

Abstract
The discovery of Helicobacter pylori has already changed the natural history of peptic ulcer disease, with most patients being cured at their first presentation. Similarly, the incidence of gastric cancer and other diseases related to H. pylori are likely to be greatly reduced in the near future. Isolation of the spiral intragastric bacterium H. pylori totally reversed the false dogma that the stomach was sterile, and it taught us that chronic infectious disease can still exist in modern society. Helicobacter pylori’s unique location, persistence, and evasion of the immune system offer important insights into the pathophysiology of the gut. Also, the fact that it was overlooked for so long encourages us to think “outside the box” when investigating other diseases with obscure etiologies. We should consider such provocative scientific ideas as bridges to the future disease control.
Key words: H. pylori diagnosis, H. pylori eradication, peptic ulcer, gastritis, gastric cancer
Introduction
Background
In recent years, research groups have concentrated on the molecular structures whereby Helicobacter pylori survives in the hostile gastric environment and persists in spite of the host’s immune response. Epidemiologic studies have also provided convincing evidence that H. pylori is associated not only with gastroduodenal diseases, including chronic gastritis, peptic ulcer diseases, low-grade gastric mucosa-associated lymphoid tissue (MALT) lymphoma, and noncardiac gastric adenocarcinoma,1–6 but also with extragut diseases.7–12
The initial advance that facilitated the explosion of research related to H. pylori was the development of microaerobic cultivation methods for this bacterium. Then, it was shown that intentional infection of a volunteer led to gastritis.13 Classical bacteriology demonstrated that H. pylori is a Gram-negative, flagellated organism that produces a number of enzymes, including catalase and urease, which help neutralize host responses and favor colonization.14,15 One of the early benefits gained from this basic research was the demonstration that urease is produced by virtually all strains of H. pylori. This led to the development of accurate diagnostic tests, including the rapid urease test and the urea breath test. A stool antigen test has also emerged as an informative, noninvasive means by which to diagnose infection via detection of bacterial antigen.16 In addition, the ability to culture H. pylori provided a diagnostic test that allowed assessment of antibiotic susceptibility.
Before the modern discovery of intragastric bacteria 25 years ago,17 several reports dealt with spiral gastric bacteria as long ago as 1890. The modern discovery of H. pylori might have been delayed because in 1954, Palmer18 denied the existence of microorganisms in the human stomach, claiming that spiral bacteria were seen only post-mortem because microorganisms could not survive in the acidic gastric environment. However, microorganisms had been described in the gastric mucosa since the 1890s.19 For example, in Japan, Kasai and Kobayashi20 of the Kitasato Institute reported propagation of a spirochete-like organism, probably Helicobacter felis, from the stomach of dogs and cats, but not from laboratory animals (Fig. 1). They showed that when rabbits infected with these spirochetes were also inoculated with the virus fixe (attenuated rabies virus), marked hemorrhagic lesions occurred in the gastric mucosa.20 They also demonstrated that spirochetes inoculated into the gastric mucosa of the mouse could be eradicated by arsaminol20 (a modern equivalent of arsaminol is thought to be bismuth, a close relative of arsenic).21,22 Seventy years then elapsed before scientists again attempted to treat gastric diseases associated with “spirochetes,” that is, Helicobacter infection.
Fig. 1.

Rokuzo Kobayashi (1887–1969), at his desk in the office of Keio University, Tokyo, Japan. Photo taken in 1935. Adopted from “Kobayashi Rokuzo” published by Dr. Rokuzo Kobayashi Memorial Association, 1971
Present status
Epidemiology
Most new H. pylori infections occur in children; however, in the absence of specific clinical signs associated with infection, the mode of transmission is difficult to define. Vomitus, saliva, and feces are the presumed sources of direct transmission, particularly in crowded housing conditions. Generally, infection correlates inversely with socioeconomic conditions,23 with lifetime infection rates in affluent countries dropping toward 10%, while rates in countries with emerging economies approach 80%–90%.16,24,25 Most infected individuals experience asymptomatic gastritis, although recurrent gastroduodenal ulceration may occur in 10%–15% of the infected population. The incidence of gastric cancer is lower, with approximately 1%–5% of infected individuals developing adenocarcinoma and even fewer experiencing gastric MALT lymphoma.1,2,26–29
Initially H. pylori depends on its urease enzyme to survive in an acid environment, and then on its spiral shape and motility to reach the gastric epithelium, where it sticks to cells through the various specific adhesion molecules. Once attached, most organisms then deliver damaging toxins to the mucosa.
Pathogenesis
The most investigated of these toxins stem from a segment of bacterial DNA referred to as the cag pathogenicity island (cag PAI). The presence of the cag PAI is usually associated with presence of the s1m1 (more harmful) subtype of the vacuolating cytotoxin VacA. Genes within the cag PAI encode proteins such as CagE, an ATPase that drives a type IV secretion apparatus that allows bacterial macromolecules, especially CagA toxin, to translocate into the host cell.3,30 The intact cag PAI of H. pylori plays a significant role in the pathogenesis of gastritis in humans, since H. pylori carrying the cag PAI is associated with increased chemokine expression in gastric mucosal specimens31 and stronger inflammation.
CagA is injected into epithelial cells by the type IV secretory system, possibly as a mechanism to reduce the stress of gastric acid on the bacterium, since its transcription is increased at low pH.32 VacA is a pore-forming protein, which causes epithelial cells to vacuolate in vitro. Retrospective analyses of samples from gastric cancer patients have suggested an association with the cagA+/vacA+-positive genotype.33 Further studies have confirmed that a risk for both cancer and peptic ulcer is associated particularly with the cagA/vacA s1m1 genotype, but rarely with strains carrying the vacA s2m2 polymorphism.34,35 However, the cagA gene does not act alone, because delivery of the CagA protein into epithelial cells appears to be dependent on a secretion system encoded for by the adjacent cluster of genes, the cag PAI.36 After the CagA protein is injected into the host cell cytoplasm, it is tyrosine phosphorylated by host Src kinases and subsequently changes the epithelial morphology.37–41 CagA in strains from distinct geographic populations appears to be phosphorylated to a different degree, resulting in graded effects on intracellular signaling.42,43
Oxygen-derived free radicals, which are released from activated neutrophils, are considered to be one of the potential toxic factors involving H. pylori-induced gastric mucosal injury, since H. pylori exhibits chemotactic activity for neutrophils.44,45 Neutrophil infiltration of the gastric mucosa leads to development of the initial lesion in H. pylori-associated gastritis and remains a hallmark of active bacterial infection. In response to the activation of neutrophils, NADPH oxidase (Nox) in the neutrophils’ cell membranes becomes activated; an electron transfer takes place from NADPH in cells to oxygen inside and outside of cells, and the oxygen molecules that receive an electron become superoxide radicals (O2−), which are rapidly converted to hydrogen per oxides (H2O2) by spontaneous dismutation, or to enzymatic superoxide dismutase (SOD) and hydroxyl radicals (•OH), which are formed nonenzymatically in the presence of Fe2+. In neutrophils, myeloperoxidase (MPO) also results in the formation of a potent oxidant, the hypochlorous anion (OCl−), from H2O2 in the presence of Cl−. This hypochlorous anion reacts with ammonia, derived from urea by H. pylori-associated urease, and yields cytotoxic oxidant, monochloramine (NH2Cl) 46 with its lipophilic property, allowing it to freely penetrate biological membranes to oxidize intracellular components47 (Fig. 2).
Fig. 2.
