

Abstract
The new paediatric European Union (EU) regulation and the consequent demand for paediatric studies on one hand and the ethical need for minimizing the burden of studies in children on the other hand necessitate optimal techniques in the assessment of safety/efficacy and use of drugs in children. Modelling and simulation (M&S) is one way to circumvent some difficulties in developing medicinal products in children. M&S allows the quantitative use of sparse sampling, characterization and prediction of pharmacokinetics/ pharmacodynamics (PK/PD), extrapolation from adults to children, interpolation between paediatric age subsets, optimal use of scientific literature and in vitro/preclinical data. Together, industry, academia and regulators recognize the usefulness of modelling and simulation in this setting. However, even if M&S is an emerging science, its integration in the EU regulatory decision making is for the time being deficient and M&S expertise is concentrated in big pharmaceutical companies and academic institutions. The European Medicines Agency, acknowledging all the above conditions, organized and hosted a Workshop on Modelling in Paediatric Medicines. The article presents the personal views of the authors on the issues presented and discussed in the workshop. We attempt to identify the regulatory framework for the use of M&S in paediatric medicinal development and to make proposals for model-based paediatric medicinal development. The objective is to open the discussion between industry, academia, paediatricians and regulators on the optimal use of M&S in paediatric medicinal development.
Keywords: modelling and simulation, paediatric drug development, regulatory science
WHAT IS ALREADY KNOWN ABOUT THE SUBJECT
The use of modelling and simulation (M&S) in paediatric drug development has been the subject of numerous scientific publications in the last decade.
Many scientific teams have elaborated on the methodology and provided successful examples of the added value of M&S in this area.
Furthermore, regulatory bodies and the US Food and Drug Administration in particular have provided guidance on good modelling and simulation practice and on the role of M&S in drug development.
WHAT THIS STUDY ADDS
This paper attempts to position M&S in the European regulatory environment based on European Medicines Agency (EMEA) guidelines.
It presents the personal views of the authors on the issues discussed in the EMEA workshop on modelling in paediatric medicines (14–15 April 2008) [1].
It proposes an algorithm for the practical implementation of M&S in paediatric drug development and a forum for further discussions.
Modelling and simulation in drug development
Modelling and simulation (M&S) is a methodology widely used to support drug development. Modelling is the science of using mathematical language to describe and quantify a system. Simulation refers to the use of these models to make quantitative predictions.
M&S methodology is based on the concept of data collection (in vitro, literature, preclinical, clinical), assumption testing (attempt to elucidate the data on the basis of mathematical/statistical models), learning (assumptions retained that are informative of the test system), prediction (simulation of data based on the underlying assumptions that can be used to predict and optimize future experimental outcome) and confirmation of model assumptions (when compared with external data). This exercise ideally follows the life cycle of a product and evolves from a purely exploratory context in the early phases of the development to a more robust regulatory framework when it is used to support claims. Similarly, as knowledge of the product accumulates the M&S evolves from a retrospective data-driven to prospective assumption-confirming and quantification exercise.
Model-based drug development involves intensive calculations, which are performed by specific algorithms. Each one of the components of the M&S methodology (data, model, algorithm, software, interpretation of the output and prediction) is equally important.
Mainly but not exclusively, pharmacokinetic/pharmacodynamic (PK/PD) models are used to support model-based drug development. The two main PK/PD modelling approaches used in drug development are the top down {classical [population (POP) or individual] PK/PD modelling}, which is data driven, and the bottom up [physiologically based pharmacokinetics (PBPK)–PD, mechanistic modelling], which originates from physiology, pharmacology and mechanistic information about the system. Both approaches are complementary and have a wide range of applications such as learning, decision making, study optimization and analysis tools in lead optimization, candidate drug selection, first in man, clinical PK/PD and safety/efficacy studies.
Good M&S practices
It is not within the scope of this paper to define an extensive list of recommendations for good modelling practice [2–4]; however, some general principles can be listed.
General strategy: it is proposed to conduct the M&S exercise throughout the drug life cycle (from discovery, throughout development to post authorization) with different steps identified to ask different questions and re-challenge and refine the model [5, 6].
For a robust M&S exercise it is considered important to define the following prospectively:
The objectives of the M&S exercise.
The data to be used for the model building.
