

Abstract
Metalloproteins contain highly specialized metal-binding sites that are designed to accept specific metal ions to maintain correct function. Though many of these sites have been modified with success, the relative paucity of functional group availability within proteinogenic amino acids can sometimes leave questions remaining about specific functions of the metal binding ligands. Attaining a more thorough analysis of individual amino acid function within metalloproteins has been realized using Expressed Protein Ligation (EPL). Here we describe our recent efforts using EPL to incorporate nonproteinogenic cysteine and methionine analogs into the Type 1 site found in Pseudomonas aeruginosa azurin.
Introduction
Protein design and engineering has contributed greatly to our understanding of protein structure and function. A common practice in the field is to use site-directed mutagenesis to replace a specific amino acid in proteins with other natural amino acids. While quite powerful, it has become increasingly evident that site-directed mutagenesis cannot address the precise role of a specific amino acid because, with limitation of 20 natural amino acids, it is often difficult to change one factor such as an electronic effect without simultaneously changing other factors such as steric effects. This limitation is even more evident in metalloprotein design and engineering, as even a smaller number of natural amino acids are capable of coordinating to metal ions (Lu, 2006b; Lu et al., 2001). More importantly, the metal-binding sites are intricately designed to accept a specific metal ion among a large number of other metal ions for binding and activity. Since geometry and ligand donor sets of metal-binding sites are much more varied than those of metal-free active sites, a subtle change of one factor can have a profound effect on the structure and activity. Therefore the need for introducing unnatural amino acids into metalloproteins for studies such as isostructural substitutions is even greater. Such an endeavor can not only define the role of amino acids more precisely, but also can produce novel metalloproteins with predicted and unprecedented properties (Lu, 2005; Lu, 2006a). As described herein, this need has recently been met by utilizing the Expressed Protein Ligation (EPL) technique.
An excellent test case for engineering of metalloprotein is the Type I (T1) blue copper proteins, a class of common electron transfer (ET) proteins involved in many important biological processes such as photosynthesis and respiration (Lu, 2003; Vila and Fernandez, 2001). The blue copper center displays much higher reduction potentials and much more efficient electron transfer properties than those of the same copper complexes found outside of the protein environment. A substantial amount of research has been applied to understanding the origin for these exceptional functional properties. Pseudomonas aeruginosa azurin is a member of type 1 (T1) or blue copper proteins. In its resting state, the azurin blue copper center contains a Cu(II) ion bound in a trigonal bipyrimidal plane by two equatorial histidines (His46, His117) and one equatorial cysteine (Cys112) (Figure 1). Similar to the majority of other blue copper proteins, the azurin T1 site has a methionine sulfur thioether (Met121) as an axial ligand. Unique to azurin, however, the backbone carbonyl from a glycine residue (Gly45) is present opposite to the methionine. The T1 site is defined by an unusually short copper to cysteine bond (2.1 Å) and this shortened bond, together with its unique metal-binding site geometry, is responsible for an intense ligand to metal charge transfer (LMCT) band resulting from the Sγ(Cys112) 3pπ → Cu(II) dx2−y2 charge transfer transition giving azurin its strong UV/Vis absorption at 625 nm and thus its intense blue color (Malmström, 1994; Pierloot et al., 1997; Solomon et al., 1992). The highly covalent nature of the LMCT between the copper and Cys ligand is also responsible for the narrow hyperfine splittings in the parallel region of the EPR spectra (A|| ~ 60 × 10−4 cm−1), smaller than those of “normal” copper coordination compounds ((A|| 160 × 10−4 cm−1) (Solomon et al., 1992).
Figure 1.
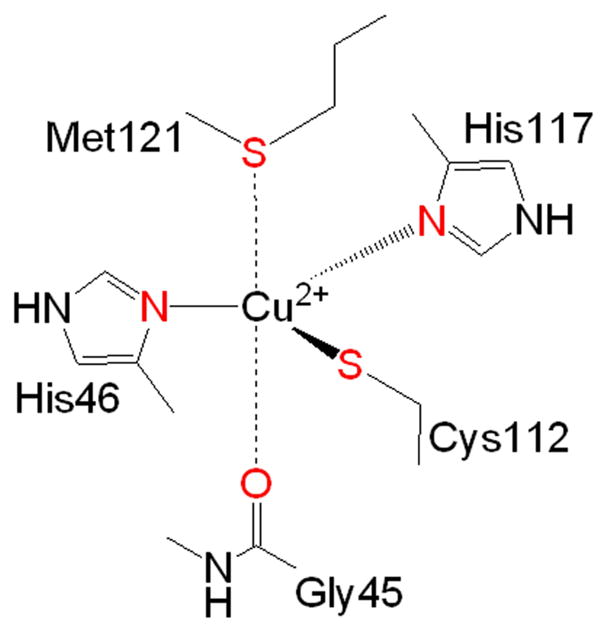
The T1 Site of Pseudomonas aeruginosa azurin
In azurin, both the equatorial Cys112 ligand and the axial Met121 ligand have been implicated in the origin of the large increase in redox potential and more efficient ET properties over “normal” copper complexes. Serving as a model protein for the study of this class of metalloproteins, the T1 site in azurin has been exhaustively modified using site-directed mutagenesis and its variants have been extensively studied using a broad range of spectroscopic and crystallographic techniques. These thorough investigations have shown that Cys112 is essential for protein function. Early studies established the loss of the azurin spectral properties when Cys112 was substituted with other natural amino acids differing greatly in size and electronics, whereas substitution of the remaining three ligands, His46, His117, and Met121 still yielded proteins dominated by the strong LMCT and blue color (Chang et al., 1991; Den Blaauwen et al., 1991; Germanas et al., 1993; Karlsson et al., 1989). However, the presence of the Cys112 thiol is not a requisite for copper binding. A single Cys112 mutant, Cys112Asp retained the ability to bind copper (Mizoguchi et al., 1992), but all of the spectral features including the distinct LMCT band at 625 nm disappeared, and the ET properties of azurin were greatly reduced (Faham et al., 1997; Mizoguchi et al., 1992). Further spectroscopic evidence demonstrated that the original T1 site had been transformed to a type 2 (T2) “normal” copper site with a bidentate bound Asp (DeBeer et al., 1999; Mizoguchi et al., 1992). While these site-directed mutagenesis studies suggest the Cys ligand is essential in maintaining blue copper structural character and efficient ET properties, the structural features of Cys that are responsible for the character and properties of the site remain unclear as mutation of Cys to other natural amino acids changes more than one factor at the time. A new methodology was needed to more precisely study the effects of Cys112 in the T1 site.