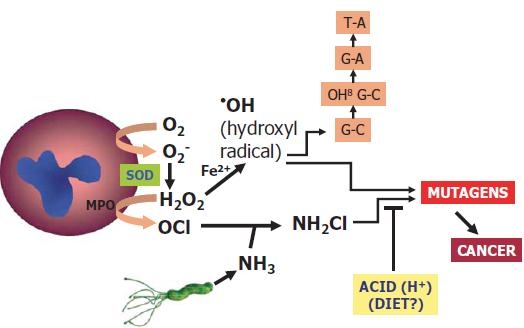
Helicobacter pylori-associated gastric oxidative injury: monochloramine pathway. MPO, myeloperoxidase; SOD, superoxide dismutase
8-Hydroxy-2-deoxyguanosine (8-OHdG) is one of the most abundant lesions in DNA induced by reactive oxygen species.48 It results from an attack by a singlet hydroxyl or oxygen radical on guanine (Fig. 2). Patients with cagA-positive H. pylori strains have higher 8-OHdG levels than both cagA-negative and H. pylori-negative patients.49 The cagA-positive patients are characterized by greater oxidative DNA damage overall and at a younger age, in the presence of multifocal atrophy.49
The preferred animal model for studying H. pylori gastritis is the Mongolian gerbil (Meriones unguiculatus).50–53 In one study, gerbils infected with a mutated strain lacking cagE exhibited a substantially reduced severity of gastritis.54 Gastric ulceration developed in all of the gerbils infected with the wild-type strain but in none of those infected with the cagE mutant strain. In addition, intestinal metaplasia and gastric cancer developed in some gerbils with wild-type infection but not in animals infected with the cagE-negative mutant.54 These findings were confirmed by showing that the repaired cagE-positive H. pylori strain induced severe gastric inflammation, gastric mucosal atrophy, and gastric ulcer formation.55 Recently, the nucleotide sequences of cDNAs encoding preproghrelin, a precursor of ghrelin (an appetite-promoting endocrine peptide produced mainly from A-like cells in the gastric fundus)52,56 and sonic hedgehog, a morphogen that may regulate and organize fundic gland epithelial differentiation, have been identified in the Mongolian gerbil.53,57 Preproghrelin and sonic hedgehog mRNA expression is significantly reduced in the gastric mucosa of Mongolian gerbils with persistent H. pylori colonization.52,53
After reaching the mucosa and attaching by means of its lectins, H. pylori applies toxin to the gastric mucosa (Fig. 3). Then, the cells change their morphology and migrate, some detach, and sometimes apoptosis occurs. The healing process involves proliferation of adjacent, more healthy cells. Gastric epithelial cell turnover is regulated by the balance between cell death (apoptosis) and cell proliferation (Fig. 3). Such a regulated cell turnover by apoptosis and proliferation is essential for maintaining healthy gastric mucosal integrity. Some cells differentiate into intestinal tissue, a transformation called intestinal metaplasia. This may be somewhat protective in that H. pylori is unable to attach to these cells, allowing inflammatory processes to become quiet in these areas. However, extensive metaplasia is associated with decreased acid secretion, which may allow other commensal bacteria to colonize and carcinogen formation to occur. Proliferation by itself is not carcinogenic, but mutagens, such as monochloramine, may be formed in the presence of ammonia and hypochlorous anion. These cause errors in transcription to occur.
Fig. 3.
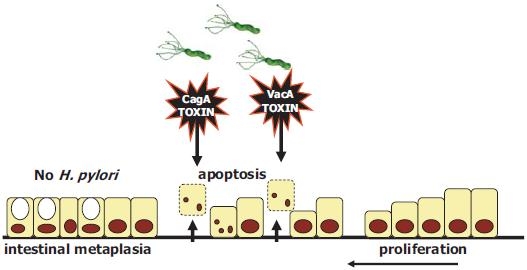
After reaching the mucosa and attaching by means of its lectins, H. pylori applies toxin to the gastric mucosa. Cells change their morphology and migrate, some detach, and sometimes apoptosis occurs. The healing process involves proliferation of adjacent, more healthy cells. Proliferation by itself is not carcinogenic, but mutagens, such as monochloramine, may be formed in the presence of ammonia and hypochlorous anion. These cause errors in transcription. Some cells differentiate into intestinal tissue, a transformation called intestinal metaplasia
At an earlier phase after bacterial inoculation, an initial transient enhancement of cell apoptosis (2–4 weeks) and a later increase in cell proliferation (16–20 weeks) occur in the H. pylori-colonized gastric mucosa of gerbils.58 While the level of apoptosis increases as H. pylori-evoked gastric mucosal inflammation advances in mice, the enhanced level of apoptosis is significantly attenuated in H. pylori-colonized gastric mucosa of gerbils with enhanced inflammation and epithelial proliferation in the gastric mucosa during longer term colonization,59 suggesting a possible link between attenuated gastric mucosal apoptosis and a specific carcinogenesis in gerbils after H. pylori colonization.60,61
Peptic ulcer disease
The strongest association of H. pylori infection is with peptic ulcer disease;62 95% of duodenal ulcers and 70% of gastric ulcers are related to H. pylori infection. Several cohort studies have estimated that the lifetime risk of ulcer disease in H. pylori-positive subjects is three to ten times that in H. pylori-negative subjects.63 Introduction of H. pylori eradication regimens has completed the evidence for a causal relation between H. pylori and peptic ulcer disease because H. pylori eradication has been shown to strongly reduce the risk of ulcer recurrence.64 In prospective controlled studies of duodenal ulcer recurrence, ulcers recurred in 55%–86% of patients in H. pylori-positive cohorts, in contrast to a rate of 3%–21% in H. pylori-negative (successfully eradicated) cohorts65–70 (Table 1). Gastric ulcers typically recurred in 46%–71% of patients in H. pylori-positive cohorts, contrasting with ulcer recurrence rates of 10% or less in H. pylori-negative (successfully eradicated) cohorts71–76 (Table 2).
Table 1.
Duodenal ulcer recurrence rate 1 year after H. pylori eradication
| Authors | Year | Noneradication group % (95% CI) | Eradication group % (95% CI) | P value |
|---|---|---|---|---|
| Marshall et al.66 | 1988 | 55 (34–75) | 21 (3–40) | 0.029 |
| Hentschel et al.65 | 1993 | 86 (76–96) | 8 (0.4–15) | <0.001 |
| Penston67 | 1994 | 67 (62–72) | 7 (5–10) | – |
| Tytgat68 | 1995 | 58 (55–61) | 2.6 (2–4) | – |
| Hopkins et al.69 | 1996 | 67 (63–72) | 6 (4–9) | 0.002 |
| Laine et al.70 | 1998 | 56 (50–61) | 20 (14–24) | <0.001 |
CI, confidence interval
Table 2.
Gastric ulcer recurrence rate 1 year after H. pylori eradication
| Authors | Years | Noneradication group % (n) | Eradication group % (n) | P value |
|---|---|---|---|---|
| Fukuda et al.72 | 1995 | 71 (42/59) | 0 (0/24) | <0.001 |
| Seppala et al.75 | 1995 | 47 (60/128) | 7 (2/29) | <0.001 |
| Sung et al.76 | 1995 | 58 (11/19) | 8 (2/26) | <0.001 |
| Kato et al.73 | 1996 | 46 (18/39) | 0 (0/11) | <0.001 |
| Axon et al.71 | 1997 | 48 (30/63) | 7 (3/44) | <0.001 |
| Lazzaroni et al.74 | 1997 | 63 (20/32) | 10 (2/20) | <0.001 |
The meta-analytic results of a North American study by Laine et al.70 have shown that H. pylori eradication markedly decreases ulcer recurrence. Nevertheless, ulcers recurred in 20% of patients in these studies within 6 months, despite a successful cure of infection and no reported use of nonsteroidal anti-inflammatory drugs (NSAIDs), suggesting that non-H. pylori, non-NSAID ulcers may be more common in the United States than previously believed.
According to a recent systematic review of 56 randomized controlled trials of treatment for peptic ulcer disease in H. pylori-positive adults,77 a 1 to 2 weeks’ course of H. pylori eradication therapy is an effective treatment. Namely, in duodenal ulcer (DU) healing, eradication therapy was superior to several weeks of ulcer healing drugs [34 trials, 3910 patients, relative risk (RR) of ulcer persisting = 0.66] or no treatment (2 trials, 207 patients, RR = 0.37). In gastric ulcer (GU) healing, no significant differences were detected between eradication therapy and administration of an ulcer healing drug (14 trials, 1572 patients, RR = 1.25). For preventing DU recurrence, no significant differences were detected between eradication therapy and maintenance therapy with an ulcer healing drug (4 trials, 319 patients, RR = 0.73), but eradication therapy was superior to no treatment (27 trials, 2509 patients, RR = 0.20). For preventing GU recurrence, eradication therapy was superior to no treatment (11 trials, 1104 patients, RR = 0.29).
The incidence of peptic ulcers has steadily decreased in Western countries in the last two decades. The decreasing prevalence of peptic ulcers is thought to result both from contraction of the formerly large pool of patients with recurrent ulcer disease by H. pylori eradication at first presentation and from the decreasing prevalence of H. pylori infection in the population.24,78,79 The latter is related to several factors, such as improved hygiene and living conditions, decreased family sizes, and the use of antimicrobial therapy for other purposes.
Helicobacter pylori and NSAIDs are the two major causes of peptic ulcer disease. Their potential interaction in the induction of ulcer disease remains controversial. A thorough analysis of interaction data has revealed that the ulcer-inducing effects of both risk factors are cumulative.80 Eradication of H. pylori in chronic NSAID users does no harm, and in some studies has decreased the incidence of ulcer disease. In a study from Hong Kong, patients starting with NSAID maintenance therapy were randomized to eradication therapy or placebo. After 6 months of follow-up, the incidence of ulcers was 12.1% in the eradication group and 34.4% in the placebo group (P = 0.0085). The corresponding probabilities of complicated ulcers after 6 months were 4.2% and 27.1% (P = 0.0026), respectively.81 These data show that, at least in an Asian population, screening and treatment for H. pylori infection significantly reduces the risk of ulcers in patients starting long-term NSAID treatment, but also that eradication alone is insufficient to totally prevent ulcers and ulcer complications. In several studies, H. pylori eradication was inferior to omeprazole maintenance treatment for prevention of recurrent NSAID-associated ulcer bleeding.82,83 An overall synthesis of these data suggests that NSAIDs and H. pylori are independent, but not necessarily additive, causes of peptic ulcer. The wise choice might be to address both issues in all patients with ulcer complications: supplement NSAID users at risk with a second agent, consider the option of selective cyclooxygenase-2 (cox-2) inhibitors, and eradicate H. pylori as well.