The assumptions and their pharmacological and physiological rationale.
The model-building methodology.
Model qualification. The qualification of the model should ideally include diagnostic plots, re-sampling techniques, sensitivity analysis, comparison with internal and external data, to ensure that the model is fit for purpose. All modelling assumptions should be challenged to ensure that interpretation is robust.
In addition to a well-established strategy, the tools (i.e. software, algorithms) used for data management and M&S should be adequately validated.
Good modelling practice will help build confidence in the modelling and simulation results and will raise the weight of evidence of a M&S exercise during the regulatory benefit–risk assessment of a medicinal product. As a rule when the stakes are high, modelling needs to be held to a high standard, and the acceptability of a M&S exercise will be based on both the objectives of the exercise and the measures taken to avoid bias.
Defining the regulatory framework for M&S in paediatrics
M&S is a methodology widely used in the exploratory context with great acceptance from both industry and regulators. However, the pivotal use of M&S, i.e. to replace clinical trials or to support regulatory claims, is controversial. This position is not particularly contentious for experimental situations where conduct of large pivotal trials is possible, i.e. where M&S can be used as an exploratory technique, but cannot substitute pivotal trials as a basis for approval. In the case of paediatrics, where ethical and practical constrains necessitate smaller trials, M&S could have a more important role in the regulatory assessment. Since there are no European regulatory guidelines on the pivotal use of M&S, we will propose a regulatory framework to support M&S as a primary degree of evidence based on current methodological guidance documents.
Regulatory science is the science of optimizing drug evaluation. The regulatory risk–benefit assessment is based on the degree of evidence that is gathered throughout the drug development process. Experimental data reflecting the likely use of a product in clinical practice (i.e. adequately powered Phase III randomized controlled trials) provide the most robust grounds for the risk–benefit assessment of a new medicinal product. Ideally, multiple pivotal trials should provide consistent positive results. However, the extent of confirmatory data needed will depend upon what is established for the product in earlier stages and what is known about related products. The minimum requirement is generally one controlled study with statistically compelling and clinically relevant results [7]. In the case where a full development programme can be conducted, M&S could provide an additional degree of evidence by compiling information from observational studies and published data and optimizing the conduct/design of the pivotal trials. Although M&S cannot usually replace pivotal trials, evidence from M&S and one pivotal trial could in some circumstances support a robust risk–benefit assessment [8, 9].
The framework for collectively analysing data from multiple sources, including bibliographic data, and using this analysis as pivotal evidence is already available in the context of meta-analysis. According to the International Conference on Harmonisation E 9 [10], ‘A confirmatory trial is an adequately controlled trial in which the hypotheses are stated in advance and evaluated’. However, in the same guideline it is stated that: ‘Under exceptional circumstances a meta analytic approach may also be the most appropriate way, or the only way, of providing sufficient overall evidence of efficacy via an overall hypothesis test. When used for this purpose the meta-analysis should have its own prospectively written protocol’. Modelling, similar to meta-analysis, if used as regulatory pivotal evidence, should preferably be performed according to a pre-specified protocol. It is assumed that at this stage the accumulated knowledge on the medicinal product would be enough to preset the assumptions to be included and confirmed by the modelling analysis. Regulators in principle do not favour retrospective analyses being used as pivotal evidence. However, we would propose that, similar to a meta-analysis, a retrospective modelling exercise could be accepted for regulatory evidence [7] provided specific prerequisites are satisfied. These are in parallel with the regulatory prerequisites of retrospective meta-analyses: a clearly specified model-building methodology, sensitivity analysis demonstrating robustness of the exercise, justification of unbiased selection of the studies, no statistically significant heterogeneity and similar structural models based on the individual studies. Consequently, the modelling exercise, when confirmatory, could be planned, executed and evaluated as a meta-analysis.
The rationale for using modelling techniques in data-limited situations, where extensive Phase III data are not obtainable, is clearly stated in the guideline on clinical trials in small populations [11]: ‘Studies with few patients are often perceived as presenting a rather simple situation: there is not much information (data) and so simple (often descriptive) analyses are all that are warranted. It seems quite counterintuitive therefore that for “simple” situations more complex approaches should be applied but this is exactly what is necessary. Crude (simple) methods may often be adequate when we have huge amounts of data – but when there are very few data, it is imperative that the most efficient and informative analytical methods should be used. Many of these methods involve “statistical modelling”. Such models usually make assumptions about the data or the form of the treatment effect. With few data, these assumptions may not be testable or verifiable. However, assumptions add to the data so that more complex statistical models give us more information than simple descriptive statistics’.
