

Abstract
(S)-2-hydroxypropylphosphonic acid epoxidase (HppE) is an O2-dependent, nonheme Fe(II)-containing oxidase that converts (S)-2-hydroxypropylphosphonic acid ((S)-HPP) to the regio-and enantiomerically specific epoxide, fosfomycin. Use of (R)-2-hydroxypropylphosphonic acid ((R)-HPP) yields the 2-keto-adduct rather than the epoxide. Here we report the chemical synthesis of a range of HPP analogs designed to probe the basis for this specificity. In past studies, NO has been used as an O2 surrogate to provide an EPR probe of the Fe(II) environment. These studies suggest that O2 binds to the iron, and substrates bind in a single orientation that strongly perturbs the iron environment. Recently, the X-ray crystal structure showed direct binding of the substrate to the iron, but both monodentate (via the phosphonate) and chelated (via the hydroxyl and phosphonate) orientations were observed. In the current study, hyperfine broadening of the homogeneous S = 3/2 EPR spectrum of the HppE-NO-HPP complex was observed when either the hydroxyl or the phosphonate group of HPP was enriched with 17O (I = 5/2). These results indicate that both functional groups of HPP bind to Fe(II) ion at the same time as NO, suggesting that the chelated substrate binding mode dominates in solution. (R)- and (S)-analog compounds that maintained the core structure of HPP but added bulky terminal groups were turned over to give products analogous to those from (R)- and (S)-HPP, respectively. In contrast, substrate analogs lacking either the phosphonate or hydroxyl group were not turned over. Elongation of the carbon chain between the hydroxyl and phosphonate allowed binding to the iron in a variety of orientations to give keto and diol products at positions determined by the hydroxyl substituent, but no stable epoxide was formed. These studies show the importance of the Fe(II)-substrate chelate structure to active antibiotic formation. This fixed orientation may align the substrate next to the iron-bound activated oxygen species thought to mediate hydrogen atom abstraction from the nearest substrate carbon.
The epoxide ring is a common structural element found in many natural products. The inherent strain energy associated with the small ring renders it reactive with enzyme nucleophiles. This has allowed many epoxide-containing compounds to be used as enzyme inhibitors and therapeutic agents. A representative example is fosfomycin ((1R, 2S)-epoxypropylphosphonic acid, 1), an antibiotic used to treat lower urinary tract infections (1, 2). The biological target of fosfomycin has been identified as UDP-α-D-GlcNAc-O-enolpyruvoyl1 transferase (3), which catalyzes the attachment of phosphoenolpyruvate to UDP-α-D-GlcNAc, a key step in the assembly of the peptidoglycan layer within the bacterial cell wall.
In most cases, the oxirane moiety of epoxide-containing natural products is biosynthesized through oxidation of the corresponding alkenes by monooxygenases with incorporation of an oxygen atom originated from dioxygen (4, 5). The synthesis of fosfomycin (1) is unusual in that the secondary hydroxyl oxygen of the immediate precursor, (S)-2-hydroxypropylphosphonic acid ((S)-HPP, 2-S), is retained in the fosfomycin product (6–9). Thus, the transformation catalyzed by (S)-2-hydroxypropylphosphonic acid epoxidase (HppE) is effectively a dehydrogenation reaction as shown in Scheme 1. The overall epoxidation reaction is a four-electron redox process involving the oxidation of substrate and NAD(P)H and the reduction of dioxygen to water.
Scheme 1.

We have expressed the HppE gene from Streptomyces wedmorensis and purified the encoded protein (8, 9). The purified HppE, after reconstitution, contains a mononuclear non-heme iron that is essential and cannot be replaced by other divalent ions (10, 11). The in vitro activity of HppE also requires NAD(P)H, dioxygen, and an auxiliary electron mediator to transfer electrons from NAD(P)H to the enzyme active site (8, 9, 11). While this mediator’s role can be fulfilled by small molecule electron carriers, such as FMN, FAD, riboflavin, and benzyl viologen (11), the physiological mediator is expected to be a reductase, which has not yet been identified. Since the only reductase gene in the three known fosfomycin biosynthetic gene clusters of S. wedmorensis (12), Pseudomonas syringae (13), and S. fradiae (14) is involved in an early step of fosfomycin biosynthesis (14, 15), the electron transfer in HppE reaction may rely on a promiscuous reductase within the cell.
The detailed mechanism of HppE is unknown, but results from several experimental approaches are consistent with formation of a radical intermediate through the action of an iron bound activated oxygen species. For example, the natural substrate, 2-S, is converted to 1 by HppE, while its epimer, (R)-HPP (2-R), is converted to a keto product, 2-oxopropylphosphonic acid (4, Scheme 1) (16). The distinct fates of these two epimers can be explained on the basis of regiospecific, radical-initiated, hydrogen atom abstraction by HppE, and this was tested by fluorine substitution at C1 of 1. It was found that (S)-[1-F2]HPP (6-S) is an inhibitor for HppE, whereas the epimeric (R)-[1-F2]HPP (6-R, Scheme 1) can still undergo turnover to the corresponding 2-keto product 7 by HppE (16). These results are most easily explained by initial radical formation at C-1 (via 3) in the S isomers and at C-2 (via 5 or 8) in the R isomers (16).
Not only does HppE exhibit highly regiospecific hydrogen atom abstraction, which is governed by the C-2 stereochemistry of HPP, it can also distinguish the two diastereotopic hydrogens at C-1 of HPP during turnover. The removal of a hydrogen atom from C-1 of 2-S by HppE had been determined to be pro-R stereospecific (17). The strict regio- as well as stereo-specificity observed for HppE reaction must stem from a specific substrate binding orientation relative to the reactive iron-oxygen species, which is responsible for the hydrogen abstraction step.
Recent crystallographic characterizations of HppE confirmed that the mononuclear metal ion in the HppE active site is coordinated by a 2-His-1-carboxylate (D/E) core as implicated by sequence alignment and mutagenesis studies (10, 18, 19). Surprisingly, the substrate, 2-S, was found to bind to the metal ion in two different configurations in the enzyme-substrate binary complex (18). As illustrated in Figure 1A, 2-S could bind to the metal ion in a monodentate mode via a phosphonate-oxygen linkage. It could also form a bidentate coordination to the metal ion using the 2-OH group and an oxygen atom from the phosphonate group as ligands (Figure 1B). The presence of two different enzyme-substrate binding modes is unusual. It is not clear whether both binding modes are catalytically relevant. It is also not known whether a similar situation applies for 2-R when it is present as the substrate.
Figure 1.
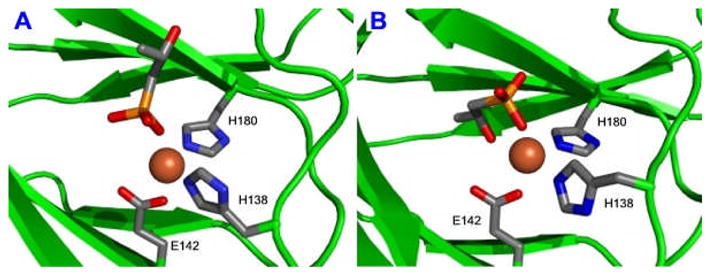
X-ray Structures of the modeled HppE active site bound with Fe(II) and 2-S in monodentate (1A) and bidentate (1B) binding modes. Models were generated on the basis of the published crystal coordinates for the S. wendomorensis enzyme (PDB entries 1ZZ7 and 1ZZ8).
It is important for mechanistic considerations to determine whether one or both of the substrate binding modes detected by X-ray crystallography applies to the functioning enzyme. The substrate chelate mode would suggest that the stereo- and regio-specific reaction is enforced through the use of multiple ligand sites on the iron, whereas these roles would shift to the overall active site structure if the monodentate binding mode applies. To learn more about the substrate-enzyme complex and to investigate the catalytic relevance of the mono- and bi-dentate coordination, we prepared a series of substrate analogues and [17O]-labeled HPPs, and studied their binding to HppE and the HppE-NO complexes. The results of this work provide significant insight into how the enzyme and substrate interact and how this interaction determines the regio-and stereo-specificity of the enzyme.
EXPERIMENTAL PROCEDURES
General
HppE from S. wedmorensis was obtained by expressing the plasmid pPL1001, which contains the corresponding gene (fom4), in E. coli BL21(DE3)/pPL1001 (9). The overproduced HppE was purified to near homogeneity as previously described (9). All reagents and solvents were purchased from commercial sources and were used without further purification unless otherwise noted. Biochemicals, including fosfomycin (1) and (S)-2-hydroxybutyric acid ((S)-HBA, 33), were acquired from Aldrich-Sigma (St. Louis, MO). Culture medium ingredients were products of Difco (Detroit, MI). Isotope-labeled water (36.8% 17O and 48.8% 18O) was obtained from Isotec Inc. (Miamisburg, OH).