Functional dyspepsia
Recently, the “Rome III” classification for functional gastrointestinal disorders was defined.84 In this classification, functional dyspepsia (FD) is included in the subcategory of functional gastroduodenal disorders.85 Four types of functional gastroduodenal disorders are distinguished: FD [B1, comprising postprandial distress syndrome (B1a) and epigastric pain syndrome (B1b)]; belching disorders (B2); functional nausea and vomiting disorders, functional vomiting, and cyclic vomiting syndrome (B3); and rumination syndrome (B4).85,86
Of patients with functional dyspepsia, 30%–60% carry H. pylori, but this prevalence is not much different from that in the unaffected population.87 Various studies have focused on the effect of H. pylori eradication in patients with both functional and uninvestigated dyspepsia. A meta-analysis of 13 randomized studies of nonulcer dyspepsia showed that when dyspepsia outcomes are dichotomized into minimal/resolved versus same/worse symptoms, H. pylori eradication is associated with an 8% RR reduction compared with placebo.88 The calculated number-needed-to-treat to cure one case of dyspepsia is 18. Apart from the focus on cure of symptoms, cost efficiency is also an issue when treating large groups of patients. Given this reported number-needed-to-treat, it remains unclear whether H. pylori eradication is a cost-effective strategy. In a Canadian primary care study, however, H. pylori eradication was reported to be more cost-effective after 1 year of follow-up than empirical diagnosis and treatment.89,90 In another interesting study, Sonnenberg and Townsend91 found that, for some reason, expenditure on many other aspects of health care decreased after patients had eradicated H. pylori.
Another Cochrane meta-analysis compared treatment strategies for uninvestigated dyspepsia.92 Individual strategies consisted of initial pharmacological therapy (with endoscopy for treatment failures), early endoscopy, testing for H. pylori and endoscopy only for those testing positive, or H. pylori eradication therapy with or without prior testing. The analysis showed that proton-pump inhibitors (PPIs) are effective in the treatment of dyspepsia. Early investigation by endoscopy or H. pylori testing benefited some patients with dyspepsia but was not cost-effective as part of an overall management strategy.92 A meta-analysis of five studies comparing prompt endoscopy followed by targeted treatment versus an H. pylori test-and-treat strategy without further investigation reported that endoscopy was associated with a small symptom benefit at 1 year (RR, 0.95), but this strategy was not cost-effective.93 In summary, H. pylori play a role in the etiology of dyspeptic symptoms; thus, H. pylori test-and-treat strategies are effective for a subgroup of patients with dyspepsia. For patients with uninvestigated dyspepsia, an H. pylori test-and-treat strategy is an appropriate option, although empirical acid-suppressive therapy can be more efficient in populations with a low H. pylori prevalence. Also, in patients with investigated FD, H. pylori eradication is a relevant option. In both situations, patients must be aware that symptom resolution may take months after completion of therapy.
Atrophic gastritis, intestinal metaplasia, and gastric cancer
Thirty years ago, Correa et al.94 proposed that gastritis could progress to gastric cancer, which made the identification of H. pylori as a major cause of gastritis even more relevant. Chronic H. pylori-induced inflammation can eventually lead to loss of the normal gastric mucosal architecture, with destruction of gastric glands and replacement by intestinal metaplasia. This process of atrophic gastritis and intestinal metaplasia occurs in approximately half of the H. pylori-colonized population, first in those subjects and at those sites where inflammation is most severe.95 Because not everyone infected with H. pylori develops gastric cancer, several modifiers have been identified. Currently, the perception is that gastric cancer arises from multiple “hits,” which include oxidative stress and environmental toxins, which increase mutation rates. Diet, bacterial factors, and genes regulating the host response likely affect the degree of oxidative stress and DNA damage.96 Host polymorphisms in the regions controlling interleukin (IL)-1 have been reported to be associated with an increased incidence of hypochlorhydria and gastric cancer,97–102 probably because these regions regulate the inflammatory response to bacterial infections. Areas of gland loss and intestinal metaplasia extend with time multifocally, and although they do not give rise to any specific symptoms, they increase the risk for gastric cancer by five-to 90-fold, depending on the extent and severity of atrophy.103
Evidence that H. pylori increases the risk of gastric cancer development via the sequence of metaplasia and atrophy originates from various studies that showed that H. pylori-positive subjects develop these conditions far more often than do uninfected controls.104 This finding is supported by data that show geographical associations between the prevalence of H. pylori and the incidence of gastric cancer.105,106 On the basis of these findings, it has been estimated that H. pylori colonization increases the risk of gastric cancer approximately tenfold, and H. pylori has been designated a class I carcinogen by WHO.107 Later case-control studies that looked more extensively into signs of previous H. pylori infection in gastric cancer patients and controls re ported even higher odds ratios, up to 68, for development of distal gastric cancer in the presence of H. pylori infection.108 This result is supported by data from animal models, most notably the Mongolian gerbil model, in which H. pylori infection induces atrophic gastritis and gastric dysplasia.60,61,109 The risk of development of atrophy and cancer in the presence of H. pylori is again related to host and bacterial factors, which influence the severity of the chronic inflammatory response and the amount of gastric mucosa involved. As such, the risk is increased not only in subjects colonized with cagA-positive strains110,111 but also in those with a genetic predisposition to higher IL-1 production in response to colonization.97 In the developed world, 60% to 80% of gastric cancers are therefore related to the long-term presence of H. pylori.
Interestingly, the incidence of gastric cancer has significantly decreased over the past decades in Western countries. This decrease preceded the aforementioned decrease in the prevalence of H. pylori. As a slow process over decades, however, this decrease is not relevant to an individual subject who is H. pylori-positive. Furthermore, in spite of the decline in gastric cancer incidence in Western countries, gastric cancer is still the fourth most common cancer in the world, as its incidence remains very high in large areas of the world, particularly in parts of East Asia and South America.112 Much research now focuses on the cancer-preventive effect of H. pylori eradication, aimed at both the general population and patients with preneoplastic conditions, particularly atrophic gastritis and intestinal metaplasia. In the case of the latter, several placebo-controlled randomized studies have now reported that H. pylori eradication can induce to some extent regression of atrophy.113–117 The effect of these interventions on gastric cancer prevention is, however, less obvious than the historical cancer incidence might suggest. In several studies, H. pylori eradication had in the first years of follow-up no significant effect on gastric cancer incidence.114,116,118 In all of these studies, the difference between the eradication and placebo groups with respect to the incidence of gastric cancer in the first 4 to 12 years after treatment was not significant. The striking observation in all of these studies, however, was that those gastric cancers that occurred after eradication treatment were confined to those subjects who already had atrophic gastritis and intestinal metaplasia at baseline, suggesting that the major cancer-preventive effect of H. pylori eradication is to be expected in subjects without those precancerous conditions; patients with those conditions may at least in part have already passed the point of no return. If confirmed by other studies, this result means that the observed regression of the severity of atrophy is of no direct relevance with respect to cancer risk. One study differed from the others in that it did observe a significant preventive effect of H. pylori eradication on gastric cancer development.119 The investigators monitored 1120 patients in a nonrandomized study for a mean of 3.4 years (range, 1–8 years) after H. pylori eradication treatment. Gastric cancer developed in 8 of 944 patients cured of infection and in 4 of 176 who had persistent infection (P = 0.04). The remarkable observation was that gastric cancers occurred only in patients with previous gastric ulcers and not in those who had duodenal ulcers (P = 0.005).119 This result is consistent with the hypothesis that gastric ulcer patients, in contrast to duodenal ulcer patients, are characterized by reduced gastric acid secretion, corpus-predominant gastritis, and accelerated progression toward atrophic gastritis and intestinal metaplasia. Similarly, previous studies have also shown that gastric ulcer patients are at higher risk for gastric cancer than are duodenal ulcer patients.26 The above-mentioned preventive studies, taken together, described cancer development in 29 (1.5%) of 1896 subjects receiving placebo and in 25 (0.9%) of 2754 subjects receiving eradication treatment, corresponding to an odds ratio of 0.54 for cancer development after H. pylori eradication compared with placebo, suggesting that H. pylori eradication may decrease the risk for development of distal gastric cancer. Further studies are being performed in this field, the results of which will allow a more precise determination of the cancer-preventive effect of H. pylori eradication, of whether atrophy and metaplasia are conditions beyond the point of no return, and of the side effects and costs of such preventive measures.
