

Abstract
Background
Elevated circulating leptin is present in human heart failure, and leptin deficiency is linked to worse outcomes in chronic ischemic injury. In the present observational study, we tested the hypothesis that cardiac leptin production and signaling are increased in the failing human heart, and that mechanical unloading with a ventricular assist device (VAD) reverses these changes.
Methods and Results
All studies were performed using human cardiac tissue obtained from 1) hearts not matched for transplantation (non-failing), 2) at time of cardiac transplant (failing), or 3) paired samples at the time of VAD implant (pre-VAD) and removal (post-VAD). Expression of brain naturetic peptide, leptin, leptin receptor and tumor necrosis factor-alpha (TNFα) mRNA was measured, and the protein expression of leptin and its receptor was examined by Western blot and immunofluorescent staining of cardiac sections. Assessment of leptin signaling was performed by measuring the phosphorylation state of the leptin receptor. The phosphorylation state of signal transducer and activator of transcription (STAT)-3 and AMP-activated kinase (AMPK) proteins were also measured. All data are expressed as mean ± stand error of the mean with statistical significance in failing relative to non-failing groups determined by student's independent t-test, and significance between pre- and post-VAD groups determined by paired t-test. In failing human hearts, the mRNA expression of leptin and its receptor were increased 5.4±0.3 (p<0.05) and 4.5±0.3 (p<0.05) fold, respectively, with similar changes in protein. The phosphorylation state of the leptin receptor and STAT-3 proteins were both increased 1.4±0.1 fold (p<0.05), and the level of phosphorylated AMPK protein was increased 1.9±0.2 fold (p<0.05). Mechanical unloading of the failing human heart with a VAD resulted in no change in TNFα expression, but a marked decrease in leptin production to 1.7±0.1% (p<0.05), and leptin receptor expression to 3.0±0.2% (p<0.05), of pre-VAD levels. Phosphorylation of the leptin receptor, STAT-3 and AMPK were also decreased to 45±7%, 75±8%, and 58±8 % of pre-VAD values, respectively (all p<0.05).
Conclusions
These results indicate that the failing human heart increases expression of leptin and its receptor, and that mechanical unloading downregulates this increase. Further, a cardioprotective role for leptin in the failing human heart is suggested through the activation of STAT-3 and AMPK signaling.
Keywords: leptin, leptin receptor, ventricular assist device, AMP kinase, heart failure
Human systolic heart failure is characterized by loss of myocyte number and decreased contractile performance of the left ventricle.1 The etiology of systolic heart failure is multi-factorial, with coronary artery disease linked to the majority of cases in the elderly.2 There are also non-ischemic causes of heart failure, resulting in viral, hypertensive and valvular cardiomyopathies.3 Regardless of the etiology, however, studies have shown that cytokines play an important role in both the progression and mitigation of cardiac remodeling. For example, whereas leptin deficient Ob/Ob mice demonstrate age-related cardiac hypertrophy,4 greater mortality in viral myocarditis,5 and worse LV function and survival in response to ischemic injury,6 repletion of leptin mitigates these deleterious effects of leptin deficiency. Additionally, cardiac leptin production and signaling are increased in mice after myocardial infarction (MI)6. In humans, serum leptin is increased after MI7 and in advanced heart failure8 independent of weight. Thus, leptin is an important cytokine that mediates beneficial effects in heart failure when present and results in deleterious effects when absent.
While serum leptin is elevated in human heart failure,8 it is unknown if leptin production or signaling is altered in the failing human heart. Further, if cardiac leptin production or signaling is altered in human heart failure, determination of whether it is a cause or consequence of pathologic changes becomes an important issue. This study was undertaken to test the hypothesis that the failing human heart produces leptin and upregulates beneficial leptin signaling pathways, including signal transducer and activator of transcription (STAT)-3 and AMP-activated protein kinase (AMPK). Additional experiments examining the effect of left ventricular mechanical unloading on cardiac leptin production and signaling were performed to test the hypothesis that alterations in leptin production and signaling are a consequence rather than a cause of myocardial dysfunction.
Methods
Human Heart Samples
Failing human heart tissue was obtained from the University of Pittsburgh after Institutional Review Board approval and with the subjects' informed consent. Transmural samples of the lateral wall of the left ventricle (LV) were obtained from failing human hearts at the time of transplant (n=10), or from the LV apex at the time of ventricular assist device (VAD) implant and removal (paired samples, n=8). Non-failing human heart tissue (NF) was obtained from the Cleveland Clinic Foundation after Institutional Review Board approval and with the subjects' informed consent. Transmural samples of the lateral wall of the LV were obtained from unmatched donor hearts that did not meet criteria for transplantation, including donor age greater than 50 years (n=2), positive toxicology screen (n=1), mild coronary artery disease (n=2), and 2+ mitral regurgitation detected on echocardiography (n=1). All cardiac tissue was placed into cold cardioplegic solution (4-8°C) and rapidly transported to the laboratory. Tissues were then divided for fixation and histology, or snap frozen in liquid nitrogen and stored at -80°C until biochemical analyses.