The need for new approaches such as M&S in the paediatric setting is acknowledged by the regulators [12]: ‘Sponsors are encouraged to explore new approaches in the development of drugs for the paediatric population’.
Proposals for model-based paediatric medicinal development
Medicinal products not authorized for paediatric use
Medicinal products intended to cure diseases occurring in both adults and paediatric patients (extrapolation from adult safety/efficacy data)
In the European Union (EU), a Paediatric Investigation Plan (PIP) should be submitted by the end of adult Phase I trials for new medicinal products. At this stage little is known about the PK, PD, efficacy and safety of the experimental drug. However, communication can be initiated with the regulatory authorities at this stage and further PIP amendments can be proposed. M&S can offer the basis for assumption testing and decision making at this early stage. Models can be refined throughout the adult and paediatric drug development. Regulators propose to consult the Food and Drug Administration (FDA) paediatric decision tree [13] before planning paediatric studies.
Paediatric decision tree (Figure 1)
Figure 1.
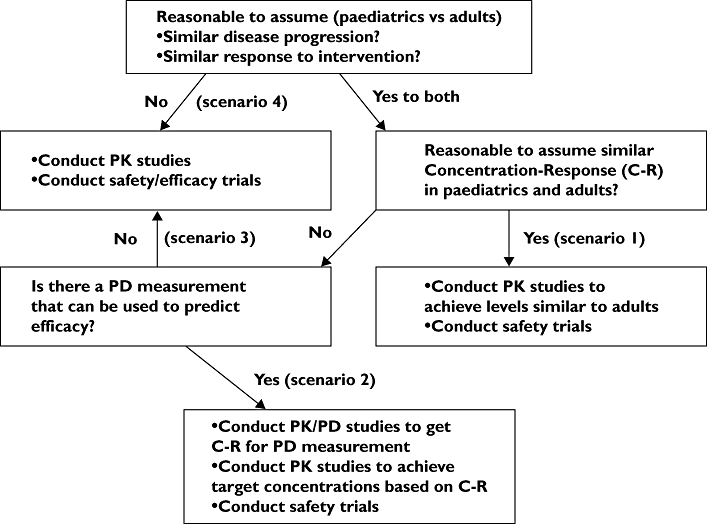
Paediatric study decision tree with identified scenarios
The paediatric decision tree established by the FDA identifies the studies required to bridge adult and paediatric data, but also data between different paediatric age groups or subsets.
Role of M&S in bridging adult and paediatric data (Figure 2)
Figure 2.
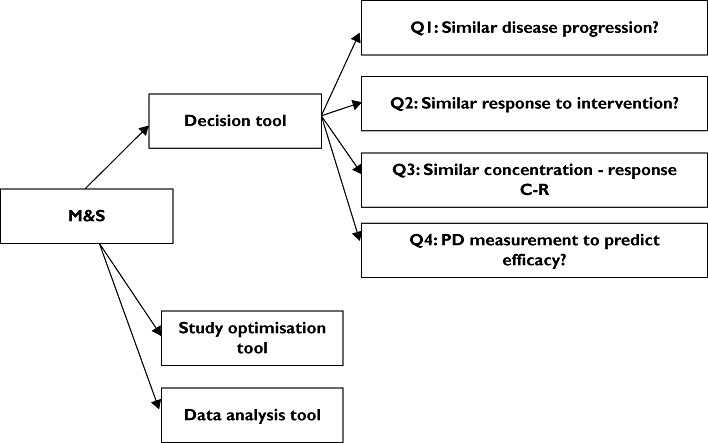
Modelling and simulation role in bridging adult and paediatric data
The role of M&S in paediatric medicinal development is summarized in Figure 2. It is proposed to use M&S as a tool to navigate through the paediatric decision tree, optimize the paediatric studies and analyse the data from clinical studies in order to address the key questions on pharmacotherapy in children.