To achieve this goal, we chose to incorporate selenocysteine (Sec or U) as an isosteric cysteine analogue. Since Se and S share similar atomic radii, electronegativity, and oxidation states, the replacement by Sec is much more conserved than replacement of Cys with other naturally occurring amino acids (Figure 2). The similarity also yields comparable oxidized analogues (e.g. disulfides and diselenides) and allows for mixed S-Se compounds (Gieselman et al., 2002; Moroder, 2005), a feature that the natural amino acid Ser cannot mimic (Wessjohann et al., 2007). However, although both Se and S are Group VIA elements with similar physical characteristics, they exhibit different reactivities. Most notably, the pKa of Sec (5.2) is much lower than that of Cys (8.6), which suggests that at physiological pH Sec remains in its anionic selenolate form rendering it more nucleophilic than Cys (Johansson et al., 2005; Moroder, 2005). Unfortunately the replacement of Cys with Sec is not easily accomplished by site directed mutagenesis and requires the use of alternative techniques for its incorporation.
Figure 2.

Comparison of the Physiochemical Properties of Cysteine and Selenocysteine.
Methods of Selenocysteine Incorporation
Several methods have been developed to incorporate selenocysteine including three biosynthetic techniques (auxotrophic substitution, use of Sec Insertion Sequences (SECIS), and employment of tRNA/synthetase pairs), one chemical synthetic technique (Native Chemical Ligation) and one semi-synthetic technique (Expressed Protein Ligation). The incorporation of seleno-amino acid analogues such as selenomethionine using E. coli auxotrophs has long been performed to solve phasing problems within protein crystallography. This methodology has also been applied to the use of cysteine auxotrophic bacteria to substitute cysteine residues with selenocysteine (Cheong et al., 2007; Neuwald et al., 1992). Unfortunately the auxotrophic method for replacement of sulfur containing amino acids with their seleno-derivatives suffers from two key limitations. First, the method replaces any cysteine found in the protein with selenocysteine and thus alters more than one single site of interest without control over which positions are being substituted. Secondly and more importantly, complete replacement of cysteine with selenocysteine in proteins using this technique has yet to be accomplished, with typical replacement percentages between 60–80% (Boschi-Muller et al., 1998; Mueller et al., 1994). Azurin has three Cys residues in the protein and thus full replacement of Cys112 (the copper binding ligand) cannot be 100% efficient, complicating characterization of the Sec mutant protein.
To overcome the inability to control both the location and level of Sec replacement using cysteine E. coli auxotrophs, the incorporation of Sec into proteins may be accomplished by the use of Sec Insertion Sequences or SECIS elements. In order to correctly recognize the UGA codon for Sec insertion and not as a stop codon, the SECIS element is used. Incorporation of selenocysteine during protein translation parallels proteinogenic amino acid translation, using a specific, tRNASec. The Sec tRNA is initially charged with a serine residue that is converted to selenocysteine by Sec synthase using selenomonophosphate as the selenium donor (Fischer et al., 2007; Su et al., 2005). The charged tRNASec is then guided into the ribosome by the elongation factor EF-Tu homolog SelB, where it undergoes GTP hydrolysis resulting in Sec incorporation at the UGA codon (Fischer et al., 2007; Hoffmann and Berry, 2005).
Recombinant selenoprotein production is relatively difficult due not only to the number of required elements needed for proper recognition of the UGA stop codon and subsequent Sec incorporation, but also due to incompatibilities between the prokaryotic and eukaryotic SECIS sequences and the inability to incorporate multiple Sec residues in a single protein. The primary incompatibility between prokaryotes and eukaryotic SECIS elements is location of and the distance between the SECIS loop and the UGA codon. In prokaryotic systems, the SECIS loop is located in the coding region at a distance of ~20 nucleotides from the UGA codon. Thus there are amino acids following the UGA codon that are needed for complete protein translation (Su et al., 2005). However the eukaryotic and archea SECIS loops are found in the 3′ untranslated region and therefore have variable distances from the UGA codon (Kryukov et al., 2003). This incompatibility hinders the usage of common prokaryotic expressions systems for recombinant expression of mammalian selenoproteins. Furthermore, only a single Sec can be inserted using this technique limiting its usefulness.
The last and most current technique for specific biological incorporation of selenocysteine (as well as other unnatural amino acids) is the nonsense suppression technique that utilizes orthogonal tRNA/synthetase pairs. By utilizing rounds of positive and negative selection, a tyrosyl amber suppressor tRNA/tRNA synthetase pair from Methanococcus jannaschii was created that allows translational incorporation of phenylselenocysteine into proteins at the desired location (Wang et al., 2007). Phenylselenocysteine however cannot be used as a ligand because the phenyl group is not readily removable. Currently no orthogonal tRNA/tRNA synthetase pair has been reported to accept and incorporate the free selenol form of selenocysteine using this methodology.