Quantitative Real time RT-PCR analysis
Total RNA was isolated and reverse transcribed as previously described6 from whole tissue homogenates. Real-time RT-PCR was performed using the DNA binding dye SYBR Green9 and previously validated primers and conditions for the real-time RT-PCR detection of human brain naturetic peptide (BNP),10 human leptin,11 human long form leptin receptor (ObR),12 human tumor necrosis factor-alpha (TNFα),13 and human GAPDH.14 Specific PCR products were confirmed by the presence of a single first derivative melting peak,15 and by the amplification product size determined by gel electrophoresis.16 Equal amplification kinetics of the target and the reference genes (GAPDH) were confirmed by serial dilutions as described.17 Quantification was then performed using the comparative Ct method (2-ΔΔCt).18
Western blotting
Protein was extracted from cardiac tissue in RIPA buffer, subjected to SDS-PAGE, and electro-transferred to PVDF membranes in equal amounts as previously described.6 All gels were run with a broad range molecular weight standard (Biorad; cat# 161-0375). Antibodies that were utilized detected leptin (Santa Cruz Biotechnology; cat #sc-842), ObR (Santa Cruz Biotechnology; cat #sc-8325), Y935-phosphorylated ObR (Santa Cruz Biotechnology; cat #sc-16419), STAT-3 (Cell Signaling; cat #9132), Y705-phosphorylated STAT-3 (Cell Signaling; cat #9131), AMPKα (Cell Signaling; cat #2532), T172-phosphorylated AMPK (Cell Signaling; cat #2531) and GAPDH (Research Diagnostics Inc; cat #RDI-TRK5G4-6C5). After transfer to membranes, blots were blocked in 5% milk plus tris buffered saline tween (TBST) for 1 hour at room temperature with agitation. Membranes were then cut at the appropriate molecular weight for probing with GAPDH and the protein of interest. X-ray films were digitized using a Visioneer OneTouch Scanner model 9220 and imported into Paperport SE software version 8.0 (Scansoft Inc.). Quantification of bands on x-ray film was performed using Image J software (NIH, USA) to determine an integrated density in pixels/unit area. Detection of GAPDH was performed on all blots to control for differences in gel loading, protein transfer, and protein quantitation. Any blots with unequal GAPDH were repeated and not used for analysis. All blots were repeated at least 3 times to assure reproducibility of results. For changes in total protein, the mean integrated density of the band of interest was determined for comparative groups, and statistical testing (t-test) performed using the integrated density values for each member of the group. For ratios of phosphorylated to total protein, the ratio of the integrated densities of the phosphorylated to total protein of interest within each comparative group were used for statistical testing (t-test). For graphical representation of the ratio data, the mean control group ratios (non-failing or pre-VAD) were arbitrarily defined as 1 for increases and 100% for decreases, and the comparative groups (failing and post-VAD) expressed as a fold or percent change relative to it, respectively.
Immunoflourescence
Cardiac tissue was harvested and fixed in 2% paraformaldehyde and cryoprotected in 30% sucrose, as previously described.6 Short-axis cryotome sections (6 micron) taken at the level of the mid-left ventricle were collected on slides and probed with either anti-leptin (Santa Cruz Biotechnology; cat #sc-842) or anti-ObR (Santa Cruz Biotechnology; cat #sc-8325) antibodies at a dilution of 1:100. A cy3-linked goat anti-rabbit (Invitrogen USA; cat #A10520) secondary antibody diluted 1:1000 was used for immunofluorescent detection. Both leptin and ObR stained sections were further stained with 488-linked phalloidin to detect sarcomeric actin and DAPI to detect nuclei as described.6 Images were acquired using an Olympus (Melville, NY, USA) Provis AX70 fluorescent microscope at ×20 magnification, digitized using a cooled charge-coupled device camera (Optronics Magnifier; East Muskogee, OK, USA) at 12-bit grey depth, and finally assembled in Adobe Photoshop 8.0 (Adobe Systems Inc., San Jose, CA, USA).
Statistical analysis
Data presented are mean±standard error of the mean (sem). Statistical significance of mean changes in LV ejection fraction (LVEF), body mass index (BMI), mRNA and protein expression between non-failing and failing samples were determined by student's unpaired t-test. Student's paired t-test was used to analyze these same data collected on pre- and post-VAD samples. Mathematical calculations, determination of p values, and t-tests were performed using the computer program Statistical Package for the Social Sciences (SPSS) v16 (SPSS, Inc, Chicago, IL, USA).
The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written.
Results
Failing human hearts demonstrate elevated BNP expression
Equal numbers of ischemic (n=5) and non-ischemic (n=5) patients were included in the failing group, which had an average left ventricular ejection fraction of 15±2% and an average age of 56±4 years old. In contrast, the LVEF of the non-failing group was preserved at 63±3% (p<0.05 versus failing). The body mass index (25.7±2.7 versus 26.4±1.8 kg/m2; p>0.05) and gender distribution (50% male, 50% female) of both non-failing and failing groups were not significantly different from one another, respectively. Consistent with increased cardiomyocyte stretch and clinical heart failure, the mRNA expression of BNP was increased 4.4±0.4 fold in failing relative to non-failing hearts (p<0.05).
Failing human hearts demonstrate increased leptin expression
Relative to non-failing samples, failing hearts showed 5.4±0.3 fold increased expression of leptin mRNA (Figure 1a). This increased leptin mRNA in failing hearts correlated with increased leptin protein expression as determined by western blot using whole tissue homogenates (Figure 1b). Cardiomyocyte-specific leptin expression was confirmed through immunofluorescent staining of sections from non-failing and failing samples (Figure 1c).
Figure 1.
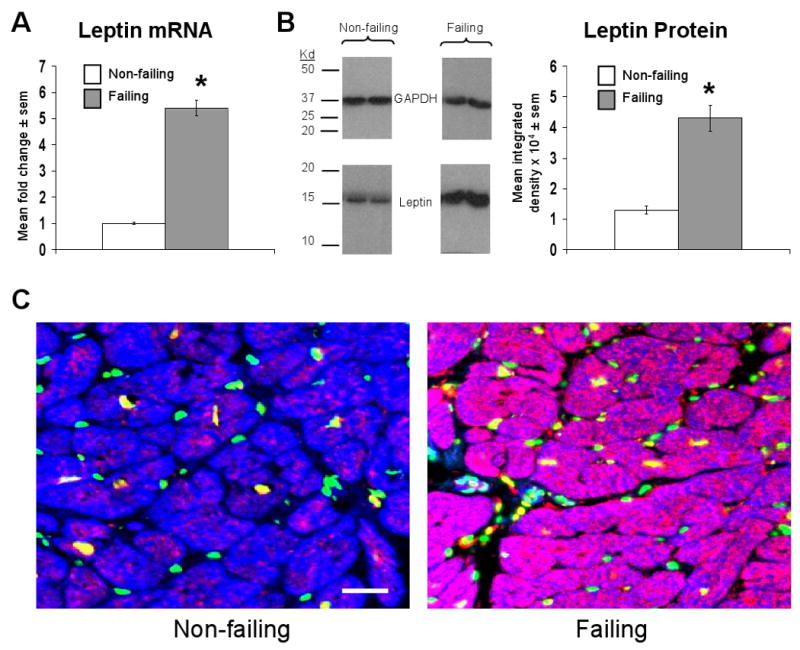
Failing human cardiac tissue demonstrates increased leptin mRNA and protein expression. A) Mean ± sem fold change in cardiac leptin mRNA in failing (n=10) relative to non-failing (n=6) samples, the latter of which were arbitrarily assigned a value of 1 after statistical calculations were performed. *p<0.05 relative to non-failing samples. B) Mean ± sem integrated density of leptin bands detected on x-ray film from failing (n=10) and non-failing (n=6) samples (right graph), with representative images and molecular weight markers shown. *p<0.05 relative to non-failing samples. C) Representative 20× power immunofluorescent staining of cardiac sections from a non-failing (left image) and failing (right image) human heart. Leptin staining is shown in red, nuclei (stained with DAPI) are shown in green, cells containing sarcomeric actin (stained with phalloidin) are shown in blue, and an accompanying scale bar (10μM) is shown in white.