Preparation of Structural Analogues of (S)-HPP (2-S)
(R)-HPP (2-R), (S)- and (R)-1,1-difluoro-HPP ([1-F2]HPP) (6-S, 6-R) were chemically synthesized as previously reported (16).
Synthesis of Isobutylphosphonic Acid (IBP, 9) (Scheme 2)
Scheme 2.

A mixture of isobutyl bromide (10, 1.37 g, 10 mmol), triethylphosphine (P(OEt)3, 4.2 mL, 20 mmol) and tetrabutylammonium iodide (Bu4NI, 50 mg, 0.14 mmol) was heated at 150 °C for 3 h, then dried under reduced pressure. The crude products were purified by flash silica gel column chromatography with hexanes/ethyl acetate (10:1 to 1:1) to give diethyl isobutylphosphonate (11) in 90% yield. A portion of 11 (0.5 g, 2.6 mmol) was then incubated with trimethylsilyl bromide (TMSBr, 0.5 mL, 3.8 mmol) and allyltrimethylsilane (0.25 mL, 1.6 mmol) for 24 h at room temperature. The reaction mixture was neutralized with NH4HCO3, extracted with water, and the aqueous fractions were pooled and lyophilized to give IBP (9) in white powder form. The overall yield was 95%. 1H NMR (300 MHz, CDCl3) δ 2.05 (1H, m), 1.66 (2H, dd, J = 6.9, 18), 1.04 (6H, d, J = 6.9). 13C NMR (75 MHz, CDCl3) δ 31.1, 21.9, 21.5 (d, J = 136.2). 31P NMR (121 MHz, CDCl3) δ 32.7.
Preparation of 17O-Labeled HPPs
(R)-HPP with 17O enriched hydroxyl group (12-R) were synthesized by the established procedure (9), except for the replacement of THF with p-toluenesulfonic acid (pTSA) and H217O in the first step. Preparation of the (S)-isomer (12-S) was also attempted. Unfortunately, the isotope enrichment of this sample was lower than that of (R)[17O]HPP. Thus, only the 12-R sample was subjected to EPR study. The (R)-HPP with 17O enriched in the phosphonate group (13-R) was prepared by dissolving chemically synthesized (R)-HPP (0.04 mmol) in 0.2 mL of [17O]H2O. The solution was incubated at room temperature for 2 weeks. The samples were lyophilized and the 17O isotope enrichment in the products was 20%, as estimated by mass spectrometry performed by the MS facility of the Department of Chemistry and Biochemistry at the University of Texas at Austin.
Synthesis of (S)- and (R)-2-Hydroxy-2-phenylethylphosphonic Acids ((S)- and (R)-HPEP, 14-S and 14-R) (Scheme 3)
Scheme 3.

(i) Ethyl (S)-(tert-Butyldimethylsilyloxy)phenylacetate (16-S)
To a solution of ethyl (S)-hydroxyphenylatetate (15-S, 5.0 g, 27.7 mmol), 4-dimethylaminopyridine (DMAP, 0.3 g, 2.5 mmol) and imidazole (4.0 g, 56 mmol) in 25 mL of dry CH2Cl2 was added portionwise a solution of t-butyldimethylsilyl chloride (TBDMSCl, 4.6 g, 30 mmol) in 25 mL of CH2Cl2 at 0 °C. After stirring at room temperature for 15 h, the mixture was diluted with CH2Cl2, washed with brine, and dried over anhydrous Na2SO4. The solvents were evaporated under reduced pressure and the residue was purified by flash silica gel column chromatography (hexanes). The yield of 16-S was 95%. 1H NMR (300 MHz, CDCl3) δ 7.48 (2H, m), 7.31 (3H, m), 5.24 (1H, s), 4.15 (2H, q, J = 7.2), 1.22 (3H, t, J = 7.2), 0.94 (9H, s), 0.13 (3H, s), 0.06 (3H, s). 13C NMR (75 MHz, CDCl3)δ 172.2, 139.3, 128.4, 128.1, 126.4, 74.5, 61.1, 25.8, 18.4, 14.2, −4.9, −5.1.
(ii) (S)-(tert-Butyldimethylsilyloxy)-2-phenylethanol (17-S)
To a solution of ethyl (S)-(tert-butyldimethylsilyloxy)phenylacetate (16-S, 7.8 g, 26.5 mmol) in 100 mL of dry CH2Cl2 was added dropwise 53 mL of diisobutylaluminium hydride (DIBAL-H) solution (1.5 M in toluene) at −78 °C. After stirring at room temperature for 4 h, the reaction was cooled to −20 °C, and 14 mL of 1:1 mixture of AcOH/H2O was added slowly. The resulting mixture was stirred at −20 °C for 30 min, then poured into a saturated NaHCO3 solution, and extracted with CH2Cl2. The combined organic extracts were dried over anhydrous Na2SO4 and the solvents were removed under reduced pressure. The residue was purified by flash silica gel column chromatography (hexanes:ethyl acetate, 5:1) to give 17-S in 67% yield. 1H NMR (300 MHz, CDCl3) δ 7.30 (5H, m), 4.76 (1H, dd, J = 6.9, 5.1), 3.59 (2H, m), 0.93 (9H, s), 0.08 (3H, s), −0.08 (3H, s). 13C NMR (75 MHz, CDCl3) δ 141.5, 128.3, 127.7, 126.3, 76.0, 69.0, 25.9, 18.3, −4.4, −4.9.
(iii) (S)-2-Bromo-1-phenylethoxy-tert-butyldimethylsilane (18-S)
To a solution of (S)-(tert-butyldimethylsilyloxy)-2-phenylethanol (17-S, 2.53 g, 10 mmol) and PPh3 (2.90 g, 11 mmol) in 20 mL of CH2Cl2 was added N-bromosuccinimide (NBS, 2.0 g, 11 mmol) at −78 °C. After stirring at the same temperature for 10 min and at room temperature for 3 h, the mixture was diluted with ether, washed with brine, and dried over anhydrous Na2SO4. The solvents were removed under reduced pressure and the residue was purified by flash silica gel column chromatography (hexanes) to give 18-S in 78% yield. 1H NMR (300 MHz, CDCl3) δ 7.36 (5H, m), 4.88 (1H, dd, J = 7.8, 5.1), 3.48 (2H, m), 0.93 (9H, s), 0.14 (3H, s), −0.06 (3H, s). 13C NMR (75 MHz, CDCl3) δ 142.3, 128.5, 128.1, 126.3, 75.4, 39.7, 25.9, 18.4, −4.6, −4.8.
(iv) Diethyl (S)-2-Phenyl-2-(tert-butyldimethylsilyloxy)ethylphosphonate (19-S)
A mixture of (S)-2-bromo-1-phenylethoxy-tert-butyldimethylsilane (18-S, 1.09 g, 3.4 mmol), P(OEt)3 (2.0 mL, 13.6 mmol) and Bu4NI (40 mg, 0.11 mmol) was heated at 120 °C for 23 h, then dried under reduced pressure. The residue was loaded onto a silica gel column, and the product was eluted with hexanes/ethyl acetate (10:1 to 1:1). Compound 19-S was isolated in 45% yield. 1H NMR (300 MHz, CDCl3) δ 7.29 (5H, m), 5.03 (1H, m), 3.92 (4H, m), 2.32 (1H, ddd, J = 16.8, 15.3, 6.9), 2.12 (1H, ddd, J = 17.4, 15.3, 5.7), 1.23 (3H, t, J = 7.2), 1.16 (3H, t, J = 7.2), 0.82 (9H, s), 0.03 (3H, s), −0.22 (3H, s). 13C NMR (75 MHz, CDCl3) δ 144.8 (d, J = 9.6), 128.3, 127.6, 126.2, 70.8, 61.3 (d, J = 4.1), 61.2 (d, J = 4.1), 37.8 (d, J = 137.0), 25.8, 18.1, 16.4, 16.3, −4.6, −4.9. 31P NMR (121 MHz, CDCl3) δ 27.9.
(v) Diethyl (S)-2-Hydroxy-2-phenylethylphosphonate (20-S)
A mixture of diethyl (S)-2-phenyl-2-(tert-butyldimethylsilyloxy)ethylphosphonate (19-S, 0.48 g, 1.2 mmol) and concentrated HCl (0.5 mL) in 5 mL of methanol was stirred for 1 h at room temperature. The solvents were removed in vacuo, and the residue was purified by flash silica gel column chromatography (hexanes:ethyl acetate, 2:1 to 1:3). The yield of 20-S was 87%. 1H NMR (300 MHz, CDCl3) δ 7.34 (5H, m), 5.09 (1H, m), 4.09 (4H, m), 3.96 (1H, d, J = 3.0), 2.19 (2H, m), 1.33 (3H, t, J = 7.5), 1.28 (3H, t, J = 6.9). 13C NMR (75 MHz, CDCl3) δ 143.5 (d, J = 16.1), 128.6, 127.8, 125.6, 68.9(d, J = 4.3), 62.2 (d, J = 6.0), 62.0 (d, J = 7.0), 36.0 (d, J = 135.8), 16.5 (d, J = 6.0), 16.4 (d, J = 6.6). 31P NMR (121 MHz, CDCl3) δ 29.9.