Incorporation of Selenocysteine into the T1 Site of Azurin
Because EPL permits the direct control over the location of amino acid substitutions with unnatural amino acids, this technique met our needs very well. Azurin is well suited for EPL as the protein is relatively small at 128 amino acids in length (Figure 3). Furthermore, three of the four copper binding ligands are found within the C-terminal 17 amino acids (H2N-CTFPGHSALMKGTLTLK-COOH) of the protein, making substitutions at these positions conveniently accessible. Therefore, azurin was prepared semi-synthetically by using a recombinantly expressed truncated protein containing the first 111 amino acids with a thioester at its C-terminus, and a synthetic peptide containing the last 17 amino acids including the replacement of Cys with Sec to afford a full length selenocysteine substituted azurin variant (C112U azurin).
Figure 3.

Amino acid sequence of Pseudomonas aeruginosa azurin. Bold residues are those involved in copper binding. The final 17 amino acids used for SPPS are italicized.
Substitution of Cys112 with selenocysteine requires a synthetic peptide with an N-terminal Sec residue that can participate directly in the ligation reaction. The use of Sec residues for EPL has been reported by several groups. In 2002 Hondal and Raines showed that replacement of Cys110 in RNase A with Sec could be accomplished using a synthetic 15 amino acid peptide containing an N-terminal Sec residue (Hondal and Raines, 2002). The EPL product protein was indistinguishable from the WT protein with similar rates of catalysis indicating that replacement of Cys with Sec did not dramatically perturb either function or protein folding. More recently, Hondal has demonstrated Sec incorporation into High Mr thioredoxins without disruption of protein folding (Eckenroth et al., 2006; Eckenroth et al., 2007). Concurrently, we developed and utilized similar methods for azurin as detailed below (Berry et al., 2002; Gieselman et al., 2001; Ralle et al., 2004).
Synthesis of Fmoc-Sec(PMB)-OH
In order to accomplish substitution of Cys112 with selenocysteine, a derivative of selenocysteine was needed that was suitable for standard Fmoc-based Solid Phase Peptide Synthesis (SPPS). The most common derivative of selenocysteine used for SPPS has been Fmoc-Sec(PMB)-OH (PMB: p-methoxybenzyl) primarily due to incompatibilities with the trityl (Trt) protecting group (Theodoropoulos et al., 1967a; Theodoropoulos et al., 1967b; Walter and Chan, 1967). Many procedures have been developed over the years to synthesize Fmoc-Sec(PMB)-OH from various starting materials. In recent years, we utilized a slightly modified and optimized scheme of our original methodology (Gieselman et al., 2001). In this route (Scheme 1) the final allyl deprotection using Pd(PPh3)4 was performed in the presence of sodium 2-ethylhexanoate as the allyl transfer reagent rather than morpholine to prevent the partial removal of the Fmoc group that was observed with the latter reagent.
Scheme 1.

Synthetic route for Fmoc-Sec(PMB)-OH from Fmoc-Ser-OH (Gieselman et al., 2001). Conditions in bold are modified and optimized conditions used for allyl deprotection
Starting from inexpensive Fmoc-Ser-OH, the precursor Fmoc-Se(PMB)-Sec-OAll compound was prepared in 3 steps (Scheme 1). The final optimized step was carried out as described by McCombie with slight modifications (Jeffrey and McCombie, 1982). Pd(PPh3)4 (556 mg, 0.48 mmol) and PPh3 (126 mg, 0.48 mmol) were combined in a dried round bottom flask and purged with Ar. The flask was covered with aluminum foil and dry CH2Cl2 (20 mL) was added. To a second round bottom flask, Fmoc-Se(PMB)-Sec-OAll (5.3 g, 9.63 mmol) and Na 2-ethylhexanoate (1.62 g, 9.73 mmol) were added, dissolved in dry CH2Cl2 (20 mL) and the contents of the flask were transferred to the Pd catalyst via cannulation under Ar pressure. The reaction was stirred at 25 °C for 20 min under Ar. After completion, the reaction was quenched with EtOAc (50 mL) and washed with 2 × 50 mL each of 2 M, 4 M, and 6 M HCl. The organic layer was then dried over MgSO4, filtered, and the solvent was evaporated to obtain yellow oil. The crude product was purified by flash chromatography using 35:1 CH2Cl2/MeOH with 0.1% AcOH (v/v) to obtain the final product as a white solid. (Rf = 0.5, 9:1 CH2Cl2/MeOH, 4.7 g, 95%). After successful synthesis of Fmoc-Sec(PMB)-OH, it was then incorporated into the C-terminal 17 amino acid sequence of azurin using standard Fmoc-based chemistry.
Solid Phase Peptide Synthesis of C-Terminal 17-mer Peptide
The C-terminal 17-mer peptide containing selenocysteine (H2N-UTFPGHSALMKGTLTLK-COOH) was synthesized on a 0.1 mmol scale with a Rainin PS3 automated synthesizer using standard Fmoc chemistry. Preloaded Fmoc-Lys(Boc)-Wang resin was first swelled in DMF (3 × 8 min, 6 mL). All Fmoc deprotections were accomplished using 20% piperidine/DMF (v/v) (3 × 3 min, 6 mL). For amino acid couplings, a 5-fold excess of Fmoc-amino acid and O-benzotriazole-N,N,N′,N′,-tetramethyl-uroniumhexafluoro-phosphate (HBTU) were combined and 0.4 M N-methylmorpholine (1 × 1.5 min) was used as the activating reagent. The activated amino acids were then transferred to the reaction vessel and coupled under N2 for 45 min.
The coupling of Fmoc-Sec(PMB)-OH was performed using 2 equivalents of 1-hydroxybenzotriazole (HOBT, 6 equiv) and N,N′-diisopropylcarbodiimide (DIC, 6 equiv) in DMF to reduce the possibility of racemization (Han et al., 1997). The N-terminal Fmoc group was then removed using 20% piperidine/DMF (v/v, 3 × 2 min, 6 mL) and the resin was washed extensively with CH2Cl2 and dried in a dessicator for 10 min. Due to the sensitivity of selenocysteine to both racemization and beta-elimination, special selenol deprotection conditions were needed during SPPS.