Failing human hearts demonstrate increased leptin receptor expression
Long form leptin receptor mRNA expression was increased 4.5±0.3 fold in failing relative to non-failing hearts (Figure 2a). This change in mRNA was paralleled by similar changes in leptin receptor protein as determined by western blot using whole heart homogenates (Figure 2b). Qualitatively, increased leptin receptor protein was localized to the cardiomyocyte in cardiac sections taken from failing hearts (Figure 2c).
Figure 2.
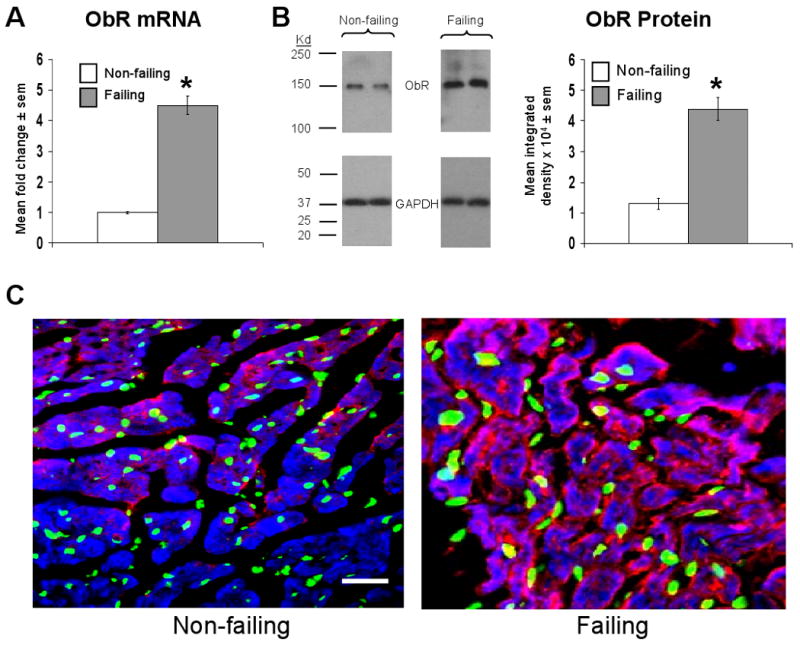
Failing human cardiac tissue demonstrates increased leptin receptor (ObR) mRNA and protein expression. A) Mean ± sem fold change in cardiac ObR mRNA in failing (n=10) relative to non-failing (n=6) samples, the latter of which were arbitrarily assigned a value of 1 after statistical calculations were performed. *p<0.05 relative to non-failing samples. B) Mean ± sem integrated density of ObR bands detected on x-ray film from failing (n=10) and non-failing (n=6) samples (right graph), with representative images and molecular weight markers shown. *p<0.05 relative to non-failing samples. C) Representative 20× power immunofluorescent staining of cardiac sections from a non-failing (left image) and failing (right image) human heart. ObR staining is shown in red, nuclei (stained with DAPI) are shown in green, cells containing sarcomeric actin (stained with phalloidin) are shown in blue, and an accompanying scale bar (10μM) is shown in white.
Failing human hearts demonstrate increased leptin receptor, STAT-3 and AMP kinase alpha phosphorylation
The ratio of phosphorylated (p) ObR to total (t) ObR was increased 1.4±0.1 fold in failing relative to non-failing hearts (Figure 3a), representing increased receptor activation. Further, the ratio of p/t STAT-3, an established downstream mediator of cardiac leptin signaling,6 was increased 1.4±0.1 fold, with a 2.3±0.2 fold increase in total STAT-3 protein (Figure 3b). Finally, the ratio of p/t AMP Kinase alpha, which has been linked to leptin signaling and metabolic substrate utilization in the heart,19 was also increased 1.9±0.2 fold, with no change in the amount of total AMP Kinase alpha protein present (Figure 3c).
Figure 3.
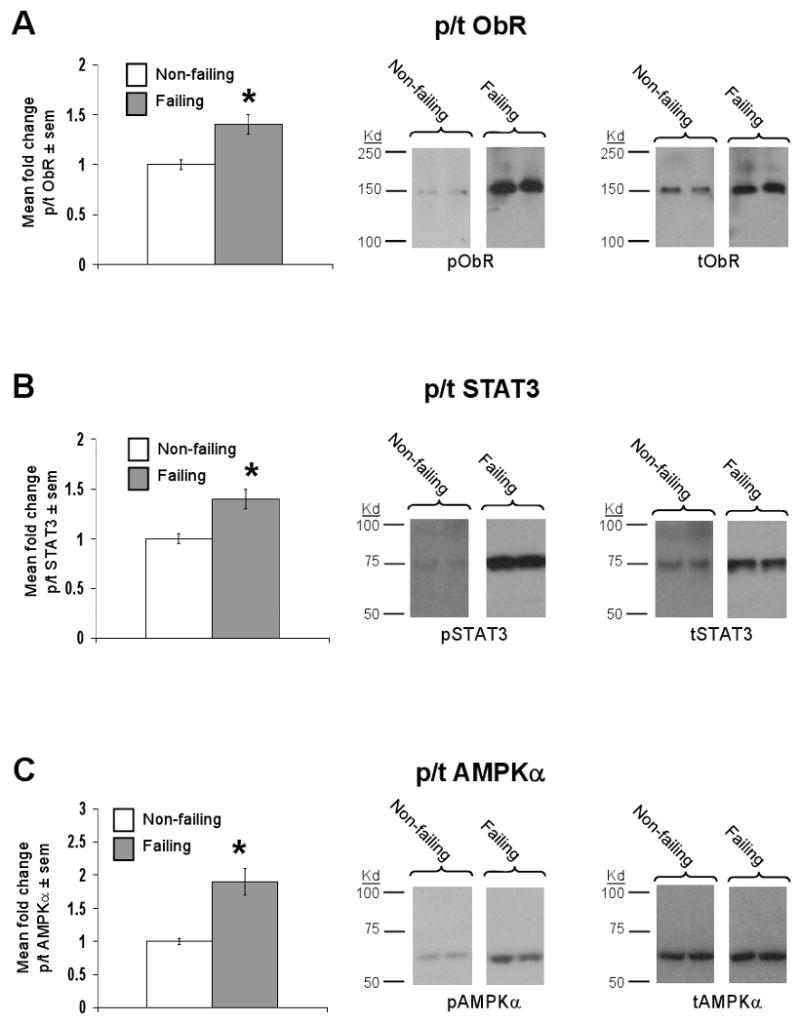
ObR signaling is upregulated in the failing human heart. Data for bar graphs are expressed as fold changes in mean values ± sem of failing (n=10) relative to non-failing (n=6) samples, which were arbitrarily assigned a value of 1 after statistical calculations were performed. *p<0.05 relative to non-failing samples. A) Mean fold change in the ratio of phosphorylated (p) to total (t) ObR protein (left graph) with representative Western blot (right panel) and molecular weight markers are shown. B) As in A, except that data for p/t STAT-3 are shown. C) As in A, except that data for p/t AMPK are shown.