The Japan Collaborative Cohort Study Group for Evaluation of Cancer Risk reported that women with a family history of gastric cancer and H. pylori infection were associated with a 5.1-fold greater risk for gastric cancer compared with women without a family history of gastric cancer and who did not have H. pylori infection.120 In contrast, family history and H. pylori infection were not found to be significant risk factors for gastric cancer in male patients in their report, where the investigation of H. pylori infection was performed only by using H. pylori antibodies. Recently, results of a large nested case-control study within a prospective cohort have been reported in Japan.121 Among 123 576 subjects that were followed up from 1990 to 2004, 511 gastric cancer cases matched to 511 controls were used in this analysis. The adjusted odds ratio of gastric cancer associated with H. pylori infection was 5.1. In addition, if anti-CagA positivity was used as an indicator of previous or current infection, the risk was reached 12.5.
Gastric MALT lymphoma
Nearly all MALT lymphoma patients are H. pylori positive,122 and H. pylori-positive subjects have a significantly increased risk for the development of gastric MALT lymphoma.123 Because of diagnostic controversies and the relative rarity of this disorder, the exact incidence in H. pylori-positive subjects is unknown, but MALT lymphomas occur in fewer than 0.1% of H. pylori-positive subjects.124 Randomized trials to determine the effect of H. pylori eradication in MALT lymphoma patients are therefore not feasible, but various case series have reported that eradication can lead to complete remission in patients with low-grade gastric MALT lymphoma.125–128 Overall, approximately 60%–80% of these patients reach complete remission following H. pylori eradication, some 10% continue to have signs of minimal residual disease, and the remainder show no response or disease progression. The variation in response among different series may be due in part to different criteria for the diagnosis of MALT lymphoma, potentially including some patients with benign lymphoid aggregates. Of those who initially reach complete remission after H. pylori eradication, 10%–35% show recurrent disease during further follow-up. For that reason, long-term follow-up of MALT lymphoma patients is mandatory.129 A major predictor of the response appears to be the presence of a t(11;18) (q21;q21) translocation. This translocation is associated with API2-MALT1 fusion, the former being involved in regulation of apoptosis and the latter resembling a caspase-like protein, with an as yet unknown biological function. Together, the fusion leads to suppression of apoptosis. Several studies have reported that MALT lymphomas with this translocation respond only rarely or not at all to H. pylori eradication.130,131
Gastroesophageal reflux disease
Gastroesophageal reflux disease (GERD) has long been considered to occur independently of H. pylori colonization; that is, it occurs with the same frequency and severity in H. pylori-positive and -negative subjects. This opinion is based on cross-sectional observations that suggest that the prevalence of H. pylori among GERD patients is similar to that among controls.132 However, further studies have suggested that H. pylori might protect against the development of GERD and by that means might be of benefit to its hosts. This slowly emerging concept derives from repeated observations of a low prevalence of H. pylori among GERD patients, particularly of the more virulent strains;133 reciprocal time and geographical trends for H. pylori prevalence compared with the incidence of GERD and its complications; a potentially increased incidence of GERD after H. pylori eradication;134 and the recognition that H. pylori-induced corpus gastritis reduces acid secretion. However, no evidence suggests that H. pylori eradication has a considerable impact on either the new development of GERD,135 the worsening of preexisting GERD when treatment has been withdrawn during disease remission,136 or preexistent GERD in remission during PPI maintenance therapy.113,117 Together, these data show that although epidemiologic data suggest that there may be an inverse relation between H. pylori and GERD, the risk for new development or worsening of preexistent GERD is not an issue when deciding whether to treat H. pylori.
Diagnosis of H. pylori infection
Diagnostic methods for H. pylori infection are categorized as invasive or noninvasive tests.137 Invasive tests involve endoscopic biopsy of gastric mucosal samples, whereas noninvasive tests do not require an endoscopic procedure. The invasive tests are histology,138 rapid urease test (RUT),139 and microaerobic bacterial culture.17 The advantage of the RUT is that it is inexpensive, easy-to-use, and rapid. The advantages of culture are its specificity of 100% (direct demonstration of the presence of H. pylori) and that it allows further characterization of the organism (determining its sensitivity to antibiotics, investigating its virulence factors, etc). Noninvasive tests are the urea breath test,140 antibody tests of sera141–143 or of urine,144,145 and the H. pylori stool antigen test.146–149
H. pylori eradication therapy
During the 20 years since the World Gastroenterology Organization (WGO-OMGE) consensus conference, international or domestic consensus conferences for H. pylori eradication have been held all over the world, including in Europe, America, and Asia (Fig. 4). Most recently, on 22 November 2006, the Japanese Society for Helicobacter Research held a “Current Consensus—2006” (Management of H. pylori infection in Japan) meeting in Tokyo. In this consensus meeting, one of the main topics was whether Japan will become the first H. pylori-free country.
Fig. 4.
Worldwide consensus conferences on H. pylori eradication therapy. ESPCG, European Society for Primary Care Gastroenterology; NIH, National Institutes of Health; ADHF, American Digestive Health Foundation; OMGE, Organisation Mondiale de Gastro-Entérologie (World Gastroenterology Organization)
Originally, in 1988, Marshall et al.66 reported that H. pylori had been eradicated in 27% of a colloidal bismuth substrate (CBS)/placebo group and in 70% of a CBS/tinidazole group. Furthermore, when H. pylori persisted, 61% of duodenal ulcers healed and 84% relapsed, whereas when H. pylori was cleared, 92% of ulcers healed and only 21% relapsed during the 12-month follow-up period.66
According to a randomized, double-blind, placebo-controlled study performed by Lind et al.,150 787 patients with proven duodenal ulcer disease were randomized to treatment twice daily for 1 week with omeprazole, 20 mg (O), plus either placebo (P) or combinations of two of the following antimicrobials: amoxicillin, 1 g (A), clarithromycin, 250 or 500 mg (C250, C500), or metronidazole, 400 mg (M). Their eradication rates for the all-patients-treated analysis were 96%, OAC500; 95%, OMC250; 90%, OMC500; 84%, OAC250; 79%, OAM; and 1%, OP. These results suggest that omeprazole-based triple therapies given twice daily for 1 week produce high eradication rates, are well-tolerated, and are associated with high patient compliance.
In terms of second-line treatment, Matsuhisa et al.151 examined the eradication rate of a PPI, amoxicillin, and metronidazole (PPI/AM) regimen in 228 patients with H. pylori infection, in whom the first-line therapy with a PPI, amoxicillin, and clarithromycin administered for 1 week had failed. Their eradication rates for PPI/AM(500) and PPI/AM(750) were 90.6% and 88.6%, respectively, suggesting that PPI/AM administered for 1 week may be a highly effective second-line regimen.
In terms of third-line regimens, Gisbert et al.152 performed a third eradication treatment with levofloxacin (500 mg), amoxicillin (1 g), and omeprazole (20 mg), each twice daily for 10 days (n = 100), which resulted in per-protocol and intention-to-treat eradication rates of 66% and 60%. Although such fluoroquinolone-based rescue therapy constitutes an encouraging empirical third-line strategy after multiple previous H. pylori eradication failures,152 a high resistance rate (47.9%) to fluoroquinolone has been observed in H. pylori strains from Japanese patients after unsuccessful H. pylori eradication. Therefore, the choice of this antibiotic for the third-line treatment should be made only after drug susceptibility testing.153,154
Side effects and contraindications for H. pylori eradication
Side effects of H. pylori eradication are reported in up to 50% of patients taking the triple agent regimen.155,156 The adverse effects are usually mild; fewer than 10% of patients stop treatment due to side effects.155 The common side effects are dysgeusia and diarrhea. Amoxicillin can cause diarrhea or an allergic reaction with skin rash. Clarithromycin can cause dysgeusia, vomiting, nausea, abdominal pain, and rarely QT prolongation. Metronidazole can cause seizures, peripheral neuropathy, and a disulfiram-like reaction with ethanol, and it can increase the toxicity of anticoagulants, lithium, and phenytoin. Acute gastrointestinal mucosal lesions have been demonstrated to be an adverse event after eradication therapy. Acute duodenal erosions were observed in 13.5% of patients who received eradication therapy;157 erosions were transient and asymptomatic in the early phase after eradication. Acute gastric erosions showed the same clinical characteristics, but acute gastric ulcers with severe symptoms rarely occurred. The acute mucosal lesions that occurred were probably attributable to an increase in acid secretion associated with the resolution of gastric mucosal inflammation.6 On the other hand, the occurrence of mild reflux esophagitis at a late phase after eradication is controversial. Helicobacter pylori infection is an environmental factor that has declined as GERD has increased.158 However, a systematic review159 of randomized trials did not show any role of H. pylori eradication in causing GERD. There was no increase in reflux symptoms in almost 3000 patients with H. pylori infection who were randomly assigned eradication therapy compared with placebo.160,161
Contraindications for H. pylori eradication are documented hypersensitivity to the eradication drugs, especially penicillin-allergy. However, alternative regimens such as bismuth/tetracycline/metronidazole combinations can be used and exhibit high cure rates.