Decision tool
M&S could help in answering the key questions identified in the paediatric decision tree.
Q1: Disease models can be used to investigate and compare disease progression between adults and paediatrics. This area is still in its infancy. Disease models can be built based on adult and paediatric bibliographic and/or primary in-house data. The comparison of disease progression between adults and paediatric populations could be improved by comparing disease progression models. Furthermore, disease progression models could give more insight into the time course of the disease and help in the construction of disease model databases that could be used to the benefit of drug development and public health.
Q2: The response to intervention could be better quantified and compared between treatment groups (paediatric or adults) with the use of disease response models. These models on top of the disease models include an assumption on the potential interaction of the drug with the natural course of the disease. This assumption could be hypothesized, data driven, or mechanism–physiology/pharmacology based. In any case, models could provide the basis for a more accurate comparison between treatment groups because they can disentangle and quantify the different effects. In addition, disease response models could be used to simulate future therapeutic outcome and combined with a PK/PD model make recommendations for dose adjustment and optimal drug development in children.
Q3: PD models could be used to investigate the concentration–effect relationship in adult and paediatrics. The models make it possible to disentangle the PK from the PD effects of the drug and enable the characterization of the shape of the concentration–effect curve. The comparison of the structural model, the statistical and error model between groups could give more insight into the differences between adults and paediatrics. Furthermore, different modelling techniques like POP–PK/PD, PBPK–PD could make optimal use of the available data and minimize the burden of studies in children needed to characterize the concentration–effect relationship.
Q4: Modelling could support and make the link between a PD biomarker and efficacy. The use of qualified biomarkers is of key importance in drug development. Models could investigate the mechanistic basis for selected biomarkers, facilitate the analysis of biomarker data and optimize the studies required for qualification. Moreover, models could estimate the quantitative relationships between PD biomarkers and efficacy, thus serving as the decision tool for whether or not to conduct efficacy trials.
Study optimization and analysis tool
The use of M&S in different scenarios identified in the paediatric decision tree will be explored.
Scenario 1
Questions to be answered by the development programme:
Paediatric dose
Safety in children
PBPK models incorporate in vitro, preclinical, bibliographic and in vivo adult data scaled with appropriate developmental factors and could be used to make predictions of the concentration profiles in different paediatric age groups. Based on simulations, a first-in-children dose could be recommended. PBPK models not only offer a platform for data gathering and prediction, but also initiate an iterative learning process that refines the in vitro, preclinical and clinical experiments and promotes the understanding of the mechanisms of drug absorption, distribution, metabolism, excretion (ADME) and the effect of maturation on all these processes [14]. The disadvantage of these models is the difficulty of extrapolating from in vitro data and the heterogeneity of the bibliographic sources. It is important to challenge assumptions made and to reflect properly the uncertainty inherent in the information on which the model is based.
POP–PK models are data-driven compartmental models that describe the dose–concentration relationship by combining a structural, statistical and random component. Similar to PBPK models, POP–PK models could incorporate extrapolation factors to predict PK in children based on adult PK data or PK data in different age subsets. Establishing these factors to account for the developmental differences in ADME is a challenging exercise involving covariate (i.e. patient-specific variables) testing algorithms [15]. In an effort to standardize this approach, the inclusion of allometric scaling [16] to account for the size effect, of a sigmoid Emax model (Hill equation) to account for the maturation effect and of a pathological variation function in clearance could be proposed [17, 18]. The volume of distribution could be also related to size by an allometric model. However, more research is required in this field and the use of M&S could enhance the understanding of these processes and the standardization of the extrapolation factors. POP–PK models can be used for paediatric dose recommendations in the context of both first-in-children and dose-finding studies, but also for the optimization of paediatric PK studies. POP–PK models enable the use of sparse sampling, lowering the burden of sampling in children. Furthermore, M&S or approaches based on Fisher information matrix [19] can be used for sampling design optimization.
The POP–PK/PD models describe quantitative and qualitative relationships between doses, exposure and PD effects. These models could be supportive in first-in-children study and in paediatric dose recommendations, but since in this scenario PK is considered surrogate for efficacy, they are used as a decision/assumption testing tool for the surrogacy of PK.