(vi) (S)-2-Hydroxy-2-phenylethylphosphonic Acid ((S)-HPEP, 14-S)
A solution of diethyl (S)-2-hydroxy-2-phenylethylphosphonate (20-S, 0.24 g, 0.9 mmol), TMSBr (0.4 mL, 3.0 mmol) and allyltrimethylsilane (0.2 mL, 1.3 mmol) in 4 mL of CH2Cl2 was stirred for 25 h at room temperature. The solvents were removed in vacuo, and the residue was vigorously stirred with 5 mL of water for 10 min. The aqueous solution was neutralized with NH4HCO3, washed with CHCl3, and then lyophilized to give product 14-S as a white powder in 96% yield. 1H NMR (300 MHz, D2O) δ 7.26 (5H, m), 4.76 (1H, m), 1.75 (2H, m). 13C NMR (75 MHz, D2O) δ 143.9 (d, J = 13.2), 128.6, 127.8, 126.1, 70.6 (d, J = 29.0), 37.5 (d, J = 126.3). 31P NMR (121 MHz, D2O) δ 20.9.
(vii) (R)-2-Hydroxy-2-phenylethylphosphonic Acid ((R)-HPEP, 14-R)
Preparation of this compound followed an identical procedure developed for the synthesis of (S)-2-hydroxy-2-phenylethylphosphonic acid (14-S) using ethyl (R)-(tert-butyldimethylsilyloxy)phenylacetate as the starting material.
Synthesis of (S)- and (R)-3-Hydroxybutylphosphonic Acid ((S)- and (R)-HBP, 21-S and 21-R) (Scheme 4)
Scheme 4.

(i) Ethyl (S)-3-(tert-Butyldiphenylsilyloxy)butyrate (23-S)
To a solution of ethyl (S)-3-hydroxybutyrate (22-S, 2.0 g, 16 mmol) and imidazole (1.3 g, 19 mmol) in 100 mL of dry CH2Cl2 was added dropwise tert-butyldiphenylsilyl chloride (TBDPSCl, 5.0 g, ~18 mmol) at 0 °C. After stirring overnight at room temperature, the mixture was diluted with CH2Cl2, washed with brine, and dried over anhydrous Na2SO4. The solvents were then removed under reduced pressure and the residue was purified by flash silica gel column chromatography (hexanes). The yield of 23-S was 92%. 1H NMR (300 MHz, CDCl3) δ 7.70 (4H, m), 7.42 (6H, m), 4.33 (1H, m), 4.15 (2H, q, J = 7.2), 2.59 (1H, dd, J = 14.7, 7.2), 2.41 (1H, dd, J = 14.7, 5.7), 1.13 (3H, t, J = 7.2), 1.06 (9H, s). 13C NMR (75 MHz, CDCl3) δ 171.9, 136.0, 135.9, 134.4, 133.9, 129.7, 129.6, 127.7, 127.6, 67.0, 51.5, 44.5, 27.0, 23.7, 19.3.
(ii) (S)-3-(tert-Butyldiphenylsilyl)butane-1,3-diol (24-S)
To a solution of ethyl (S)-3-(tert-butyldiphenylsilyloxy)butyrate (23-S, 5.4 g, 15 mmol) in 100 mL of dry CH2Cl2 was added dropwise DIBAL-H (34 mL, 1.5 M solution in toluene) at −78 °C. After stirring at room temperature for 2 h, the mixture was cooled to −20 °C, quenched by the addition of 10 mL of saturated NH4Cl solution, then poured into saturated NaHCO3 solution, and extracted with CH2Cl2. The organic extracts were combined and dried over anhydrous Na2SO4. The solvents were removed under reduced pressure, and the residue was purified by flash silica gel column chromatography (hexanes:ethyl acetate, 10:1 to 3:1). Compound 24-S was isolated in 55% yield. 1H NMR (300 MHz, CDCl3) δ 7.73 (4H, m), 7.36 (6H, m), 4.13 (1H, m), 3.86 (1H, m), 3.71 (1H, m), 2.24 (1H, br s), 1.84 (1H, m), 1.67 (1H, m), 1.11 (3H, d, J = 6.3), 1.08 (9H, s). 13C NMR (75 MHz, CDCl3) δ 135.9, 135.8, 134.2, 133.7, 129.8, 129.7, 127.7, 127.6, 68.8, 60.0, 40.7, 27.0, 23.0, 19.2.
(iii) (S)-1-Bromo-3-(tert-butyldiphenylsilyloxy)butane (25-S)
To a solution of (S)-3-(tert-butyldiphenylsilyl)butane-1,3-diol (24-S, 3.3 g, 10.1 mmol) and triphenylphosphine (PPh3, 3.2 g, 12.3 mmol) in 25 mL of CH2Cl2 was added N-bromosuccinimide (2.0 g, 11.2 mmol) at −78 °C. After stirring for 2 h at room temperature, the mixture was diluted with ether, washed with brine, and dried over anhydrous Na2SO4. The solvents were removed under reduced pressure and the residue was purified by flash silica gel column chromatography (hexanes) to give 25-S in 56% yield. 1H NMR (300 MHz, CDCl3) δ 7.72 (4H, m), 7.41 (6H, m), 4.04 (1H, m), 3.49 (2H, m), 2.10 (1H, m), 1.95 (1H, m), 1.08 (12H, m). 13C NMR (75 MHz, CDCl3) δ 135.9, 134.5, 133.8, 129.7, 129.6, 127.7, 127.5, 68.1, 42.6, 30.2, 27.1, 23.3, 19.4.
(iv) Dibenzyl (S)-3-(tert-Butyldiphenylsilyloxy)butylphosphonate (26-S)
To a solution of dibenzylphosphite (0.66 g, 2.5 mmol) in dry DMF (3 mL) was added NaH (2.5 mmol) at room temperature. After stirring for 10 min, (S)-1-bromo-3-(tert-butyldiphenylsilyloxy)butane (25-S, 0.50 g, 1.3 mmol) in dry DMF (2 mL) was added. The resulting mixture was stirred overnight, and then diluted with CH2Cl2, washed with water, and dried over anhydrous Na2SO4. The solvents were evaporated under reduced pressure, and the residue was flash chromatographed on silica gel (hexanes:ethyl acetate, 3:1 to 1:1) to give 26-S in 82% yield. 1H NMR (300 MHz, CDCl3) δ 7.65 (4H, m), 7.32 (16H, m), 5.00 (4H, m), 3.85 (1H, m), 1.99-1.67 (4H, m), 1.04 (9H, s), 1.00 (3H, d, J = 6.3). 13C NMR (75 MHz, CDCl3) δ 136.6, 136.5, 135.9, 135.8, 134.4, 134.0, 129.7, 129.6, 128.6, 128.3, 127.9, 127.8, 127.6, 127.5, 69.0 (d, J = 19.6), 67.0 (d, J = 6.3), 31.6 (d, J = 5.1), 27.0, 22.6, 21.6 (d, J = 141.9), 19.3. 31P NMR (121 MHz, CDCl3) δ 35.1.
(v) Dibenzyl (S)-3-Hydroxybutylphosphonate (27-S)
A mixture of dibenzyl (S)-3-(tert-butyldiphenylsilyloxy)butylphosphonate (26-S, 0.31 g, 0.5 mmol) and concentrated HCl (0.35 mL) in 3 mL of methanol was stirred for 13 h at room temperature. The solvents were removed in vacuo, and the residue was purified by flash silica gel column chromatography (hexanes:ethyl acetate, 2:1 to 1:2). The yield of 27-S was 72%. 1H NMR (300 MHz, CD3OD) δ 7.34 (10H, m), 4.99 (4H, m), 3.65 (1H, m), 2.02-1.52 (4H, m), 1.08 (3H, d, J = 5.7). 13C NMR (75 MHz, CD3OD) δ 136.3, 128.4, 128.3, 127.9, 67.4, 66.9, 66.7, 31.1, 21.9, 21.5 (d, J = 138.2). 31P NMR (121 MHz, CD3OD) δ 35.6.