Deprotection of Selenocysteine-containing 17-mer Peptide
Several studies have been performed using a multitude of reagents in order to successfully and fully deblock selenols- and thiols- protected with PMB but many of these reagents are harsh and do not result in full deblocking of the thiol/selenol and can promote deselenization (Hunter and Komives, 1995; Otaka et al., 1991; Yajima et al., 1988). A review of these techniques was published explaining the advantages and disadvantages of each of these techniques (Hondal, 2005). Recently a new, milder technique for deblocking selenols protected with PMB was developed using 2,2′-dithiobis(5-nitropyridine) (Harris et al., 2007). A modified version of this procedure was used in our work to afford fully deblocked peptide during the cleavage from the resin.
For a 0.1 mmol scale peptide synthesis, 2,2′-dithiobis(5-nitropyridine) (1.3 mol equiv, 41 mg) was added to the dried resin-bound peptide and 2% (v/v) triisopropylsilane (100 μL) and ddH2O (100 μL) were added followed by the addition of 2.5% (v/v) thioanisole (125 μL). To the mixture, 5 mL of neat TFA was added and the reaction was stirred vigorously at 25 °C for 2 h. The reaction was filtered through a coarse grain Büchner funnel and the resin was washed with neat TFA (3 × 5 mL). The TFA was then removed via rotary evaporation to obtain a yellow oil. The peptide was precipitated from the oil by the addition of cold Et2O and pelleted from the solution by centrifugation at 4000 rpm for 5 min. The pellet was then washed with Et2O (3 × 30 mL) with centrifugation steps between each washing. The precipitated peptide was then dissolved in a minimal amount of 1:1 MeCN/H2O and lyophilized overnight. The crude lyophilized product was purified by preparative RP-HPLC on a Waters C18 column (130 mm × 25 mm) with a water-acetonitrile solvent system using a gradient of 20–70% of solvent B over 20 min (A: H2O, 0.1% TFA; B: CH3CN 80% in H2O v/v, 0.1% TFA). Peptides were lyophilized after purification to obtain final product as a pale yellow solid and analyzed using MALDI-TOF-MS; Calculated. 1852.06 (SeH), 2006.21 (Se(pys)), 3704.1 (Dimer) Observed. 1852.08 (SeH), 3703.6 (Dimer). The typical yield for a 0.1 mmol scale synthesis was 70–80 mg (~95% coupling efficiency). The mass spectrum displayed the typical isotopic distribution pattern expected for a peptide containing one selenium atom (Figure 4).
Figure 4.

MALDI-TOF-MS for A) Deprotected C112U Azurin 17mer B) Simulated data
To accomplish incorporation of selenocysteine into azurin using EPL, the first 111 amino acids of azurin were recombinantly expressed as an Intein-Chitin binding domain (CBD) fusion using NEB vector PtxB1 intein splicing technology. After expression and intein mediated cleavage with various thiol reagents, the desired recombinant thioester could not be isolated due to its insolubility and facile thioester hydrolysis. Therefore ligations were performed on the column with the final 17 amino acid peptide in the presence of N-methylmercaptoacetamide (NMA).
General procedure for EPL of Azurin(1–111)- Intein-CBD and the C-terminal 17-mer peptide
The azurin(1–111)- Intein-CBD fusion was over-expressed in BL21 E. coli in LB media for 8 h at 37 °C. The cells from 5 mL starter culture were used to inoculate 2 L of LB and the cells were grown at 37 °C for 16 h at 210 rpm. Protein expression was induced at ~16 h with 0.3 mM IPTG and induction was continued for 4 h at 37 °C. The cells were then harvested at 9000 rpm, frozen at −20 °C and used when needed.
For ligations, the frozen cell stock was resuspended in a lysis buffer (20 mM HEPES, pH 8.0, 250 mM NaCl, 1 mM EDTA, 1 mM PMSF, 0.1% Triton-x-100) and was lysed twice with a French press. The cell lysate was then treated with 2 M urea for 10 min and the lysate was centrifuged at 13,000 rpm for 30 min. The resulting supernatant was added to chitin resin (80 mL) pre-equilibrated in 20 mM HEPES, pH 8.0, 250 mM NaCl, 1 mM EDTA (Buffer 1). The mixture was shaken gently at 4 °C for 1–2 h and poured into a column. The column was sealed with a septum and the headspace of the column was purged with Ar. The column was then washed with 3 column volumes buffer 1 containing 2 M urea under Ar pressure followed by 5–7 column volumes of buffer 1.
The 17-mer peptide (0.675 mM, 80 mg) and 2 equiv tris-(2-carboxyethyl)phosphine TCEP (1.35 mM, 27 mg) were dissolved in 40 mL of Buffer 1 containing 75 mM NMA under Ar and transferred to the column under Ar. The chitin resin was then suspended in the above buffer and agitated gently at 4 °C for 66 h to promote mixing and reaction. After ligation, the column was eluted and washed with 1 column volume of buffer 1. The eluent was centrifuged at 14,000 rpm for 30 min and the supernatants were combined and concentrated using 10,000 MWCO centricon concentration spin tubes at 5,000 rpm and exchanged into 50 mM MOPS buffer, pH 6.6 via PD10 (G25 sephadex) columns. The desalted sample was then titrated with incremental additions of freshly made 20 mM CuSO4 solution in ddH2O that had been pretreated with chelex. The titration was performed at 4 °C until maximal absorbance at 675 nm was reached. Samples were then purified using a Pharmacia Q HiTrap HP column (5 mL) which had been washed with 10 column volumes of buffer B (1 M NaCl in H2O) and then equilibrated with 10 column volumes of buffer A (50 mM MOPS, pH 6.6). The sample was then concentrated and analyzed using MALDI-TOF-MS and ESI-MS (C112U Apo-Azurin Calculated: 13999, Observed: 14000. C112U Azurin + Cu Calculated: 14055, Observed: 14054).