Mechanical unloading of the failing human heart decreases BNP and leptin mRNA expression, and downregulates leptin receptor, STAT-3 and AMPK activation
Paired heart samples analyzed in this subsequent study came from 8 male patients who received ventricular assist devices (VAD) prior to heart transplant. The LVEF prior to VAD support was 13.6±2.1% and the average length of VAD support was 35±12 days. As with the first set of failing hearts, the etiology of heart failure was equally distributed amongst ischemic (n=4) and non-ischemic (n=4) subgroups. The cardiac mRNA expression of BNP and leptin were decreased to 1.7±0.1% and 3.0±0.2% of their initial values, respectively (Figure 4a and 4b), whereas the cardiac mRNA expression of TNFα was unchanged (Figure 4c). Consistent with a downregulation of cardiac leptin and AMPK signaling, the ratio of p/t ObR (Figure 5a), p/t STAT-3 (Figure 5b), and p/t AMPKα (Figure 5c) were decreased to 45±7%, 75±8%, and 58±8 % of pre-VAD values respectively, with no change in total protein levels, after mechanical unloading.
Figure 4.

Mechanical unloading of the failing human heart downregulates the expression of BNP and leptin, but not TNFα mRNAs. Data for bar graph are expressed as percent changes in mean values ± sem of post-VAD (n=8) relative to paired pre-VAD (n=8) samples, which were arbitrarily assigned a value of 100% after statistical calculations were performed. *p<0.05 relative to failing, pre-VAD samples. A) Percent change in BNP mRNA. B) As in A, except that leptin mRNA was measured. C) As in A, except that TNFα mRNA was measured.
Figure 5.
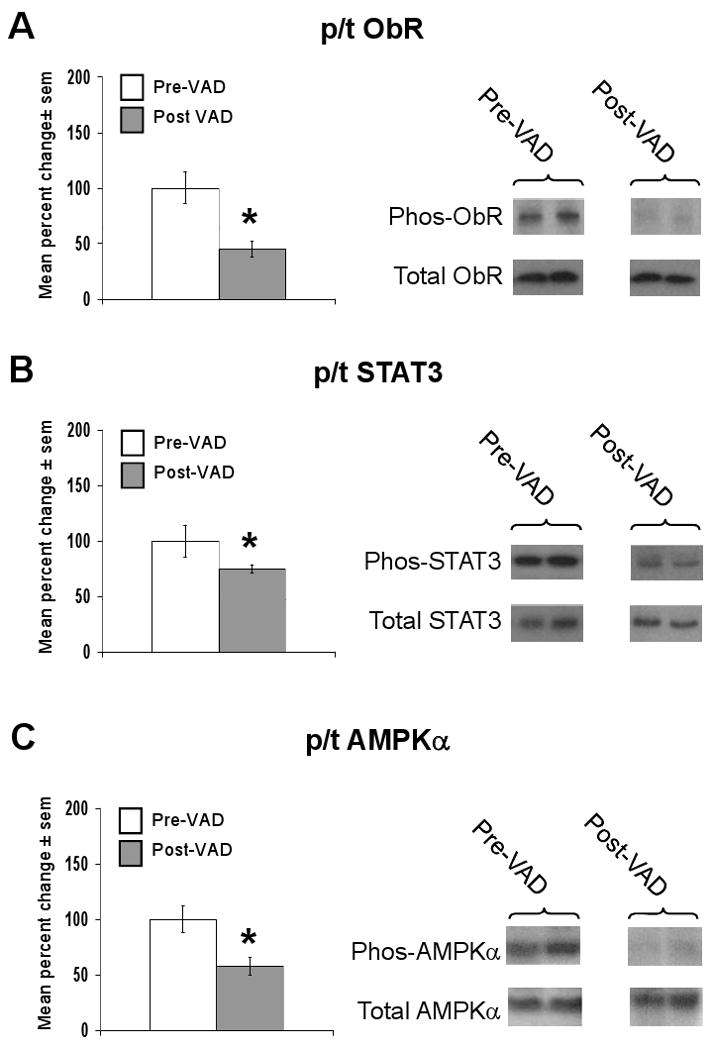
Mechanical unloading of the failing human heart downregulates cardiac leptin, STAT-3 and AMPK signaling. Data for bar graph are expressed as percent changes in mean values ± sem of post-VAD (n=8) relative to paired pre-VAD (n=8) samples, which were arbitrarily assigned a value of 100% after statistical calculations were performed. *p<0.05 relative to failing, pre-VAD samples. A) Percent change in the ratio of phosphorylated (p) to total (t) ObR protein (left graph) with representative Western blot (right panel) are shown. B) As in A, except that data for p/t STAT-3 are shown. C) As in A, except that data for p/t AMPK are shown.