(vi) (S)-3-Hydroxybutylphosphonic acid ((S)-HBP, 21-S)
A mixture of 10% Pd/C (80 mg) and dibenzyl (S)-3-hydroxybutylphosphonate (27-S, 140 mg, 0.42 mmol) in 10 mL of methanol was vigorously stirred for 30 min under a hydrogen atmosphere at room temperature. The mixture was filtered through a short celite column. The solvents were then removed under reduced pressure, the residue was dissolved in water, and neutralized with NH4HCO3. The aqueous solution was lyophilized to give 21-S as a white powder in 96% yield. 1H NMR (300 MHz, D2O) δ 3.58 (1H, m), 1.34 (4H, m), 0.92 (3H, d, J = 6.0). 13C NMR (75 MHz, D2O) δ 68.3 (d, J = 17.9), 32.1 (d, J = 4.0), 24.2 (d, J = 134.6), 21.3. 31P NMR (121 MHz, D2O) δ 26.2.
(R)-3-Hydroxybutylphosphonic acid ((R)-HBP, 21-R)
This compound was synthesized by the same procedure as described for 3-(S)-hydroxybutylphosphonic acid (21-S) using ethyl (R)-3-(tert-butyldiphenylsilyloxy)butyrate as the starting material in the first step.
Characterization of Turnover Products Generated in the Incubation of Substrate Analogues with HppE
Chemically synthesized substrate analogues were individually examined for their competence as substrates by the in vitro HppE assay (9). The products generated in the incubation mixture were purified by a P-2 gel filtration column with H2O as the eluent. The desired fractions were pooled and lyophilized, and the purified products were dissolved in D2O for NMR analysis. For 1,2-oxy-2-phenyl-ethylphosphonic acid (28) derived from 14-S: 1H NMR (300 MHz, D2O) δ 7.15 (5H, m), 3.78 (1H, m), 2.81 (1H, dd, J = 19.2, 3); 13C NMR (75 MHz, D2O) δ 136.4, 128.5, 128.4, 125.7, 58.9 (d, J = 170.0), 57.1; 31P NMR (121 MHz, D2O) δ 9.9. For 2-oxo-2-phenyl-ethylphosphonic acid (29) derived from 14-R: 1H NMR (300 MHz, D2O) δ 7.20–7.91 (2H, m), 7.48–7.52 (1H, m), 7.36–7.40 (2H, m), 3.30 (2H, d, J = 20.5); 31P NMR (121 MHz, D2O) δ 10.8. For (3R)-3,4-dihydroxybutylphosphonic acid (31) derived from 21-S: 1H NMR (500 MHz, D2O) δ 3.44–3.49 (1H, m), 3.39 (1H, dd, J = 11.5, 3.5), 3.29 (1H, dd, J = 11.5, 6.5), 1.41–1.52 (1H, m), 1.30–1.40 (2H, m), 1.16–1.28 (1H, m); 13C NMR (75 MHz, D2O) δ 70.3, 68.2, 33.1(d, J = 4.0), 24.3 (d, J = 134.6); 31P NMR (121 MHz, D2O) δ 23.3. For 3-oxo-butylphosphonic acid (32) derived from 21-R: 1H NMR (500 MHz, D2O) δ 2.57 (2H, t, J = 7.5), 2.08 (3H, s), 1.42–1.48 (2H, m); 13C NMR (75 MHz, D2O) δ 215.6, 38.3, 28.9, 22.6 (d, J = 133.0); 31P NMR (121 MHz, D2O) δ 22.5.
Preparation of NO Samples of FeII-HppE Bound with Substrate or Substrate Analogues
Double distilled H2O (ddH2O) and Tris·HCl (50 mM, pH 7.5) buffer were made anaerobic by repetitive vacuum and bubbling with argon that had been passed over a column of copper catalyst (BASF Inc.) heated at 150 °C for approximately 3 h. The Fe(II) stock solution was prepared by adding 1 mL of the above ddH2O through a gas-tight syringe into a serum vial containing pre-weighted Fe(NH4)2(SO4)2·6H2O (99.997%, Aldrich) under anaerobic conditions. Anaerobic solutions of metal-free apoprotein, substrate and substrate analogues were prepared in reaction vials by repeated cycles of vacuum and flushing with argon and then transferred to the EPR sample tubes under an argon atmosphere. Nitric oxide (NO) was introduced through a gas-tight syringe to the headspace of the quartz EPR tubes or UV-vis cuvettes containing the ferrous enzymes prepared under anaerobic conditions. An argon flush was maintained above samples to protect them from oxidation by O2 and to minimize an anomalous EPR signal near g = 2 which derives from NO.
The enzyme-substrate nitrosyl ternary complexes were made by adding substrate (or substrate analogues) and then NO (or vise versa) to the enzyme in its reduced state. For the isotope-labeling experiments, 17OH-labeled substrates were first added to the enzyme, followed by NO gas. Control samples were prepared in parallel using natural-abundance substrate. EPR samples were frozen by slow immersion in liquid N2 for latter use.
EPR Spectroscopy
The 17O-EPR spectra were recorded at 9.6 GHz frequency using a Bruker Elexsys E500 spectrometer. The EPR data of various substrate analogues were acquired using a Bruker EMX spectrometer and a high sensitivity cavity at 9.38 GHz. Both spectrometers were equipped with an Oxford Instruments liquid helium cryostat. Temperature was controlled by a digitalized Oxford temperature controller. A Bruker ER035M Gauss Meter was used during measurements to assist the g-value determination. EPR spectra of the S = 3/2 complexes were analyzed according to the spin Hamiltonian:
where B0 is the magnetic field, D is the axial zero-field splitting parameter, and E/D indicates the degree of rhombic distortion in the electronic environment. Changes in the E/D term are diagnostic of changes in the environment of the iron. Computer simulation of experimental EPR data was performed using SimFonia (Bruker).
RESULTS
Electronic Absorption Spectra of Fe(II)-HppE Nitrosyl Complexes
As a result of using DTT and EDTA in the protein purification procedure, the as-purified HppE is essentially an apo-protein (9) and it has little UV-vis absorbance above 300 nm (Figure 2, curve A). Upon anaerobic reconstitution with a stoichiometric amount of Fe(NH4)2(SO4)2, the resultant Fe(II)-HppE remained colorless above 300 nm (same as curve A in Figure 2). However, the addition of NO to the ferrous enzyme yielded a pale yellow complex with two absorption maxima at 438 nm (ε ≈ 750 M−1cm−1 per iron) and 640 nm (ε ≈ 250 M−1cm−1 per iron) (Figure 2, curve B). These two bands can be assigned, based on previous studies of the ferrous-nitrosyl complexes of various enzymes and model compounds, to the electronic absorption of the NO− → Fe3+ charge transfer and the Fe3+ ligand field transition, respectively (20). This observation provided initial evidence for binding of NO to the metal center. The visible spectrum also has a shoulder at 354 nm that was observed to vary in intensity depending on the concentration of NO. A similar spectral feature at 340 nm was previously noted in the spectrum of the nitrosyl complex of isopenicillin N synthase (IPNS) and was ascribed to nonspecific interactions of NO with protein (21).
Figure 2.

Electronic absorption spectra of Fe(II)-HppE nitrosyl complexes. (A) as-isolated HppE (200 μM) (metal-free); (B) HppE (180 μM) reconstituted with Fe(II) and exposed to NO; and (C) to sample B was added (S)-HPP (4 mM).
When the Fe(II)-HppE nitrosyl binary complex was subjected to repeated cycles of vacuum and flushing with argon, the pale-yellow color gradually faded due to loss of NO, and the enzyme eventually returned to the colorless state. Interestingly, the addition of NO to Fe(II)-HppE in the presence of 2-S led to a ternary complex with an optical spectrum exhibiting maxima at 438 (ε ≈ 400 M−1cm−1 per iron), and 680 nm (ε ≈ 300 M−1cm−1 per iron) (Figure 2, curve C). The large change in absorptivity of the 438 nm Fe-nitrosyl band indicates significant changes in the metal coordination environment upon substrate binding.
EPR Spectra of Fe(II)-HppE Nitrosyl Complexes
Previously EPR studies showed that the spectrum of the nitrosyl complex of Fe(II)-reconstituted HppE is composed of two superimposed S = 3/2 species (Table 1) with distinct relaxation properties (9). Upon addition of 2-S, most of the heterogeneity disappears and a single species (S = 3/2) with increased rhombicity, E/D = 0.066, having principal g-values of 4.42, 3.63, and 1.97 prevails (Figure 3) (9). The observed spectral change is independent of the addition order of 2-S and NO to the EPR sample, and is consistent with the change of the electronic spectrum observed upon substrate binding to the Fe(II)-HppE nitrosyl complex (Figure 2) (9). These results suggest that 2-S binds near or directly to the active site Fe(II) leading to reorganization of the metal coordination core. The sensitivity of the EPR spectrum to changes in the structure or environment of the Fe-NO complex increases as the rhombicity increases in the range of 0 to ~0.2 and then decreases as E/D approaches the limiting value of 0.33. Thus, the observation of an EPR spectrum from only one species with an intermediate size E/D value shows that the iron center has become quite homogeneous.