Future prospects
Interaction between gastric stem cells and H. pylori
Cells of the gastric mucosa undergo constant renewal, the rate depending on the health of the tissue. The current evidence162 suggests that a single stem cell in every gastric gland indirectly gives rise to a clone of differentiated cells, by production of committed progenitor cells. It is most likely that this stem cell occupies a niche in the isthmus composed of mesenchymal cells and extracellular matrices. This environment regulates the function of the epithelial stem cell via mesenchymal-epithelial cross talk. Recent studies in gnotobiotic mouse models of chronic atrophic gastritis163 have shown that parietal cell loss results in amplification of multi- and oligopotential gastric stem cells, which express sialylated glycan receptors recognized by H. pylori adhesions, SabA,164 HpaA,165 and HP0721.166 DNA microarray studies indicate that 6%–7% of H. pylori genes are differentially expressed in response to a shift in pH from 7 to 5.167 A large group of genes downregulated at pH 5, in particular, encode outer membrane proteins, including Hop family members that are predicted to be outer membrane porins and adhesins. Loss of acid-producing parietal cells increases bacterial adhesin expression and increases the proportion of stem cells with adhesin receptors.168 Another very provocative study suggests that in the mouse model of gastric cancer evoked with Helicobacter felis (cat Helicobacter), infection arises because malignant stem cells emerge from a pool of bone marrow cells that are recruited into areas of chronic gastritis.169
Other diseases of interest possibly influenced by H. pylori170
Idiopathic thrombocytopenic purpura (ITP) is an autoimmune disease that leads to thrombocytopenia as a result of increased thrombocytolysis due to the appearance of autoantibodies to platelets. The acute type frequently occurs in children, often preceded by a viral infection. It develops rapidly and shows spontaneous remission. Chronic-type ITP occurs most commonly in adults and reportedly correlates with H. pylori infection. In 1988, Gasbarrini et al.7 reported that H. pylori eradication therapy to eight patients with H. pylori-positive ITP improved thrombocytosis after the therapy. Veneri et al.,171 who administered H. pylori eradication therapy to 43 patients with H. pylori-positive ITP, reported the therapy to be successful in 41 of the 43 and recognized continuous improvement in 20 (48.8%) of these 41 patients on long-term observation. According to another report by the same authors,172 the frequencies of HLA-DRB 111, HLA-DRB 114, and HLA-DQB 103 as host factors were high in H. pylori-positive ITP, while the frequency of HLA-DRB 103 as a host factor was higher in the H. pylori-negative ITP group than in the H. pylori-positive ITP group, and HLA-DQB 103 showed favorable platelet reactivity after eradication therapy in H. pylori-positive ITP.172 Inaba et al.173 administered H. pylori eradication therapy to 35 patients with chronic ITP (25 H. pylori-positive ITP patients and 10 H. pylori-negative ITP patients), and succeeded in eradicating the bacteria in all of the patients with H. pylori-positive ITP. They reported that the platelet count was improved in 11 (44%) of the 25 patients, while the platelet count was unchanged in the 10 H. pylori-negative ITP patients. We also administered bacterial eradication therapy to 37 patients with chronic ITP (26 H. pylori-positive ITP and 11 H. pylori-negative ITP patients) and succeeded in eradicating the bacteria in all of the H. pylori-positive ITP patients; we found the platelet count to be improved in 16 (62%) of the 26 patients with H. pylori-positive ITP, while the platelet count was unchanged in the 11 patients with H. pylori-negative ITP.12 In other words, there was no improvement with bacterial eradication in H. pylori-negative ITP, and the effects of the antibacterial agent itself in terms of increasing the platelet count was negligible.
Many other reports show a relation between H. pylori infection and human diseases such as iron-deficiency anemia8,174–181 and chronic urticaria.182–184
Final Message
While substantial progress has been made in our understanding of the role of genetic and environmental factors in the etiology of H. pylori-associated disease, there is still much to be learned about the epidemiology and pathogenesis of H. pylori. Although much knowledge of the potential association of H. pylori infection with human diseases has been accumulated, much of it is rather anecdotal or uncontrolled, and studies have not been consistently replicated in other centers. It is certainly true that further evidence-based studies should be performed to confirm H. pylori associations and the beneficial effects of eradication. On the other hand, eradication of H. pylori is rather simple these days and may be of no consequence in the treatment of patients with severe chronic diseases. In countries such as Japan where dreaded diseases such as gastric cancer seem to stem from H. pylori, the twenty-first century may be the time to totally eradicate this troublesome organism from the entire population.
References
- 1.Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345:784–9. doi: 10.1056/NEJMoa001999. [DOI] [PubMed] [Google Scholar]
- 2.Correa P. Bacterial infections as a cause of cancer. J Natl Cancer Inst. 2003;95:E3. doi: 10.1093/jnci/95.7.e3. [DOI] [PubMed] [Google Scholar]
- 3.Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest. 2004;113:321–33. doi: 10.1172/JCI20925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parsonnet J, Forman D. Helicobacter pylori infection and gastric cancer—for want of more outcomes. JAMA. 2004;291:244–5. doi: 10.1001/jama.291.2.244. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki H, Miura S, Suzuki M, Terada S, Nakamura M, Tsuchiya M. Gastric mucosal injury: microcirculation and Helicobacter pylori. Keio J Med. 1994;43:1–8. doi: 10.2302/kjm.43.1. [DOI] [PubMed] [Google Scholar]
- 6.Suzuki H, Masaoka T, Nomura S, Hoshino Y, Kurabayashi K, Minegishi Y, et al. Current consensus on the diagnosis and treatment of H. pylori-associated gastroduodenal disease. Keio J Med. 2003;52:163–73. doi: 10.2302/kjm.52.163. [DOI] [PubMed] [Google Scholar]
- 7.Gasbarrini A, Franceschi F, Tartaglione R, Landolfi R, Pola P, Gasbarrini G. Regression of autoimmune thrombocytopenia after eradication of Helicobacter pylori. Lancet. 1998;352:878. doi: 10.1016/S0140-6736(05)60004-9. [DOI] [PubMed] [Google Scholar]
- 8.Barabino A, Dufour C, Marino CE, Claudiani F, Alessandri A. Unexplained refractory iron-deficiency anemia associated with Helicobacter pylori gastric infection in children: further clinical evidence. J Pediatr Gastroenterol Nutr. 1999;28:116–9. doi: 10.1097/00005176-199901000-00027. [DOI] [PubMed] [Google Scholar]
- 9.Kountouras J, Deretzi G, Zavos C, Karatzoglou P, Touloumis L, Nicolaides T, et al. Association between Helicobacter pylori infection and acute inflammatory demyelinating polyradiculoneuropathy. Eur J Neurol. 2005;12:139–43. doi: 10.1111/j.1468-1331.2004.00977.x. [DOI] [PubMed] [Google Scholar]
- 10.Pasceri V, Patti G, Cammarota G, Pristipino C, Richichi G, Di Sciascio G. Virulent strains of Helicobacter pylori and vascular diseases: a meta-analysis. Am Heart J. 2006;151:1215–22. doi: 10.1016/j.ahj.2005.06.041. [DOI] [PubMed] [Google Scholar]
- 11.Kountouras J, Tsolaki M, Gavalas E, Boziki M, Zavos C, Karatzoglou P, et al. Relationship between Helicobacter pylori infection and Alzheimer disease. Neurology. 2006;66:938–40. doi: 10.1212/01.wnl.0000203644.68059.5f. [DOI] [PubMed] [Google Scholar]