Characterization of Selenocysteine Azurin
This procedure resulted in a C112U azurin mutant that was identical to that previously reported but with slightly better yield (~0.4 mg/L cell growth) than the previously reported yield (~0.25 mg/L cell growth) (Berry et al., 2002; Ralle et al., 2004). The C112U mutant protein demonstrated distinct differences from the WT protein. The first characteristic change was a red shift in the UV-Vis spectrum from 625 nm for WT to 677 nm for the C112U mutant likely resulting from the lower ionization energy of Se when compared to S. More dramatically are the changes noted in the EPR spectra between the two proteins (Figure 5). A marked increase of the parallel hyperfine splitting (100 G) of the C112U mutant compared to WT azurin (56 G) suggests that the Se-Cu bond is less covalent in nature than the S-Cu bond. Furthermore, the observed increased rhombic character of the site with a larger increase in gy than in the gz tensor is consistent with increased axial interaction or loss of Cu(II) interaction with the selenolate. This finding was expected as Se is softer than S and thus would prefer coordination with Cu(I) rather than Cu(II). Surprisingly however, no dramatic change was noted in the reduction potential of the site when the ligand was changed from Cys to Sec (Berry et al., 2002).
Figure 5.

Spectroscopic characterization of C112U azurin compared to WT azurin A) UV-Vis spectrum B) EPR spectrum
Tuning the Blue Copper Reduction Potential Using Methionine Analogs
In addition to substitution of the Cys ligand, several studies have reported replacement of the axial methionine ligand (Figure 1). A broad range of amino acid substitutions have been utilized and the variants typically still retained the native structure and function. Though a full set of saturated mutational data of the axial methionine (M121) to all other natural amino acids in azurin has been reported (Chang et al., 1991; Karlsson et al., 1991), precise elucidation of the role of this ligand remains challenging because mutations to other natural amino acids changed more than one factor at the time. To circumvent the limitations of proteinogenic amino acids, an early attempt for the incorporation of unnatural amino acids at position 121 has been reported (Frank et al., 1985). Using a P. aeruginosa strain exposed to rounds of selenomethionine (SeMet) selection, an expression system was created to substitute Met with SeMet. This methodology serves as an effective means for the incorporation of SeMet. However, absolute control over the location of incorporation could not be completed resulting in fully substituted SeMet azurin. Though useful, the inability to individually replace Met121 with SeMet could not be achieved, demonstrating the need for techniques such as EPL to more precisely control the nature of the axial ligand.
Recently, our lab has reported the substitution of the axial methionine with nonproteinogenic amino acids using EPL (Berry et al., 2003; Garner et al., 2006). By incorporation of a broad range of methionine analogs at the Met121 position, new insights have been obtained that were not possible by natural amino acid substitution.
Use of EPL for Incorporation of Methionine Analogs
As the final 17 amino acid sequence contains all of the copper binding ligands except His46, incorporation of methionine analogs used similar methodology as the incorporation of cysteine analogues into the azurin T1 site. Furthermore, the same protein thioester truncate of azurin containing the N-terminal 111 amino acids was utilized.
General Procedure for the SPPS of Methionine Analogue 17-mer Peptides
The C-terminal 17-mer peptides containing methionine analogues (H2N-CTFPGHSALxKGTLTLK-COOH, x= Met, SeMet,, OxoMethionine (OxM), Leu, Nle, DifluoroMethionine (DFM), or TrifluoroMethionine(TFM)) were synthesized on a 0.1 mmol scale with an Advanced Chemtech Model 90 peptide synthesizer using standard Fmoc chemistry. Preloaded Fmoc-Lys(Boc)-Wang resin was swelled in DMF and all Fmoc deprotections were accomplished using 20% piperidine/DMF (v/v). For the standard amino acid couplings, a 4-fold excess of Fmoc-amino acid and O-benzotriazole-N,N,N′,N′,-tetramethyl-uroniumhexafluoro-phosphate (HBTU) were combined and 0.4 M N-methylmorpholine (1 × 1.5 min) was used as the activating reagent. The activated amino acids were then transferred to the reaction vessel and coupled under N2. The coupling of the methionine analogs was performed using 1.5 equivalents of 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU). The N-terminal Fmoc protection was removed from the peptide using 20% piperidine/DMF (v/v) and the resin was dried overnight prior to cleavage.
General Procedure for the Peptide Cleavage from the Resin
To the product of a 0.1 mmol synthesis of peptide 5% (v/v) ddH2O, thioanisole, and anisole (500 μL each) were added. To the mixture, 10 mL of neat TFA was added and the reaction was stirred at RT for 2 h. The reaction was filtered through a coarse grain Büchner funnel and the resin was washed with neat TFA (3 × 5 mL). The TFA was then removed via rotary evaporation and the peptide was precipitated by the addition of cold Et2O. The crude peptide was then purified by preparative RP-HPLC on a Waters C18 column (130 mm × 25 mm) with a water-acetonitrile solvent system using a gradient of 20–70% of solvent B over 20 min (A: H2O, 0.1% TFA; B: CH3CN 80% in H2O v/v, 0.08% TFA). Peptides were lyophilized after purification to obtain final products as a white solids and were analyzed using MALDI-TOF-MS.