Toxicity/adverse events (AE) models ideally combine quantitative and qualitative information on the dose, exposure, PD and AE relationships. The prediction of pharmacological toxicity is possible based on the models and the available preclinical and adult clinical data. However, the inclusion in the models of adverse causality based on pharmacological activity other than that intended requires a higher degree of assumptions and is a data-demanding exercise. Toxicity/AE models apart from first-in-children and paediatric dose recommendations could be used to optimize the design of the safety trials by simulating different scenarios and incorporating information from preclinical juvenile toxicity models to account for developmental toxicity effects.
Scenario 2
Questions to be answered by the development programme:
Concentration–response in children
Paediatric dose
Safety in children
PBPK–PD models could be used for first-in-children prediction. The advantage of this approach is that PD measurement can be directly linked with the concentration in the site where the pharmacological action of the drug is expected (biophase). Thereafter physiological/developmental changes in the blood–biophase barrier (e.g. blood–brain barrier) can be accounted for in the model and coupled with differences in, for example, receptor expression and affinity (tested in vitro) in order to predict the developmental effect on PK/PD. For first-in-children predictions, full characterization of the ADME toxicological and PD profile of a drug is ideally needed, or at least a stable minimal base on top of which assumptions can be constructed. Throughout paediatric drug development PBPK models can be used as an interface for understanding the mechanism of action and the processes involved in the therapeutic/toxic effects and for planning future studies.
POP–PK/PD models incorporating size and maturation effects in both PK and PD parameters could also be used for first-in-children prediction and PK/PD study optimization. In POP–PK/PD models it is harder to disentangle the effect of maturation and size in the PK and PD parameters compared with PBPK–PD models. However, assumptions of possible PK/PD relationships, based on in vitro and bibliographic data, can be tested and different ‘what if’ scenarios can be simulated, helping the optimization of future PK/PD studies in terms of study design, number of individuals needed, sampling measurement times.
Kinetic (K)–PD models[20]: PD developed for the description of drug action kinetics in the absence of drug concentration measurements. Because blood samples for drug measurements are not needed, these models are very useful in paediatric studies, by reducing their invasiveness. Paediatric dose recommendations could be based on K-PD models if the PK measures are not obtainable (i.e. lack of systemic exposure). The drawback of K–PD models is that they incorporate many physiological processes in one simple function. Thereafter their potential to extrapolate between different populations and doses can be limited. K–PD models also need extensive and precise PD measurements.
Toxicity/AE models are also useful in this context.
Scenario 3
Questions to be answered by the development programme:
Paediatric dose
Efficacy and safety in children
The absence of PD biomarkers makes the development of PD models difficult. However PK efficacy/toxicity models could be developed.
As described in the previous scenarios, both PBPK and POP–PK models could be used to support first-in-children administration.
POP–PK modelling could be used for sparse sampling and for sampling design optimization, but also to support the paediatric dose-finding exercise.
Disease progression models and response models could be used to optimize the pivotal trials in paediatrics. These models could help identify the optimal duration of the pivotal studies and maximize the assay sensitivity of such studies.
Clinical trial simulation, the virtual clinical testing based on PBPK, POP–PK/PD, disease progression, response, toxicity/AE models, demographics, compliance models, placebo or active comparator effect models, could help investigate different PK and safety, efficacy studies scenarios. Thus the design of the clinical trials will be optimized in a way to answer the specific needs of paediatric pharmacotherapy.
Statistical modelling could be used to increase the power of Phase III studies. Disease, response models could help identifying the structural components of the statistical models that will be used in the hypothesis testing.
Scenario 4
Questions to be answered by the development programme:
Paediatric dose
Efficacy and safety in children
Leverage of prior knowledge from adult studies is not possible, but M&S could be used to combine information from in vitro, preclinical and bibliographic data. Furthermore, M&S can support rational drug development as a decision, study optimization and analysis tool.
Medicinal products intended to cure diseases predominantly or exclusively affecting paediatric patients
Similarly to Scenario 4, extrapolation from adult data is not possible. However, all the advantages of the model-based drug development described above are applicable here.
Medicinal products authorized for paediatric use. Development of a paediatric formulation
A bioequivalence (BE) study in healthy adult volunteers (HV) is the preferred model to bridge paediatric data from the previous formulation to the new one. The applicant should justify that the study results can be extrapolated to children [12, 21]. However, depending on the data available in children with the authorized formulation and the quality attributes of the new formulation, additional studies could be requested.