Table 1.
EPR properties of Fe(II)-HppE nitrosyl complexes
| Complex | Compound | g// | E/D | Relative Percentage |
|---|---|---|---|---|
| HppE-Fe(II)·(S)-HPP·NO | 2-S | 4.42, 3.63 | 0.066 | > 95% |
| HppE-Fe(II)·(R)-HPP·NO | 2-R | 4.42, 3.63 | 0.066 | > 95% |
| HppE-Fe(II)·(S)-HFPP·NO | 6-S | 4.42, 3.63 | 0.066 | > 95% |
| HppE-Fe(II)·(R)-HFPP·NO | 6-R | 4.42, 3.63 | 0.066 | > 95% |
| HppE-Fe(II)·HBA·NO | 33 | 4.26, 3.80 | 0.038 | 93% |
| HppE-Fe(II)·IBP·NO | 9 | 4.45, 3.61 | 0.070 | 63% |
| 4.17, 3.89 | 0.024 | 33% | ||
| HppE-Fe(II)·fosfomycin·NO | 1 | 4.47, 3.60 | 0.073 | 54% |
| 4.23, 3.84 | 0.033 | 40% | ||
| HppE-Fe(II)·(S)-HPEP·NO | 14-S | 4.37, 3.69 | 0.056 | 43% |
| 4.21, 3.88 | 0.028 | 52% | ||
| HppE-Fe(II)·(R)-HPEP·NO | 14-R | 4.53, 3.51 | 0.085 | 29% |
| 4.37, 3.70 | 0.056 | 26% | ||
| 4.21, 3.87 | 0.028 | 26% | ||
| HppE-Fe(II)·(S)-HBP·NO | 21-S | 4.48, 3.57 | 0.075 | 26% |
| 4.31, 3.76 | 0.046 | 46% | ||
| 4.17, 3.89 | 0.023 | 23% | ||
| HppE-Fe(II)·(R)-HBP·NO | 21-R | 4.40, 3.66 | 0.061 | 63% |
| 4.18, 3.89 | 0.024 | 34% | ||
Figure 3.

EPR spectra of Fe(II)-HppE·HPP nitrosyl complexes. Blue line: Fe(II)-HppE (250μM) mixed with 2-S (2.5 mM) and exposed to NO; Black line: Fe(II)-HppE (250 μM) mixed with 2-R (2.5 mM) and exposed to NO. Instrumental parameters: temperature 2 K, microwave frequency 9.6 GHz, microwave power 0.6 mW, modulation amplitude 5 G, time constant 0.02 s, and a sweep rate of 50 G/s.
EPR Spectra of Fe(II)-HppE Complexed with Nitric Oxide and Stereoisomers of HPP and [1-F2]HPP
To gain more insight into substrate binding to HppE in the presence of NO, the EPR spectra of Fe(II)-HppE nitrosyl complex coordinated with 2-R and the fluorinated substrate analogues 6-S and 6-R were studied. It was found that the EPR spectrum of 2-R-bound Fe(II)-HppE nitrosyl complex is nearly superimposable on that of 2-S-bound complex (Figure 3). This suggests that 2-S and 2-R bind to the HppE active site in a similar manner. Very similar spectra were also obtained when 6-S and 6-R were bound (Table 1). Apparently, the stereochemistry at C-2 and the fluorine substitution at C-1 have little effect on the electronic symmetry of the enzyme-substrate-nitrosyl complex, suggesting that the same functional groups are bound in the same ligand sites.
EPR Spectra of Fe(II)-HppE Complexed with Nitric Oxide and HPP Analogues Lacking 2-OH or Phosphonate Group
To probe the role of the hydroxyl and phosphonate groups of HPP in binding to the metal center, two substrate analogues, (S)-2-hydroxybutyric acid ((S)-HBA, 33, which has the phosphonate group replaced by a carboxylic group, and isobutylphosphonic acid (IBP, 9) which carries a methyl group instead of a hydroxyl group at C-2, were prepared. Neither compound is a substrate for HppE based on the in vitro activity assay (9). Each of them was added to the nitrosyl complex of Fe(II)-HppE for EPR analysis, and the resulting EPR spectra are shown in Figure 4. A minor resonance at g = 4.07 was present in all spectra and is assigned to a small amount of denatured enzyme.2
Figure 4.

EPR spectra of Fe(II)-HppE complexed with NO and different substrate analogues. (A) (S)-HBA (33); (B) IBP (9); (C) fosfomycin (1). Instrumental parameters are the same as shown in Figure 3. Gray lines represent simulated spectra.
The spectrum of the nitrosyl complex of 33-HppE (Figure 4A) exhibits an E/D value for the dominant species of 0.038 that is much smaller than those detected for the 2-S and 2-R complexes, indicating that the iron environment is not the same as observed for true substrates (Table 1). Nevertheless, the (S)-HBA spectrum is dominated by one species like those of the 2-S and 2-R complexes, suggesting that this molecule binds predominantly in one binding orientation. Thus, the carboxylate group appears to be an effective substitute for phosphonate group in this role. In contrast, the spectrum of the nitrosyl complex of the enzyme with 9 is comprised of two major EPR species (Figure 4B): one (g = 4.45, 3.61; E/D = 0.070) has a slightly larger E/D value than the corresponding spectrum of the 2-S complex, while the other (g = 4.17, 3.89; E/D = 0.024) has a much smaller E/D value (Table 1). Since 9 is an achiral molecule, the two sets EPR signals cannot be an artifact due to the presence of two stereoisomers in the active site. Instead, they are likely to be the result of binding of 9 to Fe(II) in two different configurations. This is made more likely by the fact that 9 only has one potential metal binding group to provide orientation, whereas each of the other binding molecules discussed thus far has two. The fact that 9 binds to HppE in two different configurations when the 2-OH group is absent is suggestive of an active role of 2-OH in metal binding. Interestingly, the EPR spectrum of 1 (which also has only one binding determinant) bound to the nitrosyl complex of Fe(II)-HppE (Figure 4C, Table 1) resembles that of 9. A free hydroxyl group at C-2 of the substrate is apparently important to metal coordination.
EPR Spectra of Fe(II)-HppE Complexed with Nitric Oxide and 17O-labeled HPP
In order to fully characterize substrate coordination to the iron center of HppE, 17O-labeled HPP was chemically synthesized and used in sample preparation. Figure 5 shows the EPR spectra of Fe(II)-HppE·HPP nitrosyl complexes prepared using (R)-HPP with 17O enriched in the 2-OH group ((R)-[17OH]HPP, 12-R). The spectrum is superimposed with a reference spectrum obtained using unlabeled 2-R for comparison. The small, but clearly discernible, linewidth broadening due to hyperfine interactions between 17O nuclear spin (I = 5/2) and {Fe-NO}7 electronic spin (S = 3/2) is obtained. As shown in Figures 5B and 5C, the 17O labeling causes broadening of 2.3 and 2.5 G of the spectral features near g = 4.42 and 3.63, respectively. Considering the low enrichment (~20%) of 17O in the sample, this change is comparable to those reported for other 17O-labeled-substrate-enzyme complexes, and thus, demonstrates direct coordination of the hydroxyl oxygen of HPP to the iron center (22–25).
Figure 5.
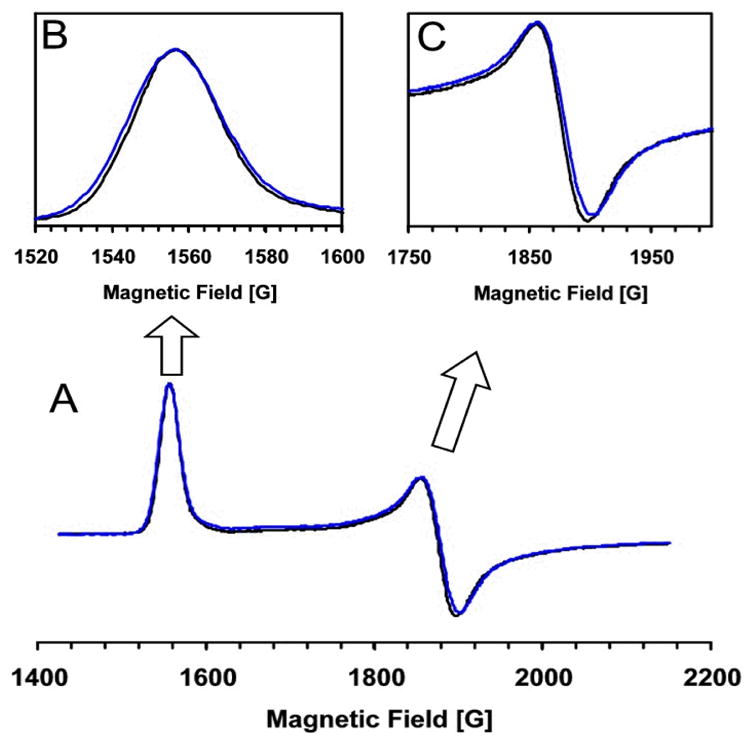
EPR Spectra of Fe(II)-HppE nitrosyl complex with (R)-[17OH]HPP (12-R). Panel A: full low field region of the spectrum; Panel B: detailed view of the g = 4.42 region; Panel C; detailed view of the g = 3.63 region. Black lines: (R)-[16OH]HPP; Blue lines: (R)-[17OH]HPP. Instrumental parameters are the same as shown in Figure 3.