Discussion
Heart failure is characterized by an increase in the production of cytokines that mediate both compensatory and pathologic responses. In human heart failure, circulating leptin is increased independent of weight,8 and previous experiments with leptin deficient mice demonstrate that leptin plays a protective role in the heart. Specifically, leptin deficiency results in worse survival and greater LV dysfunction in chronic ischemic injury,6 increased cardiac hypertrophy4 and apoptosis20 with aging, and greater mortality in viral myocarditis.5 In each of these studies,4-6, 20 leptin-repletion restored measures of morbidity and mortality to wildtype levels. While weight was controlled for in the ischemia,6 hypertrophy4 and apoptosis20 studies, we cannot exclude the possibility that other deleterious consequences of the leptin-deficient state outside of obesity contributed to the worse outcomes seen. For example, Ob/Ob mice demonstrate a diabetic phenotype with altered cardiac glucose and fatty acid metabolism that is exacerbated further by their obesity.21 Therefore, even in lean Ob/Ob mice, diabetes that develops secondary to leptin deficiency may contribute to the worse outcomes seen after myocardial infarction. Nevertheless, a beneficial cardiac response to leptin in the setting of myocardial injury also likely requires activation of known cardioprotective signaling pathways. Our data demonstrate that cardiac leptin production, and leptin receptor, STAT-3 and AMPK activation, are increased in human failing hearts and decreased in response to mechanical unloading. These data suggest that the expression of leptin in the failing myocardium is potentially compensatory and is regulated by mechanical signaling and/or wall stress.
In humans, leptin receptor is present in normal and hypertrophied hearts,22 and the increased systemic leptin levels seen in the setting of myocardial infarction7 and heart failure8 are likely secondary to increased production by fat. However, a major finding in our study was that the failing human heart, independent of BMI, upregulates leptin production and signaling. This is in agreement with our prior study6 demonstrating increased cardiac leptin production and signaling after chronic ischemic injury in lean wildtype mice. Studies also demonstrate that systemic leptin deficiency is associated with worse outcomes in the setting of cardiac injury independent of weight,6 and that restoration of signaling in leptin-deficient mice,5, 6 or increasing circulating leptin levels in lean wildtype mice,23 both by administration of exogenous leptin, improves cardiac function at 4 weeks post-myocardial infarction. Combined, these data suggest that systemic leptin plays an important role in conserving heart function in the setting of myocardial infarction, and that exogenous administration may be useful therapy for improving cardiac function in patients with heart failure.
Systemic leptin production is decreased in response to caloric restriction and increased with caloric loading.24, 25 However, the effect of caloric restriction or loading on cardiac leptin production is unknown. Studies that have examined cardiac leptin production demonstrate increases in response to cardiac injury.6, 26 It is interesting to note that obesity, and its accompanying hyperleptinemia, are established risk factors for the development of hypertension, heart failure, and coronary artery disease, yet overweight and obese patients with these cardiovascular diagnoses have more favorable short and long term prognoses.27 Clinically, this ‘obesity paradox’ might be explained by the hyperleptinemia of obesity, and selective resistance to the metabolic actions of leptin centrally, but not in cardiac tissue.28 Indeed, our data demonstrate increased cardiac leptin production and peripheral activation of leptin signaling in the failing human heart. Thus, both systemic and cardiac production of leptin may contribute to more favorable short and long term outcomes in the setting of myocardial injury and heart failure.
The benefits of leptin in heart failure are likely dependent on the activation of cardiac leptin signaling. Based on this, we hypothesized that the mechanism of improvement likely involves the activation of beneficial leptin signaling pathways in the heart, including STAT-3 and AMPK. Indeed, our finding of increased STAT-3 and AMPK activation in failing human hearts is supportive of this hypothesis, and is consistent with the known cardioprotective nature of the STAT-329, 30 and AMPK31, 32 proteins in heart failure. While we cannot exclude the possibility that cell types other than the cardiomyocyte are contributing to the changes seen in leptin production and signaling in the whole heart homogenates examined in this study, our immunoflourescent data is at least suggestive that the cardiomyocyte is the predominant source of these proteins in the heart. It may also explain why failing hearts, which demonstrate a mean 15% LVEF, eventually require transplantation despite increased leptin, STAT-3 and AMPK signaling. Clearly elevated cardiac leptin expression and signaling are insufficient to prevent end-stage heart failure development, but may reflect a persistent compensatory mechanism that has greater beneficial effects at an earlier stage of less severe heart failure.
Intact leptin production and signaling were also seen prior to mechanical unloading of the LV with a VAD. Mechanical unloading of the failing human heart reverses mitochondrial metabolic dysfunction,33 improves baseline contractility of the LV,34 decreases cardiomyocyte hypertrophy35 and apoptosis,36 and increases VEGF and other angiogenic growth factors.37 VAD support also results in a reduction in the expression of genes that are important markers of cardiac remodeling, including BNP.38 Consistent with this, in our series of pre- and post-VAD patients, cardiac production of BNP decreased with mechanical unloading. Similarly, cardiac mRNA levels of leptin decreased post-VAD, and decreased levels of leptin receptor, STAT-3 and AMPK phosphorylation were seen. In contrast, the expression of TNFα remained unchanged, suggesting that our observed decrease in leptin production and signaling was not a non-specific response to mechanical unloading. It is possible, however, that a mild degree of ischemia that accompanies decompensated heart failure, regardless of etiology, might be contributory to the changes we see in p/t AMPK ratios. Nevertheless, our finding of decreased STAT-3 and AMPK activation with mechanical unloading suggests that the cardiac production of leptin may influence the response of the failing heart to alterations in workload.
Although our data suggest a beneficial role for leptin in failing human hearts, there are several limitations to our study that deserve comment. First, our analysis was limited to the activation state of STAT-3 and AMPK. There are other known cardiac leptin signaling pathways that may mediate beneficial responses in heart failure that were not examined here, including Akt39 and phosphatidylinositol-3-kinase.40 Second, because this study was conducted with acutely collected human tissue, it is largely observational in its findings and we are unable to assess responses in the failing human heart to leptin-specific agonist or antagonist challenge. Third, heart failure is a complex process involving the production of many cytokines and the activation of numerous intracellular signaling pathways. For example, in addition to leptin, the activation of STAT-3 and AMPK in the failing heart could also arise from leptin-independent pathways, including interleukin-641 and adiponectin,42 respectively. Additionally, we acknowledge that diverse changes in cardiac STAT-3 phosphorylation have been reported in the failing human heart,43-45 and that it remains unclear whether leptin can activate AMPK in both skeletal46 and cardiac striated muscle.47 However, our observation of increased leptin production in the failing human heart is consistent with activation of downstream components of known leptin signaling pathways, including STAT-3 and AMPK. Finally, we did not measure serum leptin or changes in wall stress or workload in the failing or mechanically unloaded human heart, although changes in these measures are anticipated based upon prior reports.7, 8, 34, 48, 49
In conclusion, our data shows increased leptin production and signaling in the failing human heart, with altered expression in response to changes in cardiac wall stress/workload. These observations complement studies reporting increased circulating leptin in human heart failure,8 as well as acutely after myocardial infarction,7 independent of weight. In vitro studies utilizing cultured cardiomyocytes50, 51 and results from experiments using animal models of leptin deficiency subjected to cardiac injury5, 6 suggest that leptin is a cardioprotective cytokine. The beneficial effect of STAT-3 and AMPK activation in the failing heart has been reported in multiple studies, 29-32 and provides a plausible pathway by which physiologic increases in leptin signaling may be beneficial to the injured heart.