- 12.Asahi A, Kuwana M, Suzuki H, Hibi T, Kawakami Y, Ikeda Y. Effects of a Helicobacter pylori eradication regimen on anti-platelet autoantibody response in infected and uninfected patients with idiopathic thrombocytopenic purpura. Haematologica. 2006;91:1436–7. [PubMed] [Google Scholar]
- 13.Marshall BJ, Armstrong JA, McGechie DB, Glancy RJ. Attempt to fulfil Koch’s postulates for pyloric Campylobacter. Med J Aust. 1985;142:436–9. doi: 10.5694/j.1326-5377.1985.tb113443.x. [DOI] [PubMed] [Google Scholar]
- 14.Kavermann H, Burns BP, Angermuller K, Odenbreit S, Fischer W, Melchers K, et al. Identification and characterization of Helicobacter pylori genes essential for gastric colonization. J Exp Med. 2003;197:813–22. doi: 10.1084/jem.20021531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo BP, Mekalanos JJ. Rapid genetic analysis of Helicobacter pylori gastric mucosal colonization in suckling mice. Proc Natl Acad Sci U S A. 2002;99:8354–9. doi: 10.1073/pnas.122244899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med. 2002;347:1175–86. doi: 10.1056/NEJMra020542. [DOI] [PubMed] [Google Scholar]
- 17.Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1:1311–5. doi: 10.1016/s0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]
- 18.Palmer ED. Investigation of the gastric mucosal spirochetes of the human. Gastroenterology. 1954;27:218–20. [PubMed] [Google Scholar]
- 19.Bizzozero G. Ueber die schlauchformigen Dursen des Magen-darmkanals und die Beziehungen ihres Epithels zu dem Oberflachenepithel der Schleimhaut. Arch Mikr Anat. 1893;42:82–152. [Google Scholar]
- 20.Kasai K, Kobayashi R. The stomach spirochete occurring in mammals. J Parasitol. 1919;6:1–10. [Google Scholar]
- 21.Saito K. Introductory remark of Dr. Rokuzo Kobayashi’s achievements. Keio J Med. 2002;51:2–3. [PubMed] [Google Scholar]
- 22.Marshall BJ. Gastric spirochaetes: 100 years of discovery before and after Kobayashi. Keio J Med. 2002;51:33–7. doi: 10.2302/kjm.51.supplement2_33. [DOI] [PubMed] [Google Scholar]
- 23.Graham DY, Malaty HM, Evans DG, Evans DJ, Jr, Klein PD, Adam E. Epidemiology of Helicobacter pylori in an asymptomatic population in the United States. Effect of age, race, and socioeconomic status. Gastroenterology. 1991;100:1495–501. doi: 10.1016/0016-5085(91)90644-z. [DOI] [PubMed] [Google Scholar]
- 24.Parsonnet J. The incidence of Helicobacter pylori infection. Aliment Pharmacol Ther. 1995;9(Suppl 2):45–51. [PubMed] [Google Scholar]
- 25.Frenck RW, Jr, Clemens J. Helicobacter in the developing world. Microbes Infect. 2003;5:705–13. doi: 10.1016/s1286-4579(03)00112-6. [DOI] [PubMed] [Google Scholar]
- 26.Hansson LE, Nyren O, Hsing AW, Bergstrom R, Josefsson S, Chow WH, et al. The risk of stomach cancer in patients with gastric or duodenal ulcer disease. N Engl J Med. 1996;335:242–9. doi: 10.1056/NEJM199607253350404. [DOI] [PubMed] [Google Scholar]
- 27.Zucca E, Bertoni F, Roggero E, Bosshard G, Cazzaniga G, Pedrinis E, et al. Molecular analysis of the progression from Helicobacter pylori-associated chronic gastritis to mucosa-associated lymphoid-tissue lymphoma of the stomach. N Engl J Med. 1998;338:804–10. doi: 10.1056/NEJM199803193381205. [DOI] [PubMed] [Google Scholar]
- 28.Fischbach W, Dragosics B, Kolve-Goebeler ME, Ohmann C, Greiner A, Yang Q, et al. Primary gastric B-cell lymphoma: results of a prospective multicenter study. The German-Austrian Gastrointestinal Lymphoma Study Group. Gastroenterology. 2000;119:1191–202. doi: 10.1053/gast.2000.19579. [DOI] [PubMed] [Google Scholar]
- 29.Ekstrom AM, Held M, Hansson LE, Engstrand L, Nyren O. Helicobacter pylori in gastric cancer established by CagA immunoblot as a marker of past infection. Gastroenterology. 2001;121:784–91. doi: 10.1053/gast.2001.27999. [DOI] [PubMed] [Google Scholar]
- 30.Naumann M, Crabtree JE. Helicobacter pylori-induced epithelial cell signalling in gastric carcinogenesis. Trends Microbiol. 2004;12:29–36. doi: 10.1016/j.tim.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki H, Mori M, Sakaguchi AA, Suzuki M, Miura S, Ishii H. Enhanced levels of C-X-C chemokine, human GROα, in H. pylori-associated gastric disease. J Gastroenterol Hepatol. 1998;13:516–20. doi: 10.1111/j.1440-1746.1998.tb00679.x. [DOI] [PubMed] [Google Scholar]
- 32.Karita M, Tummuru MK, Wirth HP, Blaser MJ. Effect of growth phase and acid shock on Helicobacter pylori cagA expression. Infect Immun. 1996;64:4501–7. doi: 10.1128/iai.64.11.4501-4507.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crabtree JE, Wyatt JI, Sobala GM, Miller G, Tompkins DS, Primrose JN, et al. Systemic and mucosal humoral responses to Helicobacter pylori in gastric cancer. Gut. 1993;34:1339–43. doi: 10.1136/gut.34.10.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basso D, Navaglia F, Brigato L, Piva MG, Toma A, Greco E, et al. Analysis of Helicobacter pylori vacA and cagA genotypes and serum antibody profile in benign and malignant gastroduodenal diseases. Gut. 1998;43:182–6. doi: 10.1136/gut.43.2.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Atherton JC, Cao P, Peek RM, Jr, Tummuru MK, Blaser MJ, Cover TL. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem. 1995;270:17771–7. doi: 10.1074/jbc.270.30.17771. [DOI] [PubMed] [Google Scholar]
- 36.Fischer W, Puls J, Buhrdorf R, Gebert B, Odenbreit S, Haas R. Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol Microbiol. 2001;42:1337–48. doi: 10.1046/j.1365-2958.2001.02714.x. [DOI] [PubMed] [Google Scholar]
- 37.Stein M, Rappuoli R, Covacci A. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc Natl Acad Sci U S A. 2000;97:1263–8. doi: 10.1073/pnas.97.3.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, et al. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science. 2002;295:683–6. doi: 10.1126/science.1067147. [DOI] [PubMed] [Google Scholar]
- 39.Selbach M, Moese S, Hauck CR, Meyer TF, Backert S. Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo. J Biol Chem. 2002;277:6775–8. doi: 10.1074/jbc.C100754200. [DOI] [PubMed] [Google Scholar]
- 40.Tanaka J, Suzuki T, Mimuro H, Sasakawa C. Structural definition on the surface of Helicobacter pylori type IV secretion apparatus. Cell Microbiol. 2003;5:395–404. doi: 10.1046/j.1462-5822.2003.00286.x. [DOI] [PubMed] [Google Scholar]
- 41.Tsutsumi R, Higashi H, Higuchi M, Okada M, Hatakeyama M. Attenuation of Helicobacter pylori CagA × SHP-2 signaling by interaction between CagA and C-terminal Src kinase. J Biol Chem. 2003;278:3664–70. doi: 10.1074/jbc.M208155200. [DOI] [PubMed] [Google Scholar]
- 42.Higashi H, Tsutsumi R, Fujita A, Yamazaki S, Asaka M, Azuma T, et al. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci U S A. 2002;99:14428–33. doi: 10.1073/pnas.222375399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Higashi H, Nakaya A, Tsutsumi R, Yokoyama K, Fujii Y, Ishikawa S, et al. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J Biol Chem. 2004;279:17205–16. doi: 10.1074/jbc.M309964200. [DOI] [PubMed] [Google Scholar]
- 44.Mai UEH, Prez-Perez GI, Allen JB, Wahl SM, Blaser MJ, Smith PD. Surface proteins from Helicobacter pylori exhibit chemotactic activity for human leukocytes and are present in gastric mucosa. J Exp Med. 1992;175:517–25. doi: 10.1084/jem.175.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Craig PM, Territo MC, Karnes WE, Walsh JH. Helicobacter pylori secretes a chemotactic factor monocytes and neutrophils. Gut. 1992;33:1020–3. doi: 10.1136/gut.33.8.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suzuki M, Miura S, Suematsu M, Fukumura D, Kurose I, Suzuki H, et al. Helicobacter pylori-associated ammonia production enhances neutrophil-dependent gastric mucosal cell injury. Am J Physiol. 1992;263:G719–25. doi: 10.1152/ajpgi.1992.263.5.G719. [DOI] [PubMed] [Google Scholar]