Role of M&S (Figure 3)
Figure 3.
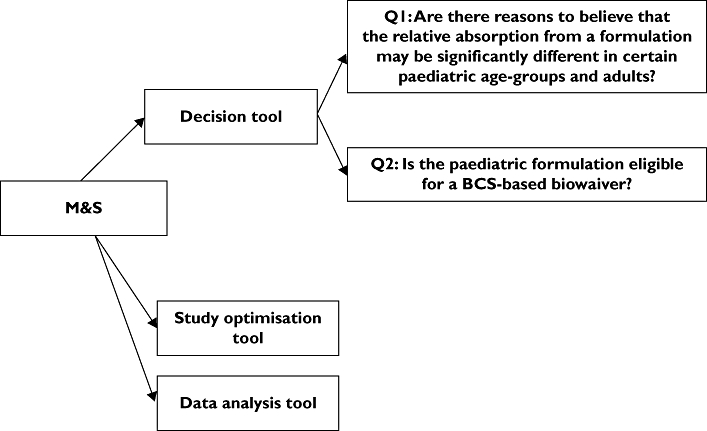
Modelling and simulation role in developing a new paediatric formulation of an already authorized in children medicinal product
Decision tool
PBPK models incorporating information on dissolution profile, solubility, permeability and the effect of maturation in gastrointestinal absorption (i.e. physiology, enzymes and transporters) could be used to compare in silico the two formulations in children and HV.
Q1: PBPK could support the evaluation of the extrapolability of the HV adult bioequivalence in paediatric age groups. The effects of covariates such as maturation, disease and nutrition on absorption could be modelled. Different ‘what if’ scenarios could be simulated in order to evaluate the effect of the above-mentioned parameters on BE, and to identify the most sensitive population. In a hypothetical scenario, if the modelling and simulation exercise concluded that demonstration of BE in HV could be extrapolated to children, a standard BE study in HV could be adequate. Otherwise, a more thorough consideration of the design of the bioequivalence exercise would be needed and additional paediatric studies would be required.
Q2: Demonstration of in silico BE based on PBPK models and BE trial simulation, in addition to the prerequisites described in the BE guideline [21], could waive the need for BE studies for a new paediatric formulation.
These applications of PBPK models are very challenging and require more research and understanding of the physiological processes and the developmental PK. The regulators would need to be exposed to these approaches before defining a regulatory framework to evaluate them, and communication of all involved bodies (industry, academia and authorities) is crucial.
Study optimization and analysis tool
PBPK, POP–PK models and BE trial simulation could be used to optimize BE studies by predicting the design and the number of individuals necessary to achieve the predefined BE margins. Furthermore, population analysis could theoretically be used to support sparse sampling in BE studies. However, the use of sparse sampling and population models in this context requires further reflection and evaluation.
Risks associated with M&S
Poor-quality M&S practices could affect decision making, study optimization and data analysis. Not conducting M&S at high standards could lead to a biased model or overestimation of the predictive power of the models. Erroneous decisions or non-optimal studies based on inappropriate M&S could have a serious impact on drug development and public health.
Following good modelling practices is the best way to circumvent these risks. Extra care should be given to the complexity of the systems to be modelled and the possibility of confounding effects. Uncertainty concerning the data used to build the model should be properly reflected. Measures should be taken to minimize and identify early the biased models, such as by extensive and continuous evaluation of the models and the definition of appropriate study designs. Understanding of the physiological and pharmacological rationale behind the model is also very important. However, sometimes given the complexity of the systems, the little scientific knowledge and the limited data available, modelling and simulation cannot offer solutions and more conventional approaches should be used.
Misuse of M&S: M&S is based on assumptions and, as such, should be supported by experimental data. It cannot and should not be used to replace data from well-conducted studies as primary degree of evidence.
To avoid the risk of misuse, close collaboration of industry academia and regulators is recommended. Defining the degree of evidence that is acceptable from a M&S exercise and the methodology to support pivotal M&S is crucial.
It is expected that the risks associated with M&S will be minimized with the accumulated expertise gained in companies, academia and regulatory bodies.