Acknowledgments
From the University of Pittsburgh Medical Center, we thank the Center for Biologic Imaging at the University of Pittsburgh for assistance with microscopy, the Research Nurse Co-ordinators within the Cardiovascular Institute and Division of Cardiothoracic Surgery for assistance in patient enrollment and clinical data, and the cardiothoracic surgeons and their support staff for assistance in acquisition of surgical specimens. From the Cleveland Clinic Foundation, we thank the cardiac transplant teams, Department of Thoracic and Cardiovascular Surgery (CCF), residents in the Department of Pathology (CCF), and Life Banc of Northeastern Ohio for helping to obtain cardiac tissues for research.
Funding Sources: The Cardiovascular Institute at the University of Pittsburgh Medical Center and NIH (HL087009-02 to K.R.M.)
Footnotes
Clinical Perspective: The obesity paradox refers to an increase risk of developing hypertension, coronary artery disease, and heart failure with increasing body mass index (BMI), but paradoxically overweight and obese patients with these cardiovascular diagnoses demonstrate more favorable short and long term prognoses versus patients with a more normal BMI. A key advance in our understanding of obesity was the discovery of leptin, a circulating, predominantly fat derived cytokine which regulates metabolism and limits appetite through centrally mediated pathways. As more fat accumulates, more leptin is produced. However, a condition of ‘central leptin resistance’ may develop in the presence of chronically elevated circulating leptin, in which leptin fails to suppress appetite, setting the stage for clinical obesity. Aside from obesity, circulating leptin is increased independent of BMI after myocardial infarction and in heart failure. In this study, we examined cardiac leptin production and signaling in failing and mechanically unloaded human hearts from overweight (BMI ∼26 kg/m2) but not obese patients. Relative to non-failing hearts, failing cardiac tissue demonstrated increased cardiac leptin production and signaling through its receptor, as well as activation of cardioprotective signal transducer and activator of transcription-3 and AMP activated protein kinase proteins. Mechanical unloading with a ventricular assist device reversed these changes. Taken together, these findings suggest that the hyperleptinemia seen in obesity, cardiac ischemia, and heart failure might contribute to the obesity paradox by mediating the activation of cardioprotective peripheral (i.e., cardiac) leptin signaling pathways to improve outcomes in the presence of central leptin resistance.
Disclosures: None
References
- 1.Mudd JO, Kass DA. Tackling heart failure in the twenty-first century. Nature. 2008;451:919–928. doi: 10.1038/nature06798. [DOI] [PubMed] [Google Scholar]
- 2.Rich MW. Epidemiology, pathophysiology, and etiology of congestive heart failure in older adults. J Am Geriatr Soc. 1997;45:968–974. doi: 10.1111/j.1532-5415.1997.tb02968.x. [DOI] [PubMed] [Google Scholar]
- 3.Hwang JJ, Dzau VJ, Liew CC. Genomics and the pathophysiology of heart failure. Curr Cardiol Rep. 2001;3:198–207. doi: 10.1007/s11886-001-0023-z. [DOI] [PubMed] [Google Scholar]
- 4.Barouch LA, Berkowitz DE, Harrison RW, O'Donnell CP, Hare JM. Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation. 2003;108:754–759. doi: 10.1161/01.CIR.0000083716.82622.FD. [DOI] [PubMed] [Google Scholar]
- 5.Kanda T, Takahasi T, Kudo S, Takeda T, Tsugawa H, Takekoshi N. Leptin deficiency enhances myocardial necrosis and lethality Minokoshi Y, Kim Yb, Peroni. Life Sci. 2004;75:1435–1447. doi: 10.1016/j.lfs.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 6.McGaffin KR, Sun CK, Rager JJ, Romano LC, Zou B, Mathier MA, O'Doherty RM, McTiernan CF, O'Donnell CP. Leptin signalling reduces the severity of cardiac dysfunction and remodelling after chronic ischaemic injury. Cardiovasc Res. 2008;77:54–63. doi: 10.1093/cvr/cvm023. [DOI] [PubMed] [Google Scholar]
- 7.Fujimaki S, Kanda T, Fujita K, Tamura J, Kobayashi I. The significance of measuring plasma leptin in acute myocardial infarction. J Int Med Res. 2001;29:108–113. doi: 10.1177/147323000102900207. [DOI] [PubMed] [Google Scholar]
- 8.Schulze PC, Kratzsch J, Linke A, Schoene N, Adams V, Gielen S, Erbs S, Moebius-Winkler S, Schuler G. Elevated serum levels of leptin and soluble leptin receptor in patients with advanced chronic heart failure. Eur J Heart Fail. 2003;5:33–40. doi: 10.1016/s1388-9842(02)00177-0. [DOI] [PubMed] [Google Scholar]
- 9.Marino JH, Cook P, Miller KS. Accurate and statistically verified quantification of relative mRNA abundances using SYBR Green I and real-time RT-PCR. J Immunol Methods. 2003;283:291–306. doi: 10.1016/s0022-1759(03)00103-0. [DOI] [PubMed] [Google Scholar]
- 10.Goetze J, Christoffersen C, Perko M, Arendrup H, Rehfeld JF, Kastrup J, Nielsen LB. Increased cardiac BNP expression associated with myocardial ischemia. FASEB J. 2003;17:1105–1107. doi: 10.1096/fj.02-0796fje. [DOI] [PubMed] [Google Scholar]
- 11.Dotsch J, Nusken KD, Knerr I, Kirschbaum M, Repp R, Rascher W. Leptin and neuropeptide Y gene expression in human placenta: ontogeny and evidence for similarities to hypothalamic regulation. J Clin Endocrinol Metab. 1999;84:2755–2758. doi: 10.1210/jcem.84.8.5892. [DOI] [PubMed] [Google Scholar]