- 47.Grisham MB, Jefferson MM, Melton DF, Thomas EL. Chlorination of endogenous amines by isolated neutrophils: ammonia-dependent bactericidal, cytotoxic, and cytolytic activities of the chloramines. J Biol Chem. 1984;259:10404–13. [PubMed] [Google Scholar]
- 48.Halliwell B, Aruoma OI. DNA damage by oxygen-derived species. Its mechanism and measurement in mammalian systems. FEBS Lett. 1991;281:9–19. doi: 10.1016/0014-5793(91)80347-6. [DOI] [PubMed] [Google Scholar]
- 49.Farinati F, Cardin R, Russo VM, Busatto G, Franco M, Rugge M. Helicobacter pylori CagA status, mucosal oxidative damage and gastritis phenotype: a potential pathway to cancer? Helicobacter. 2003;8:227–34. doi: 10.1046/j.1523-5378.2003.00149.x. [DOI] [PubMed] [Google Scholar]
- 50.Suzuki H, Mori M, Seto K, Kai A, Kawaguchi C, Suzuki M, et al. H. pylori-associated gastric pro- and anti-oxidant formation in Mongolian gerbils. Free Radic Biol Med. 1999;26:679–84. doi: 10.1016/s0891-5849(98)00248-2. [DOI] [PubMed] [Google Scholar]
- 51.Suzuki H, Mori M, Seto K, Nagahashi S, Kawaguchi C, Kai A, et al. Ethanol intake preceding Helicobacter pylori inoculation promotes gastric mucosal inflammation in Mongolian gerbils. J Gastroenterol Hepatol. 1999;14:1062–9. doi: 10.1046/j.1440-1746.1999.01999.x. [DOI] [PubMed] [Google Scholar]
- 52.Suzuki H, Masaoka T, Hosoda H, Ota T, Minegishi Y, Nomura S, et al. Helicobacter pylori infection modifies gastric and plasma ghrelin dynamics in Mongolian gerbils. Gut. 2004;53:187–94. doi: 10.1136/gut.2003.021568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suzuki H, Minegishi Y, Nomoto Y, Ota T, Masaoka T, Brink GR, et al. Down-regulation of a morphogen (sonic hedgehog) gradient in the gastric epithelium of Helicobacter pylori-infected Mongolian gerbils. J Pathol. 2005;206:186–97. doi: 10.1002/path.1763. [DOI] [PubMed] [Google Scholar]
- 54.Ogura K, Maeda S, Nakao M, Watanabe T, Tada M, Kyutoku T, et al. Virulence factors of Helicobacter pylori responsible for gastric diseases in Mongolian gerbil. J Exp Med. 2000;192:1601–10. doi: 10.1084/jem.192.11.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Israel DA, Salama N, Arnold CN, Moss SF, Ando T, Wirth HP, et al. Helicobacter pylori strain-specific differences in genetic content, identified by microarray, influence host inflammatory responses. J Clin Invest. 2001;107:611–20. doi: 10.1172/JCI11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suzuki H, Masaoka T, Hosoda H, Nomura S, Ohara T, Kangawa K, et al. Plasma ghrelin concentration correlates with the levels of serum pepsinogen I and pepsinogen I/II ratio—a possible novel and non-invasive marker for gastric atrophy. Hepatogastroenterology. 2004;51:1249–54. [PubMed] [Google Scholar]
- 57.Nishizawa T, Suzuki H, Masaoka T, Minegishi Y, Iwasaki E, Hibi T. Helicobacter pylori eradication restored sonic hedgehog expression in the human stomach. Hepatogastroenterology 2007 (in press) [PubMed]
- 58.Peek RMJ, Moss SF, Tham KT, Perez-Perez GI, Wang S, Miller GG, et al. Helicobacter pylori CagA+ strains and dissociation of gastric epithelial cell proliferation from apoptosis. J Natl Cancer Inst. 1997;89:863–8. doi: 10.1093/jnci/89.12.863. [DOI] [PubMed] [Google Scholar]
- 59.Suzuki H, Miyazawa M, Nagahashi S, Seto K, Kai A, Suzuki M, et al. Attenuated apoptosis in H. pylori-colonized gastric mucosa of Mongolian gerbils in comparison with mice. Dig Dis Sci. 2002;47:90–9. doi: 10.1023/a:1013219621422. [DOI] [PubMed] [Google Scholar]
- 60.Watanabe T, Tada M, Nagai H, Sasaki S, Nakao M. Helicobacter pylori infection induces gastric cancer in Mongolian gerbils. Gastroenterology. 1998;115:642–8. doi: 10.1016/s0016-5085(98)70143-x. [DOI] [PubMed] [Google Scholar]
- 61.Honda S, Fujioka T, Tokieda M, Satoh R, Nishizono A, Nasu M. Development of Helicobacter pylori-induced gastric carcinoma in Mongolian gerbils. Cancer Res. 1998;58:4255–9. [PubMed] [Google Scholar]
- 62.Marshall BJ, McGechie DB, Rogers PA, Glancy RJ. Pyloric Campylobacter infection and gastroduodenal disease. Med J Aust. 1985;142:439–44. doi: 10.5694/j.1326-5377.1985.tb113444.x. [DOI] [PubMed] [Google Scholar]
- 63.Nomura A, Stemmermann GN, Chyou PH, Perez-Perez GI, Blaser MJ. Helicobacter pylori infection and the risk for duodenal and gastric ulceration. Ann Intern Med. 1994;120:977–81. doi: 10.7326/0003-4819-120-12-199406150-00001. [DOI] [PubMed] [Google Scholar]
- 64.Rauws EA, Tytgat GN. Cure of duodenal ulcer associated with eradication of Helicobacter pylori. Lancet. 1990;335:1233–5. doi: 10.1016/0140-6736(90)91301-p. [DOI] [PubMed] [Google Scholar]
- 65.Hentschel E, Brandstatter G, Dragosics B, Hirschl AM, Nemec H, Schutze K, et al. Effect of ranitidine and amoxicillin plus metronidazole on the eradication of Helicobacter pylori and the recurrence of duodenal ulcer. N Engl J Med. 1993;328:308–12. doi: 10.1056/NEJM199302043280503. [DOI] [PubMed] [Google Scholar]
- 66.Marshall BJ, Goodwin CS, Warren JR, Murray R, Blincow ED, Blackbourn SJ, et al. Prospective double-blind trial of duodenal ulcer relapse after eradication of Campylobacter pylori. Lancet. 1988;2:1437–42. doi: 10.1016/s0140-6736(88)90929-4. [DOI] [PubMed] [Google Scholar]
- 67.Penston JG. Review article: Helicobacter pylori eradication—understandable caution but no excuse for inertia. Aliment Pharmacol Ther. 1994;8:369–89. doi: 10.1111/j.1365-2036.1994.tb00304.x. [DOI] [PubMed] [Google Scholar]
- 68.Tytgat GN. Peptic ulcer and Helicobacter pylori: eradication and relapse. Scand J Gastroenterol Suppl. 1995;210:70–2. doi: 10.3109/00365529509090275. [DOI] [PubMed] [Google Scholar]
- 69.Hopkins RJ, Girardi LS, Turney EA. Relationship between Helicobacter pylori eradication and reduced duodenal and gastric ulcer recurrence: a review. Gastroenterology. 1996;110:1244–52. doi: 10.1053/gast.1996.v110.pm8613015. [DOI] [PubMed] [Google Scholar]
- 70.Laine L, Hopkins RJ, Girardi LS. Has the impact of Helicobacter pylori therapy on ulcer recurrence in the United States been overstated? A meta-analysis of rigorously designed trials. Am J Gastroenterol. 1998;93:1409–15. doi: 10.1111/j.1572-0241.1998.452_a.x. [DOI] [PubMed] [Google Scholar]
- 71.Axon AT, O’Morain CA, Bardhan KD, Crowe JP, Beattie AD, Thompson RP, et al. Randomised double blind controlled study of recurrence of gastric ulcer after treatment for eradication of Helicobacter pylori infection. BMJ. 1997;314:565–8. doi: 10.1136/bmj.314.7080.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fukuda Y, Yamamoto I, Okui M, Tonokatsu Y, Tamura K, Shimoyama T. Combination therapy with mucosal protective agent for the eradication of Helicobacter pylori. Eur J Gastroenterol Hepatol. 1995;7(Suppl 1):S45–7. [PubMed] [Google Scholar]
- 73.Kato M, Asaka M, Kudo M, Sukegawa M, Katagiri M, Koshiyama T, et al. Effects of lansoprazole plus amoxycillin on the cure of Helicobacter pylori infection in Japanese peptic ulcer patients. Aliment Pharmacol Ther. 1996;10:821–7. doi: 10.1046/j.1365-2036.1996.52194000.x. [DOI] [PubMed] [Google Scholar]
- 74.Lazzaroni M, Perego M, Bargiggia S, Maconi G, Fiocca R, Solcia E, et al. Helicobacter pylori eradication in the healing and recurrence of benign gastric ulcer: a two-year, double-blind, placebo controlled study. Ital J Gastroenterol Hepatol. 1997;29:220–7. [PubMed] [Google Scholar]