General procedure for EPL of Azurin(1–111)- Intein-CBD and the C-terminal Peptides Containing Methionine Analogs
The azurin(1–111)- Intein-CBD fusion was over-expressed as described above but in BLR1 E. coli at 30 °C and without IPTG induction. The ligation procedure was performed as previously described but with the following differences. As buffer, 20 mM HEPES, pH 8.0, 250 mM NaCl, 1 mM EDTA (buffer 2) was used. Though the stringent anaerobic environment needed for selenocysteine ligation was not needed for ligation of the Met analog peptides, the column headspace was still purged with Ar prior to ligation. The column was equilibrated with buffer 2 prior to adding the 17-mer peptide (10 mg) pre-dissolved in 4 mL of buffer 1 containing 50 mM NMA. The peptide mixture was then transferred to the column and shaken gently at 4 °C for 60 h. After ligation the column was eluted, the eluent was treated and concentrated exactly as before but exchanged into 50 mM ammonium acetate buffer, pH 5.1 and titrated with incremental additions of freshly made CuSO4 solution (10 mM) in ddH2O. The titration was performed at 4 °C until maximal absorbance at 625 nm was reached. Samples were then purified via FPLC using anion exchange chromatography.
Characterization of Methionine Analogs of Azurin
The incorporation of methionine analogues into the azurin T1 site demonstrated that the use of isosteric structures reduces the perturbation of the site. UV-vis spectroscopy of all of the variants resulted in < 6 nm shifts in the S(Cys)-Cu charge transfer (CT) band in the electronic absorption spectra and < 8 G changes in the copper hyperfine coupling constants (A||) in the X-band EPR spectra.
In contrast to the minimal spectral feature changes that are visible from the substitution of the axial Met with isostructural amino acids, substantial deviations from the WT reduction potential are evident. It is well known that Met plays an important role in fine-tuning the redox potentials of the blue copper center, but the molecular origin remains unclear because conclusions drawn from site-directed mutagenesis depend heavily on the choice of natural amino acids used to replace Met. The incorporation of isostructural unnatural amino acids using EPL has made it possible to deconvolute different factors affecting the reduction potentials of the blue copper center. A careful analysis, including a plot of observed reduction potentials of the WT azurin and its variants and the corresponding hydrophobicity of the axial ligand side chain defined by the log of the partition coefficient (logP) (Figure 6), revealed hydrophobicity as the dominant factor in tuning the reduction potentials of blue copper centers by axial ligands (Berry et al., 2003). Extension of such a finding in azurin to other type 1 blue copper proteins established a linear correlation between hydrophobicity of the axial ligand and the reduction potential of all type 1 copper centers, independent of the protein scaffold, experimental conditions, measurement techniques and steric modifications (Garner et al., 2006).
Figure 6.

Reduction potential vs log P of azurin M121 variants. Unnatural amino acid labels: OxM=Oxomethionine, SeM=Selenomethionine, DFM=Difluoromethionine, TFM=Trifluoromethionine, Nle=Norleucine
Conclusion
In this work the specific functionalities of the cysteine ligand and the axial methionine ligand in azurin have been extensively probed utilizing unnatural amino acid substitutions via EPL. The incorporation of Sec to replace Cys created a variant in which the overall function and redox activity of azurin remained relatively unperturbed and for which noticeable spectroscopic changes can be attributed to the differences in the electronic characteristics between sulfur and selenium. A thorough investigation of the axial methionine by replacement with a broad range of electronic and hydrophobic mimics of Met have definitively shown that the axial methionine is a key determinant in the magnitude of the reduction potential of the T1 site found in azurin. Variants with more hydrophobic side-chains demonstrated large increases in reduction potential whereas variants that are highly electron withdrawing or that have charged side-chains display lower reduction potentials when compared to the WT protein. We have shown that engineering of metalloproteins can be accomplished using EPL to gain a better understanding of protein function and to alter the coordination sphere of the metal ion to fine tune the desired properties of the engineered variant.
Acknowledgments
We wish to thank Dr. Mark Nilges for help with EPR experiments and Mr. Furong Sun for technical assistance with Mass Spec analysis. The materials described here is based upon work supported by the National Science Foundation under Award No. CHE 05-52008 (to Y.L.), and the National Institutes of Health under Award No. GM58822 (to W.A.V.).
References
- Berry SM, Gieselman MD, Nilges MJ, van der Donk WA, Lu Y. An Engineered Azurin Variant Containing a Selenocysteine Copper Ligand. J Am Chem Soc. 2002;124:2084–2085. doi: 10.1021/ja0169163. [DOI] [PubMed] [Google Scholar]