- 12.Meller M, Qui C, Vadachkoria S, Abetew DF, Luthy DA, Williams MA. Changes in placental adipocytokine gene expression associated with gestational diabetes mellitus. Physiol Res. 2006;55:501–512. doi: 10.33549/physiolres.930830. [DOI] [PubMed] [Google Scholar]
- 13.Olsen T, Goll R, Cui G, Husebekk A, Vonen B, Birketvedt GS, Florholmen J. Tissue levels of tumor necrosis factor-alpha correlates with grade of inflammation in untreated ulcerative colitis. Scand J Gastroenterol. 2007;42:1312–1320. doi: 10.1080/00365520701409035. [DOI] [PubMed] [Google Scholar]
- 14.Murphy R, Watt KO, Cameron-Smith D, Gibbons CJ, Snow RJ. Effects of creatinine supplementation on housekeeping genes in human skeletal muscle using real-time RT-PCR. Physiol Genomics. 2003;12:163–174. doi: 10.1152/physiolgenomics.00060.2002. [DOI] [PubMed] [Google Scholar]
- 15.Ririe KM, Rasmussen RP, Wittwer CT. Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal Biochem. 1997;245:154–160. doi: 10.1006/abio.1996.9916. [DOI] [PubMed] [Google Scholar]
- 16.Lekanne-Deprez RH, F A, Ruijter JM, Moorman AF. Sensitivity and accuracy of quantitative real-time polymerase chain reaction using SYBR green I depends on cDNA synthesis conditions. Anal Biochem. 2002;309:63–69. doi: 10.1016/s0003-2697(02)00021-0. [DOI] [PubMed] [Google Scholar]
- 17.Liu W, Saint DA. Validation of a quantitative method for real time PCR kinetics. Biochem Biophys Res Commun. 2002;294:347–353. doi: 10.1016/S0006-291X(02)00478-3. [DOI] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:204–208. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 19.Bouhidel O, Pons S, Souktani R, Zini R, Berdeaux A, Ghaleh B. Myocardial ischemic postconditioning against ischemia-reperfusion is impaired in ob/ob mice. Am J Physiol Heart Circ Physiol. 2008;295:H1580–1586. doi: 10.1152/ajpheart.00379.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barouch LA, Gao D, Lei C, Miller KL, Xu W, Phan AC, Kittleson MM, Minhas KM, Berkowitz DE, Wei C, Hare JM. Cardiac myocyte apoptosis is associated with increased DNA damage and decreased survival in murine models of obesity. Circ Res. 2006;98:119–124. doi: 10.1161/01.RES.0000199348.10580.1d. [DOI] [PubMed] [Google Scholar]
- 21.Mazumder PK, O'Neil BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, Boudina S, Abel DE. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53:2366–2374. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- 22.Perego L, Pizzocri P, Corradi D, Maisano F, Paganelli M, Fiorina P, Barbieri M, Morabito A, Paolisso G, Folli F, Pontiroli AE. Circulating leptin correlates with left ventricular mass in morbid (grade III) obesity before and after weight loss induced by bariatric surgery: a potential role for leptin in mediating human left ventricular hypertrophy. J Clin Endocrinol Metab. 2005;90:4087–4093. doi: 10.1210/jc.2004-1963. [DOI] [PubMed] [Google Scholar]
- 23.Abe Y, O K, Kawamura T, Wada H, Kita T, Shimatsu A, Hasegawa K. Leptin induces elongation of cardiac myocytes and causes eccentric left ventricular dilation with elongation. Am J Physiol Heart Circ Physiol. 2007;292:H2387–2396. doi: 10.1152/ajpheart.00579.2006. [DOI] [PubMed] [Google Scholar]
- 24.Frederich RC, Lollmann B, Hamann A, Napolitano-Rosen A, Kahn BB, Lowell BB, Flier JS. Expression of ob mRNA and its encoded protein in rodents. Impact of nutrition and obesity. J Clin Invest. 1995;96:1658–1663. doi: 10.1172/JCI118206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kinzig KP, Hargrave SL, Tao EE. Central and peripheral effects of chronic food restriction and weight restoration in the rat. Am J Physiol Endocrinol Metab. 2009;296:E282–290. doi: 10.1152/ajpendo.90523.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Purdham DM, Zou MX, Rajapurohitam V, Karmazyn M. Rat heart is a site of leptin production and action. Am J Physiol Heart Circ Physiol. 2004;287:H2877–2884. doi: 10.1152/ajpheart.00499.2004. [DOI] [PubMed] [Google Scholar]
- 27.Lavie CJ, Milani RV, Ventura HO. Obesity and cardiovascular disease. J Am Coll Cardiol. 2009;53:1925–1932. doi: 10.1016/j.jacc.2008.12.068. [DOI] [PubMed] [Google Scholar]
- 28.Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev. 2008;88:389–419. doi: 10.1152/physrev.00017.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hilfiker-Kleiner D, Hilfiker A, Fuchs M, Kaminski K, Schaefer A, Schieffer B, Hillmer A, Schmiedl A, Ding Z, Podewski E, Podewski E, Poli V, Schneider MD, Schulz R, Park JK, Wollert KC, Drexler H. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res. 2004;95:187–195. doi: 10.1161/01.RES.0000134921.50377.61. [DOI] [PubMed] [Google Scholar]
- 30.Hilfiker-Kleiner D, Hilhinfer A, Drexler H. Many good reasons to have STAT3 in the heart. Pharmacol Ther. 2005;107:131–137. doi: 10.1016/j.pharmthera.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 31.Russell RR, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ, Young LH. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Young LH, Li J, Baron SJ, Russell RR. AMP-activated protein kinase: a key stress signaling pathway in the heart. Trends Cardiovasc Med. 2005;15:110–118. doi: 10.1016/j.tcm.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 33.Lee SH, Doliba N, Osbakken M, Oz M, Mancini D. Improvement of myocardial mitochondrial function after hemodynamic support with left ventricular assist devices in patients with heart failure. J Thorac Cardiovasc Surg. 1998;116:344–349. doi: 10.1016/s0022-5223(98)70136-9. [DOI] [PubMed] [Google Scholar]