Discussion
In this article the authors have presented a pragmatic approach to the use of M&S in paediatrics. Some of these methods are in their infancy, others are supported by data and well established. The use of M&S is often questioned, but this is mainly due to the gaps in our knowledge of pharmacological/physiological systems or to the complexity of these systems and the influence of extrinsic factors. Models can be considered as scattered pieces of a giant puzzle (i.e. all the physiological and pharmacological processes in the human body) that could potentially be completed in the future with great benefit to the child and adult populations. For this, however, close collaboration of health authorities, industry, academia, paediatricians and patient organizations is needed. The contribution of patient organizations and clinicians in collecting and sharing data with the other health stakeholders is also important. Companies, consortia and learned societies are encouraged to consult the regulatory authorities when they develop a model for a specific use in order to gain the feedback of the agencies but also to promote the awareness of the regulators towards M&S. The EU Regulation provides that companies, consortia and learned societies can obtain the regulator's scientific opinion through European Medicines Agency (EMEA)/Committee for Medicinal Products for Human Use scientific advice, which is a forum for such discussions. In this context the EMEA has published recently a new procedure, the qualification of novel methodologies for drug development [22]. The objective of this process is either to qualify (qualification opinion) or to propose studies (qualification advice) for qualification of specific use of novel methodologies, such as M&S, in the context of research and drug development.
Conclusion
The regulators encourage the use of M&S in paediatric drug development. M&S can be used as a decision, analysis and optimization tool. It is also a learning tool that facilitates the understanding of the physiological and pharmacological mechanisms, which is of major importance in this maturing population. M&S is not independent from primary data and should not be considered as waiving the need to conduct studies in children. The degree of evidence expected from a model in the regulatory benefit–risk assessment is directly proportional to the measures envisaged to demonstrate the robustness of the M&S exercise. The implementation of M&S in paediatric medicinal development and regulatory thinking needs further discussion, and the EMEA could host such communication under the novel methodologies qualification process.
Competing interests
Nothing to declare.
The authors thank the participants in the Workshop on Modelling in Paediatric Medicines. Special thanks to the following individuals who reviewed the document and submitted comments: Professor Nick Holford (University of Auckland, New Zealand), Professor Geoff Tucker, University of Sheffield and Simcyp Ltd, UK, Professor Michel Tod (Université de Lyon, France), Mr Robert Hemmings (MHRA, UK), Professor Spiros Vamvakas (Deputy Head of Sector Scientific Advice and Orphan Drugs, EMEA), and in particular to Dr Agnès Saint Raymond (Head of Sector Scientific Advice and Orphan Drugs, EMEA).
REFERENCES
- 1.EMEA. EMEA Workshop on Modelling in Paediatric Medicines. London: EMEA; pp. 14–15. April 2008 Available at http://www.emea.europa.eu/htms/human/paediatrics/workshops.htm (last accessed 1 May 2009. [Google Scholar]
- 2.Holford NHG, Kimko HC, Monteleone JPR, Peck CC. Simulation of Clinical Trials. Annu Rev Pharmacol Toxicol. 2000;40:209–34. doi: 10.1146/annurev.pharmtox.40.1.209. [DOI] [PubMed] [Google Scholar]
- 3.Guideline on Reporting the Results of Population Pharmacokinetic Analyses. CHMP/EWP/185990/06. Available at http://www.emea.europa.eu/pdfs/human/ewp/18599006enfin.pdf (last accessed 1 May 2009.
- 4.Guidance for Industry Population Pharmacokinetics. Available at http://www.fda.gov/CDER/guidance/1852fnl.pdf (last accessed 1 May 2009.
- 5.Sheiner LB, Steimer JL. Pharmacokinetic/pharmacodynamic modeling in drug development. Annu Rev Pharmacol Toxicol. 2000;40:67–95. doi: 10.1146/annurev.pharmtox.40.1.67. [DOI] [PubMed] [Google Scholar]
- 6.Sheiner LB. Learning versus confirming in clinical drug development. Clin Pharmacol Ther. 1997;61:275–91. doi: 10.1016/S0009-9236(97)90160-0. [DOI] [PubMed] [Google Scholar]
- 7.Points to Consider on Application with 1. Meta-analyses; 2. One Pivotal study. CPMP/EWP/2330/99). Available at http://www.emea.europa.eu/pdfs/human/ewp/233099fen.pdf (last accessed 1 May 2009.