- Berry SM, Ralle M, Low DW, Blackburn NJ, Lu Y. Probing the Role of Axial Methionine in the Blue Copper Center of Azurin with Unnatural Amino Acids. J Am Chem Soc. 2003;125:8760–8768. doi: 10.1021/ja029699u. [DOI] [PubMed] [Google Scholar]
- Boschi-Muller S, Muller S, Van Dorsselaer A, Bock A, Branlant G. Substituting selenocysteine for active site cysteine 149 of phosphorylating glyceraldehyde 3-phosphate dehydrogenase reveals a peroxidase activity. FEBS Lett. 1998;439:241–245. doi: 10.1016/s0014-5793(98)01377-5. [DOI] [PubMed] [Google Scholar]
- Chang TK, Iverson SA, Rodrigues CG, Kiser CN, Lew AYC, Germanas JP, Richards JH. Gene Synthesis, Expression, and Mutagenesis Of the Blue Copper Proteins Azurin and Plastocyanin. Proc Natl Acad Sci USA. 1991;88:1325–1329. doi: 10.1073/pnas.88.4.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong JJ, Hwang I, Rhee S, Moon T, Choi Y, Kwon HB. Complementation of an E. coli cysteine auxotrophic mutant for the structural modification study of 3′(2′),5′-bisphosphate nucleotidase. Biotechnol Lett. 2007;29:913–918. doi: 10.1007/s10529-007-9324-7. [DOI] [PubMed] [Google Scholar]
- DeBeer S, Kiser CN, Mines GA, Richards JH, Gray HB, Solomon EI, Hedman B, Hodgson KO. X-ray Absorption Spectra of the Oxidized and Reduced Forms of C112D Azurin from Pseudomonas aeruginosa. Inorg Chem. 1999;38:433–438. doi: 10.1021/ic9804622. [DOI] [PubMed] [Google Scholar]
- Den Blaauwen T, Van de Kamp M, Canters GW. Type I and II copper sites obtained by external addition of ligands to a His117Gly azurin mutant. J Am Chem Soc. 1991;113:5050–5052. [Google Scholar]
- Eckenroth B, Harris K, Turanov AA, Gladyshev VN, Raines RT, Hondal RJ. Semisynthesis and Characterization of Mammalian Thioredoxin Reductase. Biochemistry. 2006;45:5158–5170. doi: 10.1021/bi0517887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckenroth BE, Lacey BM, Lothrop AP, Harris KM, Hondal RJ. Investigation of the C-Terminal Redox Center of High-Mr Thioredoxin Reductase by Protein Engineering and Semisynthesis. Biochemistry. 2007;46:9472–9483. doi: 10.1021/bi7004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faham S, Mizoguchi TJ, Adman ET, Gray HB, Richards JH, Rees DC. Role of the active-site cysteine of Pseudomonas aeruginosa azurin. Crystal structure analysis of the CuII(Cys112Asp) protein. J Biol Inorg Chem. 1997;2:464–469. [Google Scholar]
- Fischer N, Paleskava A, Gromadski KB, Konevega AL, Wahl MC, Stark H, Rodnina MV. Towards Understanding Selenocysteine Incorporation into Bacterial Proteins. Biol Chem. 2007;388:1061–1067. doi: 10.1515/BC.2007.108. [DOI] [PubMed] [Google Scholar]
- Frank P, Licht A, Tullius TD, Hodgson KO, Pecht I. A selenomethionine-containing azurin from an auxotroph of Pseudomonas aeruginosa. J Biol Chem. 1985;260:5518–5525. [PubMed] [Google Scholar]
- Garner DK, Vaughan MD, Hwang HJ, Savelieff MG, Berry SM, Honek JF, Lu Y. Reduction Potential Tuning of the Blue Copper Center in Pseudomonas aeruginosa Azurin by the Axial Methionine as Probed by Unnatural Amino Acids. J Am Chem Soc. 2006;128:15608–15617. doi: 10.1021/ja062732i. [DOI] [PubMed] [Google Scholar]
- Germanas JP, Di Bilio AJ, Gray HB, Richards JH. Site saturation of the histidine-46 position in Pseudomonas aeruginosa azurin: Characterization of the His46Asp copper and cobalt proteins. Biochemistry. 1993;32:7698–7702. doi: 10.1021/bi00081a014. [DOI] [PubMed] [Google Scholar]
- Gieselman MD, Xie L, van der Donk WA. Synthesis of a Selenocysteine-Containing Peptide by Native Chemical Ligation. Org Lett. 2001;3:1331–1334. doi: 10.1021/ol015712o. [DOI] [PubMed] [Google Scholar]
- Gieselman MD, Zhu Y, Zhou H, Galonic D, van der Donk WA. Selenocysteine Derivatives for Chemoselective Ligations. Chembiochem. 2002;3:709–716. doi: 10.1002/1439-7633(20020802)3:8<709::AID-CBIC709>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Han Y, Albericio F, Barany G. Occurrence and Minimization of Cysteine Racemization during Stepwise Solid-Phase Peptide Synthesis. J Org Chem. 1997;62:4307–4312. doi: 10.1021/jo9622744. [DOI] [PubMed] [Google Scholar]
- Harris KM, Flemer S, Hondal RJ. Studies on deprotection of cysteine and selenocysteine side-chain protecting groups. J Pept Sci. 2007;13:81–93. doi: 10.1002/psc.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann PR, Berry MJ. Selenoprotein Synthesis: A Unique Translational Mechanism Used by a Diverse Family of Proteins. Thyroid. 2005;15:769–775. doi: 10.1089/thy.2005.15.769. [DOI] [PubMed] [Google Scholar]
- Hondal RJ. Incorporation of Selenocysteine into Proteins Using Peptide Ligation. Protein Pept Let. 2005;12:757–764. doi: 10.2174/0929866054864319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondal RJ, Raines RT. Semisynthesis of proteins containing selenocysteine. Methods Enzymol. 2002;347:70–83. doi: 10.1016/s0076-6879(02)47009-7. [DOI] [PubMed] [Google Scholar]
- Hunter MJ, Komives EA. Deprotection of S-Acetamidomethyl Cysteine-Containing Peptides by Silver Trifluoromethanesulfonate Avoids the Oxidization of Methionines. Anal Biochem. 1995;228:173–177. doi: 10.1006/abio.1995.1333. [DOI] [PubMed] [Google Scholar]
- Jeffrey PD, McCombie SW. Homogeneous, palladium(0)-catalyzed exchange deprotection of allylic esters, carbonates and carbamates. J Org Chem. 1982;47:587–590. [Google Scholar]