- 34.Burkhoff D, Holmes J, Madigan J, Barbone A, Oz MC. Left ventricular assist device-induced reverse ventricular remodeling. Prog Cardiovasc Dis. 2000;43:19–26. doi: 10.1053/pcad.2000.7190. [DOI] [PubMed] [Google Scholar]
- 35.Zafeiridis A, Jeevanandam V, Houser SR, Margulies KB. Regression of cellular hypertrophy after left ventricular assist device support. Circulation. 1998;98:656–662. doi: 10.1161/01.cir.98.7.656. [DOI] [PubMed] [Google Scholar]
- 36.Baba HA, Stypmann J, Grabellus F, Kirchhof P, Sokoll A, Schäfers M, Takeda A, Wilhelm MJ, Scheld HH, Takeda N, Breithardt G, Levkau B. Dynamic regulation of MEK/Erks and Akt/GSK-3beta in human end-stage heart failure after left ventricular mechanical support: myocardial mechanotransduction-sensitivity as a possible molecular mechanism. Cardiovasc Res. 2003;59:390–399. doi: 10.1016/s0008-6363(03)00393-6. [DOI] [PubMed] [Google Scholar]
- 37.Rastogi S, Gupta RC, Mishra S, Morita H, Tanhehco EJ, Sabbah HN. Long-term therapy with the acorn cardiac support device normalizes gene expression of growth factors and gelatinases in dogs with heart failure. Heart Lung Transplant. 2005;24:1619–1625. doi: 10.1016/j.healun.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 38.Blaxall BC, Tschannen-Moran MB, Milano CA, Koch WJ. Differential gene expression and genomic patient stratification following left ventricular assist device support. J Am Coll Cardiol. 2003;41:1096–1106. doi: 10.1016/s0735-1097(03)00043-3. [DOI] [PubMed] [Google Scholar]
- 39.Smith CC, Mocanu MM, Davidson SM, Wynne AM, Simpkin JC, Yellon DM. Leptin, the obesity-associated hormone, exhibits direct cardioprotective effects. Br J Pharmacol. 2006;149:5–13. doi: 10.1038/sj.bjp.0706834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tajmir P, Ceddia RB, Li RK, Coe IR, Sweeney G. Leptin increases cardiomyocyte hyperplasia via extracellular signal-regulated kinase and phosphatidylinositol 3-kinase-dependent pathways. Endocrinology. 2004;145:1550–1555. doi: 10.1210/en.2003-1128. [DOI] [PubMed] [Google Scholar]
- 41.Kurdi M, Booz GW. Can the protective actions of JAK-STAT in the heart be exploited therapeutically? Parsing the regulation of interleukin-6-type cytokine signaling. J Cardiovasc Pharmacol. 2007;50:126–141. doi: 10.1097/FJC.0b013e318068dd49. [DOI] [PubMed] [Google Scholar]
- 42.Gonon AT, Widergren U, Bulhak A, Salehzadeh F, Persson J, Sjöquist PO, Pernow J. Adiponectin protects against myocardial ischaemia-reperfusion injury via AMP-activated protein kinase, Akt, and nitric oxide. Cardiovasc Res. 2008;78:116–122. doi: 10.1093/cvr/cvn017. [DOI] [PubMed] [Google Scholar]
- 43.Zolk O, Ng Ll, O'Brien RJ, Weyand M, Eschenhagen T. Augmented Expression of Cardiotrophin-1 in Failing Human Hearts Is Accompanied by Diminished Glycoprotein 130 Receptor Protein Abundance. Circulation. 2002;106:1442–1446. doi: 10.1161/01.cir.0000033117.39335.df. [DOI] [PubMed] [Google Scholar]
- 44.Podewski EK, Hilfiker-Kleiner D, Hilfiker A, Morawietz H, Lichtenberg A, Wollert KC, Drexler H. Alterations in Janus kinase (JAK)-signal transducers and activators of transcription (STAT) signaling in patients with end-stage dilated cardiomyopathy. Circulation. 2003;107:798–802. doi: 10.1161/01.cir.0000057545.82749.ff. [DOI] [PubMed] [Google Scholar]
- 45.Ng DC, Court NW, dos Remedios CG, Bogoyevitch MA. Activation of signal transducer and activator of transcription (STAT) pathways in failing human hearts. Cardiovasc Res. 2003;57:333–346. doi: 10.1016/s0008-6363(02)00664-8. [DOI] [PubMed] [Google Scholar]
- 46.Minokoshi Y, Kim Yb, Peroni OD, Fryer LG, Müller C, Carling D, Kahn BB. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415:339–343. doi: 10.1038/415339a. [DOI] [PubMed] [Google Scholar]
- 47.Atkinson LL, Fischer MA, Lopaschuk GD. Leptin activates cardiac fatty acid oxidation independent of changes in the AMP-activated protein kinase-acetyl-CoA carboxylase-malonyl-CoA axis. J Biol Chem. 2002;277:29424–29430. doi: 10.1074/jbc.M203813200. [DOI] [PubMed] [Google Scholar]
- 48.Xydas S, Rosen RS, Ng C, Mercando M, Cohen J, DiTullio M, Magnano A, Marboe CC, Mancini DM, Naka Y, Oz MC, Maybaum S. Mechanical unloading leads to echocardiographic, electrocardiographic, neurohormonal, and histologic recovery. J Heart Lung Transplant. 2006;25:7–15. doi: 10.1016/j.healun.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 49.Maybaum S, Mancini D, Xydas S, Starling RC, Aaronson K, Pagani FD, Miller LW, Margulies K, McRee S, Frazier OH, Torre-Amione G, LVAD Working Group Cardiac improvement during mechanical circulatory support: a prospective multicenter study of the LVAD Working Group. Circulation. 2007;(115):2497–2505. doi: 10.1161/CIRCULATIONAHA.106.633180. [DOI] [PubMed] [Google Scholar]
- 50.Eguchi M, Liu Y, Shin EJ, Sweeney G. Leptin protects H9c2 rat cardiomyocytes from H2O2-induced apoptosis. FEBS J. 2008;275:3136–3144. doi: 10.1111/j.1742-4658.2008.06465.x. [DOI] [PubMed] [Google Scholar]
- 51.Erkasap N, Ikizler M, Shneyvays V, Zinman T, Mamedova LK, Uyar R, Shainberg A. Leptin protects the cardiac myocyte cultures from hypoxic damage. Life Sci. 2006;78:1098–1102. doi: 10.1016/j.lfs.2005.06.039. [DOI] [PubMed] [Google Scholar]