(R)-HPP with 17O enriched in the phosphonate group was prepared by incubating unlabeled 2-R in [17O]-enriched water for several weeks. Binding of this (R)-[P17O3]HPP (13-R, 36.8% enrichment of 17O) to the Fe(II)-HppE nitrosyl complex led to the apparent hyperfine broadening in each spectral resonance (Figure 6). Based on the change in width at half height of the resonance near g = 4.42, the broadening is approximately 11 G, the largest reported for interactions of this type. This demonstrates the direct coordination between the phosphonate oxygen and the iron center. Since the EPR spectrum of the nitrosyl complex of the enzyme-substrate complexes shows that there is only one species present, the hyperfine broadening observed when either the OH or the phosphonate substituent is labeled with 17O shows that these groups simultaneously bind to the iron. Moreover, the presence of NO as a ligand shows that three ligand sites are simultaneously occupied by exogenous ligands in the complex frozen from solution.
Figure 6.

EPR Spectra of Fe(II)-HppE·nitrosyl complex with (R)-[P17O3]HPP (13-R). Black dotted line: (R)-[P16O3]HPP; Blue solid line: (R)-[P17O3]HPP. Instrumental parameters are the same as shown in Figure 5. Spectra are aligned to give approximately the same number of integrated spins of the E/D = 0.066 species. The minor resonance at g = 4.07 in the spectrum of the (R)-[P17O3]HPP (13-R) containing sample is due to a minor amount of denatured enzyme.2
Activity Assay and EPR Analysis of Other Substrate Analogues
Four other HPP analogues were synthesized and used to study substrate binding in HppE active site (Scheme 5). The phenyl substituted (S)- and (R)-HPEP (14-S and 14-R) are both substrates for HppE. They could be converted to the corresponding epoxide (28) and ketone (29) product, albeit at much reduced rates. Their addition to the nitrosyl complex of Fe(II)-HppE led to complex EPR spectra with multiple overlapped signals in the g = 4 region (Figure 7). Table 1 summarizes the relative spin quantitation of each of the EPR signals based on simulations. In contrast to the nearly identical spectra arising from the nitrosyl complexes of HppE bound with 2S and 2R, those from the complexes with (S)-HPEP (14-S) and (R)-HPEP (14-R) are quite different (Figure 7A and 7B). It appears that many binding orientations are possible for each isomer, likely stemming from the bulk of the phenyl group.
Scheme 5.

Figure 7.
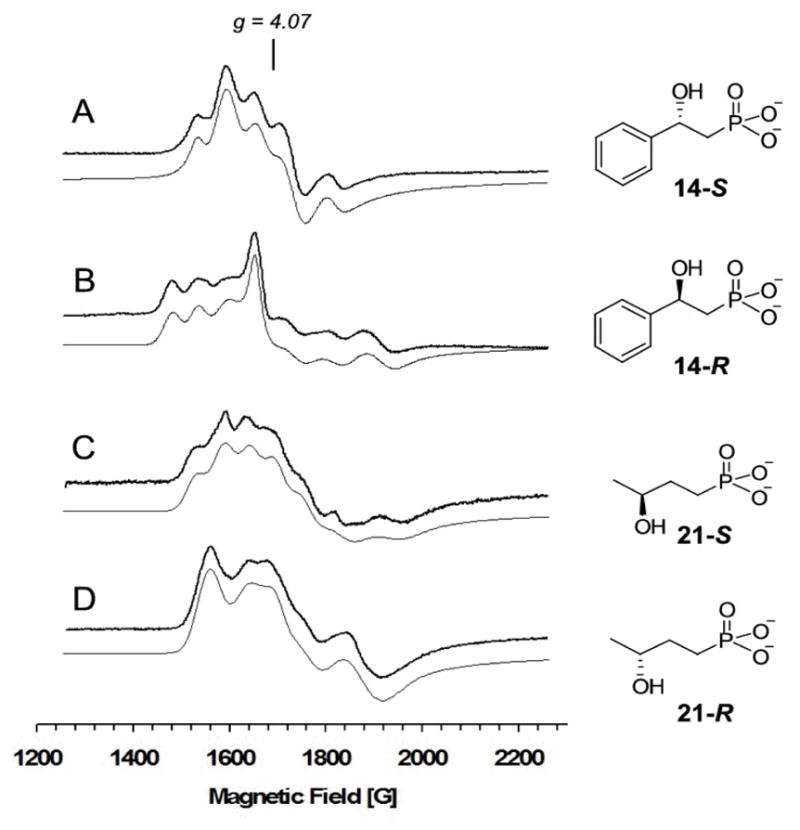
EPR spectra of Fe(II)-HppE·nitrosyl complexes with different substrate analogues. (A) (S)-HPEP (14-S); (B) (R)-HPEP (14-R); (C) (S)-HBP (21-S); (D) (R)-HBP (21-R). Instrumental parameters are the same as shown in Figure 3. Gray lines represent simulated spectra.
(S)- and (R)-HBP (21-S and 21-R) which contain an extra methylene unit between the hydroxyl and phosphonate groups were also prepared. The increased distance between these two iron-binding groups is expected to have significant impact on the regio- as well as stereo-selectivity of turnover. Reaction of HBP with HppE showed that both isomers could serve as substrates. The product of (S)-HBP (21-S) is a diol derivative (31), while that of (R)-HBP (21-R) is a keto-phosphonate (32). The former may derive from hydrolysis of an epoxide precursor (30, Scheme 5). In both cases, the oxidation occurred at C-4 or C-3, instead of at C-1 or C-2. These results imply a more crucial role of the hydroxyl group in determining the regiochemistry of the reaction. The EPR spectra of Fe(II)-HppE nitrosyl complexes with 21-S or 21-R (Figures 7C and 7D) are different from those with 2-S or 2-R, consistent with the different products that are generated.
DISCUSSION
Substrate Binding to HppE in the Presence of NO
Nitric oxide has been used extensively as a probe for the oxygen-binding site in non-heme iron enzymes because of its similarity to O2 in size and binding mode as well as its ability to bind to ferrous ions to form EPR-visible S = 1/2 (26–29) or S = 3/2 (21, 23,30–36){FeNO}7 complexes (37). Using this probe, we have conducted optical and EPR spectral studies of the reduced form of HppE, and demonstrated that the ferrous center is simultaneously accessible to its substrates and NO. Both the electronic absorption and EPR data revealed a substantial reorganization of the metal coordination environment upon substrate binding. The EPR spectra show that the metal center of HppE in solution becomes homogeneous when substrate binds, suggesting that not all of the substrate binding orientations observed in the crystal structure are relevant to catalysis. The critical roles of hydroxyl and phosphonate groups of HPP in binding to the iron center of HppE were investigated by labeling them with 17O in the true substrate and replacing them with other functional groups in substrate analogues. Our results demonstrated that both functional groups are needed for the formation of the homogeneous EPR species with E/D value of 0.066 (Figure 3). Thus, a bidentate substrate coordination involving both the hydroxyl and phosphonate groups is likely to be the active form of the substrate complex which can then bind O2 (or its surrogate NO) in an adjacent position in the iron coordination.
The direct ligation of substrate to iron was demonstrated by the observation that the EPR resonances were broadened by the hyperfine interactions between the iron-nitrosyl center and the 17O nucleus (I = 5/2) in substrate either enriched in the hydroxyl oxygen or the phosphonate oxygen (Figures 5 and 6). Such isotope broadening is possible only if the coupled nuclei are connected through one bond, since the interactions through space or through multiple bonds are too weak to be detected by EPR spectroscopy at X-band frequency. These results provide the most compelling evidence for the direct coordination of both the hydroxyl and the phosphonate groups of HPP to the active site iron of HppE in the presence of NO. Thus, although both monodentate and bidentate binding of 2-S to the metal center were observed in the crystal structure of enzyme-substrate binary complex (18), only the bidentate binding mode is present in the ternary complex of Fe(II)-HppE bound with NO and (S)-HPP in solution under more physiologically relevant conditions.