- Johansson L, Gafvelin G, Arner ESJ. Selenocysteine in proteins--properties and biotechnological use. Biochim Biophys Acta. 2005;1726:1–13. doi: 10.1016/j.bbagen.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Karlsson BG, Aasa R, Malmstrom BG, Lundberg LG. Rack-induced bonding in blue copper proteins: Spectroscopic properties and reduction potential of the azurin mutant Met-121 --> Leu. FEBS Lett. 1989;253:99–102. [Google Scholar]
- Karlsson BG, Nordling M, Pascher T, Tsai LC, Sjolin L, Lundberg LG. Cassette mutagenesis of Met121 in azurin from Pseudomonas aeruginosa. Protein Eng. 1991;4:343–349. doi: 10.1093/protein/4.3.343. [DOI] [PubMed] [Google Scholar]
- Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, Gladyshev VN. Characterization of Mammalian Selenoproteomes. Science. 2003;300:1439–1443. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- Lu Y. Cupredoxins in Comprehensive Coordination Chemistry II: From Biology to Nanotechnology. Elsevier; Oxford: 2003. [Google Scholar]
- Lu Y. Design and engineering of metalloproteins containing unnatural amino acids or non-native metal-containing cofactors. Curr Opin Chem Biol. 2005;9:118–126. doi: 10.1016/j.cbpa.2005.02.017. [DOI] [PubMed] [Google Scholar]
- Lu Y. Biosynthetic Inorganic Chemistry. Angew Chem, Int Ed. 2006a;45:5588–5601. doi: 10.1002/anie.200600168. [DOI] [PubMed] [Google Scholar]
- Lu Y. Metalloprotein and Metallo-DNA/RNAzyme Design: Current Approaches, Success Measures, and Future Challenges. Chem Rev. 2006b;45:9930–9940. doi: 10.1021/ic052007t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Berry SM, Pfister TD. Engineering Novel Metalloproteins: Design of Metal-Binding Sites into Native Protein Scaffolds. Inorg Chem. 2001;101:3047–3080. doi: 10.1021/cr0000574. [DOI] [PubMed] [Google Scholar]
- Malmström BG. Rack-Induced Bonding In Blue-Copper Proteins. Eur J Biochem. 1994;223:711–718. doi: 10.1111/j.1432-1033.1994.tb19044.x. [DOI] [PubMed] [Google Scholar]
- Mizoguchi TJ, Di Bilio AJ, Gray HB, Richards JH. Blue to type 2 binding. Copper(II) and cobalt(II) derivatives of a Cys112Asp mutant of Pseudomonas aeruginosa azurin. J Am Chem Soc. 1992;114:10076–10078. [Google Scholar]
- Moroder L. Isosteric Replacement of Sulfur with Other Chalcogens in Peptides and Proteins. J Pept Sci. 2005;11:187–214. doi: 10.1002/psc.654. [DOI] [PubMed] [Google Scholar]
- Mueller S, Senn H, Gsell B, Vetter W, Baron C, Boeck A. The Formation of Diselenide Bridges in Proteins by Incorporation of Selenocysteine Residues: Biosynthesis and Characterization of (Se)2-Thioredoxin. Biochem. 1994;33:3404–3412. doi: 10.1021/bi00177a034. [DOI] [PubMed] [Google Scholar]
- Neuwald AF, Krishnan BR, Brikun I, Kulakauskas S, Suziedelis K, Tomcsanyi T, Leyh TS, Berg DE. cysQ, a gene needed for cysteine synthesis in Escherichia coli K-12 only during aerobic growth. J Bacteriol. 1992;174:415–425. doi: 10.1128/jb.174.2.415-425.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otaka A, Koide T, Shide A, Fujii N. Application of dimethylsulphoxide(DMSO)/trifluoroacetic acid(TFA) oxidation to the synthesis of cystine-containing peptide. Tetrahedron Lett. 1991;32:1223–1226. [Google Scholar]
- Pierloot K, De Kerpel JOA, Ryde U, Roos BO. Theoretical Study of the Electronic Spectrum of Plastocyanin. J Am Chem Soc. 1997;119:218–226. [Google Scholar]
- Ralle M, Berry SM, Nilges MJ, Gieselman MD, vanderDonk WA, Lu Y, Blackburn NJ. The Selenocysteine-Substituted Blue Copper Center: Spectroscopic Investigations of Cys112SeCys Pseudomonas aeruginosa Azurin. J Am Chem Soc. 2004;126:7244–7256. doi: 10.1021/ja031821h. [DOI] [PubMed] [Google Scholar]
- Solomon EI, Baldwin MJ, Lowery MD. Electronic Structures of Active Sites in Copper Proteins: Contributions to Reactivity. Chem Rev. 1992;92:521–542. [Google Scholar]
- Su D, Li Y, Gladyshev VN. Selenocysteine insertion directed by the 3′-UTR SECIS element in Escherichia coli. Nucleic Acids Res. 2005;33:2486–2492. doi: 10.1093/nar/gki547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoropoulos D, Schwartz IL, Walter R. New synthesis of L-selenocysteine derivatives and peptides. Tetrahedron Lett. 1967a;8:2411–2414. [Google Scholar]
- Theodoropoulos D, Schwartz IL, Walter R. Synthesis of Selenium-Containing Peptides. Biochemistry. 1967b;6:3927–3932. doi: 10.1021/bi00864a039. [DOI] [PubMed] [Google Scholar]
- Vila AJ, Fernandez CO. Handbook on Metalloproteins. New York, NY: 2001. Copper in Electron transfer Proteins. [Google Scholar]
- Walter R, Chan WY. Syntheses and pharmacological properties of selenium isologs of oxytocin and deaminooxytocin. J Am Chem Soc. 1967;89:3892–3898. doi: 10.1021/ja00991a037. [DOI] [PubMed] [Google Scholar]
- Wang J, Schiller SM, Schultz PG. A Biosynthetic Route to Dehydroalanine-Containing Proteins. Angew Chem Int Ed. 2007;46:6849–6851. doi: 10.1002/anie.200702305. [DOI] [PubMed] [Google Scholar]
- Wessjohann LA, Schneider A, Abbas M, Brandt W. Selenium in chemistry and biochemistry in comparison to sulfur. Biol Chem. 2007;388:997–1006. doi: 10.1515/BC.2007.138. [DOI] [PubMed] [Google Scholar]
- Yajima H, Fujii N, Funakoshi S, Watanabe T, Murayama E, Otaka A. New strategy for the chemical synthesis of proteins. Tetrahedron. 1988;44:805–819. [Google Scholar]