi: 10.1007/s00018-002-8518-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Asanuma K, Kobayashi D, Furuya D, Tsuji N, Yagihashi A, Watanabe N. A role for survivin in radioresistance of pancreatic cancer cells. Jpn J Cancer Res. 2002;93:1057–1062. doi: 10.1111/j.1349-7006.2002.tb02483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chakravarti A, Zhai GG, Zhang M, Malhotra R, Latham DE, Delaney MA, Robe P, Nestler U, Song Q, Loeffler J. Survivin enhances radiation resistance in primary human glioblastoma cells via caspase-independent mechanisms. Oncogene. 2004;23:7494–7506. doi: 10.1038/sj.onc.1208049. [DOI] [PubMed] [Google Scholar]
- 47.Notarbartolo M, Cervello M, Dusonchet L, Cusimano A, D'Alessandro N. Resistance to diverse apoptotic triggers in multidrug resistant HL60 cells and its possible relationship to the expression of P-glycoprotein, Fas and of the novel anti-apoptosis factors IAP (inhibitory of apoptosis proteins) Cancer Lett. 2002;180:91–101. doi: 10.1016/s0304-3835(01)00834-5. [DOI] [PubMed] [Google Scholar]
- 48.Kirby S, Gertler SZ, Mason W, Watling C, Forsyth P, Aniagolu J, Stagg R, Wright M, Powers J, Eisenhauer EA. Phase 2 study of T138067-sodium in patients with malignant glioma: Trial of the National Cancer Institute of Canada Clinical Trials Group. Neuro-oncol. 2005;7:183–188. doi: 10.1215/S1152851704000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakahara T, Takeuchi M, Kinoyama I, Minematsu T, Shirasuna K, Matsuhisa A, Kita A, Tominaga F, Yamanaka K, Kudoh M, Sasamata M. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007;67:8014–8021. doi: 10.1158/0008-5472.CAN-07-1343. [DOI] [PubMed] [Google Scholar]
- 50.Rubenstein SM, Baichwal V, Beckmann H, Clark DL, Frankmoelle W, Roche D, Santha E, Schwender S, Thoolen M, Ye Q, Jaen JC. Hydrophilic, pro-drug analogues of T138067 are efficacious in controlling tumor growth in vivo and show a decreased ability to cross the blood brain barrier. J Med Chem. 2001;44:3599–3605. doi: 10.1021/jm000478d. [DOI] [PubMed] [Google Scholar]