Substrate Binding and Catalytic Stereospecificity
The metal site of the active form of the substrate complex of HppE that emerges from the X-ray crystallography and the current studies consists of a coordinately saturated Fe center with 2-OH and phosphonate of HPP, the 2-His-1-carboxylate residues (H138, E142, H180) of HppE and NO as the binding ligands. This substrate bidentate-coordination model is consistent with the stereospecificity determined for the epoxidation reaction. It has been shown in the crystal form of HppE that bidentate binding of substrate induces a large conformational change around the active site leaving a small channel at the interface of the α- and β-domains as the passage for O2 (18). Indeed, this channel leads to the only open coordination site on the metal in the bidentate 2-S complex where O2 could bind. An oxygen species bound to the open site would have only access to the pro-Rα-H, which is removed during the conversion of 2-S to 1 (17).
As mentioned above, binding of 2-S and 2-R in the ternary complex is not affected by the chirality at C-2, and both bind in a bidentate manner to the metal center. Because of this rigid binding mode, the outcome of turnover for each enantiomer is solely governed by the stereochemistry of the 2-OH group of HPP. As molecular oxygen is predicted to bind to the only open site of the metal center in the bidentate substrate enzyme complex, an iron-oxygen species at this site must have exclusive access to the relevant hydrogen at C-1 of 2-S or at C-2 of 2-R in a regiospecific manner. The extent of regioselectivity may simply be a function of the proximity of the reactive iron-oxygen species to the targeted hydrogen being abstracted in each isomer.
Shown in Scheme 6 is a substrate-metal-O2 ternary complex model highlighting the proposed distance-dependent regio- and stereospecificity of the HppE reaction. This model explains how the pro-Rα-H in 2-S or the β-H in 2-R is reached by the reactive iron-oxygen intermediate (for simplicity, only the case of Fe(III)-peroxo species (35, Scheme 7) is shown here). Such a regio-as well as stereo-specific hydrogen atom abstraction leads to distinctive radical intermediates (3 versus 5) and consequently, different turnover products (1 versus 4) for these two substrate isomers. Similar observations were also made on the turnover of 21-S and 21-R catalyzed by HppE (Scheme 5). It was found that the oxidation occurs close to the hydroxyl group instead of the phosphonate group in 21-S and 21-R. These results strongly indicate a significant role of the hydroxyl group in HBP, as well as in other HppE substrates, in determining the regiospecificity and stereospecificity of HppE-catalyzed reactions.
Scheme 6.

Scheme 7.

Mechanistic Implications
Scheme 7 presents a mechanistic hypothesis based on the current and previous studies. Although the details of the catalytic mechanism remain to be elucidated, it is generally believed to involve an enzyme-bound substrate radical that is generated via hydrogen abstraction by an activated iron-oxygen species. As depicted in Scheme 7, the initial steps involve substrate binding and activation of dioxygen by the Fe(II) center, which in the presence of exogenous electron(s) from NADH, can produce various oxidative iron-oxygen intermediates, such as Fe(III)-superoxo (35), Fe(III)-hydroperoxo (36), or Fe(IV)-oxo (37). One of these intermediates is expected to be responsible for the abstraction of a C1 hydrogen atom from enzyme-bound 2-S (in 34) to generate a substrate radical intermediate (38, 39, or 40). Radical induced homolytic cleavage of the Fe O bond in this intermediate will produce 1 and also regenerate the iron core.
The direct coordination between active site iron of HppE and the hydroxyl oxygen of 2-S, or 2-R, is likely important to HppE catalysis by facilitating a fast electron transfer from the substrate radical intermediate to the iron center. Given that epoxide 1 and ketone 4 are exclusively produced from 2-S and 2-R, respectively (16), a rapid electron transfer process may be necessary to shorten the lifetime of the radical intermediates and thus prevent potential radical rearrangements. Moreover, a fast electron transfer through the bond connecting the iron center and the hydroxyl oxygen of 2-S may be the key in determining the reaction course toward epoxidation. In the absence of the above Fe-O bond between enzyme and substrate, reaction may proceed to give hydroxylation product, since epoxidation in this case would require the electron be transferred through space, a much slower process.
Conclusion
This study has established a clear picture for the binding of substrates and substrate analogues to HppE in the presence of dioxygen analogue, nitric oxide. By using isotope-labeled substrate, the bidentate mode of substrate binding to the active site iron in the solution state has been fully established. The further characterization of HppE activity using chemically synthesized mechanistic probes has also shown that the binding of the hydroxyl group of the substrates determines the site of oxidation. The combined bioorganic and EPR spectroscopic characterizations have resulted in more detailed understanding of the enzyme-substrate interactions and insight into the stereospecificity of the enzyme action. This study has also provided support for the previously proposed catalytic mechanism of HppE.
Acknowledgments
We thank Ms. Tingfeng Li for helping the measurement of the EPR spectra of some of the substrate analogues.
Footnotes
Abbreviations: ACCO, 1-Aminocyclopropane-1-carboxylate oxidase; CS, clavaminate synthase; DTT, dithiothreitol; EDTA, ethylenediaminetetraacetate; EPR, electron paramagnetic resonance; GlcNAc, N-acetyl-α-D-glucosamine; HBA, 2-hydroxybutyric acid; HBP, 3-hydroxybutylphosphonic acid; HPEP, 2-hydroxy-2-phenylethylphosphonic acid; HPP, 2-hydroxypropylphosphonic acid; HppE, (S)-2-hydroxypropylphosphonic acid epoxidase; IPNS, isopenicillin N synthase; NO, nitric oxide; UDP, uridine 5′-diphosphate.
The intensity of the g = 4.07 signal was dependent on NO concentration. When excess NO (3 psi) was introduced to Fe(II)-HppE, the enzyme became denatured. Gentle shaking caused the enzyme to re-dissolved but the EPR spectrum exhibited the g = 4.07 signal, which is not observed for the nitrosyl complex of the active enzyme.
This work was supported in part by the National Institutes of Health Grants (GM40541 to H.-w.L. and GM24689 to J.D.L.) and the ORAU Faculty Enhancement Award for Life Sciences (to A.L.).
References
- 1.Christensen BG, Leanza WJ, Beattie TR, Patchett AA, Arison BH, Ormond RE, Kuehl FA, Jr, Albers-Schonberg G, Jardetzky O. Phosphonomycin: structure and synthesis. Science. 1969;166:123–125. doi: 10.1126/science.166.3901.123. [DOI] [PubMed] [Google Scholar]
- 2.Hendlin D, Stapley EO, Jackson M, Wallick H, Miller AK, Wolf FJ, Miller TW, Chaiet L, Kahan FM, Foltz EL, Woodruff HB, Mata JM, Hernandez S, Mochales S. Phosphonomycin, a new antibiotic produced by strains of streptomyces. Science. 1969;166:122–123. doi: 10.1126/science.166.3901.122. [DOI] [PubMed] [Google Scholar]
- 3.Seto H, Kuzuyama T. Bioactive natural products with carbon-phosphorus bonds and their biosynthesis. Nat Prod Rep. 1999;16:589–596. doi: 10.1039/a809398i. [DOI] [PubMed] [Google Scholar]
- 4.Ortiz de Montellano PR. Cytochrome P450: Structure, Mechanism, and Biochemistry. Plenum Press; New York: 1995. [Google Scholar]
- 5.Sono M, Roach MP, Coulter ED, Dawson JH. Heme-containing oxygenases. Chem Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 6.Hammerschmidt F. Biosynthesis of natural products with a phosphorus-carbon bond. Part 8 On the origin of the oxirane oxygen atom of fosformycin in Streptomyces fradiae. J Chem Soc, Perkin Trans. 1991;1:1993–1996. [Google Scholar]