- 8.Peck CC, Rubin DB, Sheiner LB. Hypothesis: a single clinical trial plus causal evidence of effectiveness is sufficient for drug approval. Clin Pharmacol Ther. 2003;73:481–90. doi: 10.1016/S0009-9236(03)00018-3. [DOI] [PubMed] [Google Scholar]
- 9.Lee H, Yim D-S, Zhou H, Peck CC. Evidence of effectiveness: how much can we extrapolate from existing studies? AAPS J. 2005;7:E467–74. doi: 10.1208/aapsj070247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.ICH Topic E 9 Statistical Principles for Clinical Trials Step 5. Note for guidance on statistical principles for clinical trials. CPMP/ICH/363/96). Available at http://www.emea.europa.eu/pdfs/human/ich/036396en.pdf (last accessed 1 May 2009.
- 11.Guideline on Clinical Trials in Small Populations. CHMP/ EWP/83561/05). Available at http://www.emea.europa.eu/pdfs/human/ewp/8356105en.pdf (last accessed 1 May 2009.
- 12.Guideline on the Role of Pharmacokinetics in the Development of Medicinal Products in the Paediatric Population. CHMP/EWP/147013/04 Corrigendum). Available at http://www.emea.europa.eu/pdfs/human/ewp/14701304en.pdf (last accessed 1 May 2009.
- 13.Guidance for Industry, Exposure–Response Relationships, Study Design, Data Analysis, and Regulatory Applications. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM072109.pdf (last accessed 1 May 2009.
- 14.Johnson TN. Modelling approaches to dose estimation in children. Br J Clin Pharmacol. 2005;59:663–669. doi: 10.1111/j.1365-2125.2005.02429.x. Available at http://www.pubmedcentral.nih.gov/picrender.fcgi?artid=1884869&blobtype=pdf (last accessed 1 May 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wahlby U, Jonsson EN, Karlsson MO. Comparison of stepwise covariate model building strategies in population pharmacokinetic–pharmacodynamic analysis. AAPS PharmSci. 2002;4 doi: 10.1208/ps040427. article 27. DOI: 10.1208/ps040427. Available at http://www.aapsj.org/articles/ps0404/ps040427/ps040427.pdf (last accessed 1 May 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.West GB, Brown JH, Enquist BJ. The fourth dimension of life: fractal geometry and allometric scaling of organisms. Science. 1999;284:1677–9. doi: 10.1126/science.284.5420.1677. [DOI] [PubMed] [Google Scholar]
- 17.Tod M, Jullien V, Pons G. Facilitation of drug evaluation in children by population methods and modelling. Clin Pharmacokinet. 2008;47:231–43. doi: 10.2165/00003088-200847040-00002. [DOI] [PubMed] [Google Scholar]
- 18.Anderson BJ, Holford NHG. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol. 2008;48:303–32. doi: 10.1146/annurev.pharmtox.48.113006.094708. [DOI] [PubMed] [Google Scholar]
- 19.Retout S, Duffull S, Mentré F. Development and implementation of the population Fisher information matrix for evaluation of population pharmacokinetic designs. Comput Methods Programs Biomed. 2001;65:141–51. doi: 10.1016/s0169-2607(00)00117-6. [DOI] [PubMed] [Google Scholar]
- 20.Jacqmin P, Snoeck E, van Schaick EA. Modelling response time profiles in the absence of drug concentrations: definition and performance evaluation of the K-PD model. J Pharmacokinet Pharmacodyn. 2007;34:57–85. doi: 10.1007/s10928-006-9035-z. [DOI] [PubMed] [Google Scholar]
- 21.Draft Guideline on the Investigation of Bioequivalence. CPMP/EWP/QWP/1401/98 Rev. 1). Available at http://www.emea.europa.eu/pdfs/human/qwp/140198enrev1.pdf (last accessed 1 May 2009.
- 22.Qualification of Novel Methodologies for Drug Development. Guidance to Applicants. EMEA/CHMP/SAWP/72894/2008. Available at http://www.emea.europa.eu/pdfs/human/sciadvice/7289408en.pdf (last accessed 1 May 2009.