- 7.Seto H, Hidaka T, Kuzuyama T, Shibahara S, Usui T, Sakanaka O, Imai S. Studies on the biosynthesis of fosfomycin. 2 Conversion of 2-hydroxypropyl-phosphonic acid to fosfomycin by blocked mutants of Streptomyces wedmorensis. J Antibiot (Tokyo) 1991;44:1286–1288. doi: 10.7164/antibiotics.44.1286. [DOI] [PubMed] [Google Scholar]
- 8.Liu P, Murakami K, Seki T, He X, Yeung SM, Kuzuyama T, Seto H, Liu H-w. Protein purification and function assignment of the epoxidase catalyzing the formation of fosfomycin. J Am Chem Soc. 2001;123:4619–4620. doi: 10.1021/ja004153y. [DOI] [PubMed] [Google Scholar]
- 9.Liu P, Liu A, Yan F, Wolfe MD, Lipscomb JD, Liu HW. Biochemical and spectroscopic studies on (S)-2-hydroxypropylphosphonic acid epoxidase: a novel mononuclear non-heme iron enzyme. Biochemistry. 2003;42:11577–11586. doi: 10.1021/bi030140w. [DOI] [PubMed] [Google Scholar]
- 10.McLuskey K, Cameron S, Hammerschmidt F, Hunter WN. Structure and reactivity of hydroxypropylphosphonic acid epoxidase in fosfomycin biosynthesis by a cation- and flavin-dependent mechanism. Proc Natl Acad Sci USA. 2005;102:14221–14226. doi: 10.1073/pnas.0504314102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan F, Munos JW, Liu P, Liu H-w. Biosynthesis of Fosfomycin, ReExamination and Re-Confirmation of a Unique Fe(II)- and NAD(P)H-Dependent Epoxidation Reaction. Biochemistry. 2006;45:11473–11481. doi: 10.1021/bi060839c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hidaka T, Goda M, Kuzuyama T, Takei N, Hidaka M, Seto H. Cloning and nucleotide sequence of fosfomycin biosynthetic genes of Streptomyces wedmorensis. Mol Gen Genet. 1995;249:274–280. doi: 10.1007/BF00290527. [DOI] [PubMed] [Google Scholar]
- 13.Kuzuyama T, Seki T, Kobayashi S, Hidaka T, Seto H. Cloning and expression in Escherichia coli of 2-hydroxypropylphosphonic acid epoxidase from the fosfomycin-producing organism, Pseudomonas syringae PB-5123. Biosci Biotechnol Biochem. 1999;63:2222–2224. doi: 10.1271/bbb.63.2222. [DOI] [PubMed] [Google Scholar]
- 14.Woodyer RD, Zhao Z, Thomas PM, Kelleher NL, Blodgett JAV, Metcalf WW, van der Donk WA, Zhao H. Heterologous production of fosfomycin and identification of the minimal biosynthetic gene cluster. Chem Biol. 2006;13:1171–1182. doi: 10.1016/j.chembiol.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 15.Woodyer RD, Li G, Zhao H, van der Donk WA. New insight into the mechanism of methyl transfer during the biosynthesis of fosfomycin. Chem Comm. 2007;28:359–361. doi: 10.1039/b614678c. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Z, Liu P, Murakami K, Kuzuyama T, Seto H, Liu H-w. Mechanistic studies of HPP epoxidase: configuration of the substrate governs its enzymatic fate. Angew Chem Int Ed. 2002;41:4529–4532. doi: 10.1002/1521-3773(20021202)41:23<4529::AID-ANIE4529>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 17.Hammerschmidt F, Kaehlig H. Biosynthesis of natural products with a phosphorus-carbon bond. 7 Synthesis of 1,1-2H2-, 2,2-2H2-, (R)- and (S)-1-2H1(2-hydroxyethyl)phosphonic acid and (R,S)-1-2H1(1,2-dihydroxyethyl)phosphonic acid and incorporation studies into fosfomycin in Streptomyces fradiae. J Org Chem. 1991;56:2364–2370. [Google Scholar]
- 18.Higgins LJ, Yan F, Liu P, Liu H-w, Drennan CL. Structural insight into antibiotic fosfomycin biosynthesis by a mononuclear iron enzyme. Nature. 2005;437:838–844. doi: 10.1038/nature03924. [DOI] [PubMed] [Google Scholar]
- 19.Yan F, Li T, Lipscomb JD, Liu A, Liu H-w. Site-directed mutagenesis and spectroscopic studies of the iron-binding site of (S)-2-hydroxypropylphosphonic acid epoxidase. Arch Biochem Biophys. 2005;442:82–91. doi: 10.1016/j.abb.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 20.Brown CA, Pavlosky MA, Westre TE, Zhang Y, Hedman B, Hodgson KO, Solomon EI. Spectroscopic and theoretical description of the electronic structure of S = 3/2 iron-nitrosyl complexes and their relation to O2 activation by non-heme iron enzyme active sites. J Am Chem Soc. 1995;117:715–732. [Google Scholar]
- 21.Chen VJ, Orville AM, Harpel MR, Frolik CA, Surerus KK, Münck E, Lipscomb JD. Spectroscopic studies of isopenicillin N synthase. A mononuclear nonheme iron(II) oxidase with metal coordination sites for small molecules and substrate. J Biol Chem. 1989;264:21677–21681. [PubMed] [Google Scholar]
- 22.Arciero DM, Lipscomb JD. Binding of 17O-labeled substrate and inhibitors to protocatechuate 4,5-dioxygenase-nitrosyl complex. Evidence for direct substrate binding to the active site Fe2+ of extradiol dioxygenases. J Biol Chem. 1986;261:2170–2178. [PubMed] [Google Scholar]
- 23.Arciero DM, Orville AM, Lipscomb JD. [17O]Water and nitric oxide binding by protocatechuate 4,5-dioxygenase and catechol 2,3-dioxygenase. Evidence for binding of exogenous ligands to the active site Fe2+ of extradiol dioxygenases. J Biol Chem. 1985;260:14035–14044. [PubMed] [Google Scholar]
- 24.Whittaker JW, Lipscomb JD. 17O-Water and cyanide ligation by the active site iron of protocatechuate 3,4-dioxygenase. Evidence for displaceable ligands in the native enzyme and in complexes with inhibitors or transition state analogs. J Biol Chem. 1984;259:4487–4495. [PubMed] [Google Scholar]
- 25.David SS, Que L., Jr Anion binding to uteroferrin. Evidence for phosphate coordination to the iron(III) ion of the dinuclear active site and interaction with the hydroxo bridge. J Am Chem Soc. 1990;112:6455–6463. [Google Scholar]
- 26.Woolum JC, Tiezzi E, Commoner B. Electron spin resonane of iron-nitric oxide complexes with amino acids, peptides and proteins. Biochim Biophys Acta. 1968;160:311–320. doi: 10.1016/0005-2795(68)90204-3. [DOI] [PubMed] [Google Scholar]
- 27.Le Brun NE, Andrews SC, Moore GR, Thomson AJ. Interaction of nitric oxide with non-haem iron sites of Escherichia coli bacterioferritin: reduction of nitric oxide to nitrous oxide and oxidation of iron(II) to iron(III) Biochem J. 1997;326:173–179. doi: 10.1042/bj3260173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boese M, Mordvintcev PI, Vanin AF, Busse R, Mèulsch A. S-nitrosation of serum albumin by dinitrosyl-iron complex. J Biol Chem. 1995;270:29244–29249. doi: 10.1074/jbc.270.49.29244. [DOI] [PubMed] [Google Scholar]
- 29.Drapier JC, Pellat C, Henry Y. Generation of EPR-detectable nitrosyl-iron complexes in tumor target cells cocultured with activated macrophages. J Biol Chem. 1991;266:10162–10167. [PubMed] [Google Scholar]
- 30.Arciero DM, Lipscomb JD, Huynh BH, Kent TA, Münck E. EPR and Mössbauer studies of protocatechuate 4,5-dioxygenase. Characterization of a new Fe2+ environment. J Biol Chem. 1983;258:14981–14991. [PubMed] [Google Scholar]
- 31.Clay MD, Cosper CA, Jenney FE, Jr, Adams MWW, Johnson MK. Nitric oxide binding at the mononuclear active site of reduced Pyrococcus furiosus superoxide reductase. Proc Natl Acad Sci USA. 2003;100:3796–3801. doi: 10.1073/pnas.0636858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harpel MR, Lipscomb JD. Gentisate 1,2-dioxygenase from Pseudomonas. Substrate coordination to active site Fe2+ and mechanism of turnover. J Biol Chem. 1990;265:22187–22196. [PubMed] [Google Scholar]
- 33.Harpel MR, Lipscomb JD. Gentisate 1,2-dioxygenase from Pseudomonas acidovorans. Methods Enzymol. 1990;188:101–107. doi: 10.1016/0076-6879(90)88019-7. [DOI] [PubMed] [Google Scholar]
- 34.Nelson MJ. The nitric oxide complex of ferrous soybean lipoxygenase-1. Substrate, pH, and ethanol effects on the active-site iron. J Biol Chem. 1987;262:12137–12142. [PubMed] [Google Scholar]
- 35.Orville AM, Lipscomb JD. Simultaneous binding of nitric oxide and isotopically labeled substrates or inhibitors by reduced protocatechuate 3,4-dioxygenase. J Biol Chem. 1993;268:8596–8607. [PubMed] [Google Scholar]
- 36.Zhang Y, Pavlosky MA, Brown CA, Westre TE, Hedman B, Hodgson KO, Solomon EI. Spectroscopic and theoretical description of the electronic structure of the S = 3/2 nitrosyl complex of non-heme iron enzymes. J Am Chem Soc. 1992;114:9189–9191. [Google Scholar]
- 37.Enemark JH, Feltham RD. Principles of structure, bonding, and reactivity for metal nitrosyl complexes. Coord Chem Rev. 1974;13:339–406. [Google Scholar]