- 75.Seppala K, Pikkarainen P, Sipponen P, Kivilaakso E, Gormsen MH. Cure of peptic gastric ulcer associated with eradication of Helicobacter pylori. Finnish Gastric Ulcer Study Group. Gut. 1995;36:834–7. doi: 10.1136/gut.36.6.834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sung JJ, Chung SC, Ling TK, Yung MY, Leung VK, Ng EK, et al. Antibacterial treatment of gastric ulcers associated with Helicobacter pylori. N Engl J Med. 1995;332:139–42. doi: 10.1056/NEJM199501193320302. [DOI] [PubMed] [Google Scholar]
- 77.Ford AC, Delaney BC, Forman D, Moayyedi P. Eradication therapy for peptic ulcer disease in Helicobacter pylori positive patients. Cochrane Database Syst Rev 2006 Apr 19;(2):CD003840 [DOI] [PubMed]
- 78.Kuipers EJ, Pena AS, van Kamp G, Uyterlinde AM, Pals G, Pels NF, et al. Seroconversion for Helicobacter pylori. Lancet. 1993;342:328–31. doi: 10.1016/0140-6736(93)91473-y. [DOI] [PubMed] [Google Scholar]
- 79.Roosendaal R, Kuipers EJ, Buitenwerf J, Uffelen C, Meuwissen SG, Kamp GJ, et al. Helicobacter pylori and the birth cohort effect: evidence of a continuous decrease of infection rates in childhood. Am J Gastroenterol. 1997;92:1480–2. [PubMed] [Google Scholar]
- 80.Huang JQ, Sridhar S, Hunt RH. Role of Helicobacter pylori infection and non-steroidal anti-inflammatory drugs in peptic-ulcer disease: a meta-analysis. Lancet. 2002;359:14–22. doi: 10.1016/S0140-6736(02)07273-2. [DOI] [PubMed] [Google Scholar]
- 81.Chan FK, To KF, Wu JC, Yung MY, Leung WK, Kwok T, et al. Eradication of Helicobacter pylori and risk of peptic ulcers in patients starting long-term treatment with non-steroidal anti-inflammatory drugs: a randomised trial. Lancet. 2002;359:9–13. doi: 10.1016/s0140-6736(02)07272-0. [DOI] [PubMed] [Google Scholar]
- 82.Chan FK, Chung SC, Suen BY, Lee YT, Leung WK, Leung VK, et al. Preventing recurrent upper gastrointestinal bleeding in patients with Helicobacter pylori infection who are taking low-dose aspirin or naproxen. N Engl J Med. 2001;344:967–73. doi: 10.1056/NEJM200103293441304. [DOI] [PubMed] [Google Scholar]
- 83.Hawkey CJ, Tulassay Z, Szczepanski L, Rensburg CJ, Filipowicz-Sosnowska A, Lanas A, et al. Randomised controlled trial of Helicobacter pylori eradication in patients on non-steroidal anti-inflammatory drugs: HELP NSAIDs study. Helicobacter Eradication for Lesion Prevention. Lancet. 1998;352:1016–21. doi: 10.1016/s0140-6736(98)04206-8. [DOI] [PubMed] [Google Scholar]
- 84.Drossman DA. The functional gastrointestinal disorders and the Rome III process. Gastroenterology. 2006;130:1377–90. doi: 10.1053/j.gastro.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 85.Tack J, Talley NJ, Camilleri M, Holtmann G, Hu P, Malagelada JR, et al. Functional gastroduodenal disorders. Gastroen terology. 2006;130:1466–79. doi: 10.1053/j.gastro.2005.11.059. [DOI] [PubMed] [Google Scholar]
- 86.Suzuki H, Nishizawa T, Hibi T. Therapeutic strategies for functional dyspepsia and the introduction of the Rome III classification. J Gastroenterol. 2006;41:513–23. doi: 10.1007/s00535-006-1847-5. [DOI] [PubMed] [Google Scholar]
- 87.Talley NJ, Hunt RH. What role does Helicobacter pylori play in dyspepsia and nonulcer dyspepsia? Arguments for and against H. pylori being associated with dyspeptic symptoms. Gastroenterology. 1997;113:S67–77. doi: 10.1016/s0016-5085(97)80016-9. [DOI] [PubMed] [Google Scholar]
- 88.Moayyedi P, Soo S, Deeks J, Delaney B, Harris A, Innes M et al. Eradication of Helicobacter pylori for non-ulcer dyspepsia. Cochrane Database Syst Rev 2005 Jan 25;(1):CD002096 [DOI] [PubMed]
- 89.Chiba N, Zanten SJ, Sinclair P, Ferguson RA, Escobedo S, Grace E. Treating Helicobacter pylori infection in primary care patients with uninvestigated dyspepsia: the Canadian adult dyspepsia empiric treatment-Helicobacter pylori positive (CADET-Hp) randomised controlled trial. BMJ. 2002;324:1012–6. doi: 10.1136/bmj.324.7344.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chiba N, Veldhuyzen Van Zanten SJ, Escobedo S, Grace E, Lee J, Sinclair P, et al. Economic evaluation of Helicobacter pylori eradication in the CADET-Hp randomized controlled trial of H. pylori-positive primary care patients with uninvestigated dyspepsia. Aliment Pharmacol Ther. 2004;19:349–58. doi: 10.1111/j.1365-2036.2004.01865.x. [DOI] [PubMed] [Google Scholar]
- 91.Sonnenberg A, Townsend WF. Costs of duodenal ulcer therapy with antibiotics. Arch Intern Med. 1995;155:922–8. [PubMed] [Google Scholar]
- 92.Delaney B, Ford AC, Forman D, Moayyedi P, Qume M. Initial management strategies for dyspepsia. Cochrane Database Syst Rev 2005 Oct 19;(4):CD001961 [DOI] [PubMed]
- 93.Ford AC, Qume M, Moayyedi P, Arents NL, Lassen AT, Logan RF, et al. Helicobacter pylori “test and treat” or endoscopy for managing dyspepsia: an individual patient data meta-analysis. Gastroenterology. 2005;128:1838–44. doi: 10.1053/j.gastro.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 94.Correa P, Cuello C, Duque E. Gastric cancer in Colombia. III. Natural history of precursor lesions. J Natl Cancer Inst. 1976;57:1027–35. doi: 10.1093/jnci/57.5.1027. [DOI] [PubMed] [Google Scholar]
- 95.Kuipers EJ, Uyterlinde AM, Pena AS, Roosendaal R, Pals G, Nelis GF, et al. Long-term sequelae of Helicobacter pylori gastritis. Lancet. 1995;345:1525–8. doi: 10.1016/s0140-6736(95)91084-0. [DOI] [PubMed] [Google Scholar]
- 96.Suzuki H, Hibi T. Oxidative stress in Helicobacter pylori-associated gastroduodenal disease. J Clin Biochem Nutr. 2006;39:56–63. [Google Scholar]
- 97.El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398–402. doi: 10.1038/35006081. [DOI] [PubMed] [Google Scholar]
- 98.Uehara A, Okumura T, Sekiya C, Okamura K, Takasugi Y, Namiki M. Interleukin-1 inhibits the secretion of gastric acid in rats: possible involvement of prostaglandin. Biochem Biophys Res Commun. 1989;162:1578–84. doi: 10.1016/0006-291x(89)90855-3. [DOI] [PubMed] [Google Scholar]
- 99.Hwang IR, Kodama T, Kikuchi S, Sakai K, Peterson LE, Graham DY, et al. Effect of interleukin 1 polymorphisms on gastric mucosal interleukin 1beta production in Helicobacter pylori infection. Gastroenterology. 2002;123:1793–803. doi: 10.1053/gast.2002.37043. [DOI] [PubMed] [Google Scholar]
- 100.Figueiredo C, Machado JC, Pharoah P, Seruca R, Sousa S, Carvalho R, et al. Helicobacter pylori and interleukin 1 genotyping: an opportunity to identify high-risk individuals for gastric carcinoma. J Natl Cancer Inst. 2002;94:1680–7. doi: 10.1093/jnci/94.22.1680. [DOI] [PubMed] [Google Scholar]
- 101.Rad R, Prinz C, Neu B, Neuhofer M, Zeitner M, Voland P, et al. Synergistic effect of Helicobacter pylori virulence factors and interleukin-1 polymorphisms for the development of severe histological changes in the gastric mucosa. J Infect Dis. 2003;188:272–81. doi: 10.1086/376458. [DOI] [PubMed] [Google Scholar]
- 102.Machado JC, Figueiredo C, Canedo P, Pharoah P, Carvalho R, Nabais S, et al. A proinflammatory genetic profile increases the risk for chronic atrophic gastritis and gastric carcinoma. Gastroenterology. 2003;125:364–71. doi: 10.1016/s0016-5085(03)00899-0. [DOI] [PubMed] [Google Scholar]
- 103.Sipponen P, Kekki M, Haapakoski J, Ihamaki T, Siurala M. Gastric cancer risk in chronic atrophic gastritis: statistical calculations of cross-sectional data. Int J Cancer. 2985;35:173–7. doi: 10.1002/ijc.2910350206. [DOI] [PubMed] [Google Scholar]
- 104.Kuipers EJ. Review article: relationship between Helicobacter pylori, atrophic gastritis and gastric cancer. Aliment Pharmacol Ther. 1998;12(Suppl 1):25–36. doi: 10.1111/j.1365-2036.1998.00009.x. [DOI] [PubMed] [Google Scholar]
- 105.Forman D, Sitas F, Newell DG, Stacey AR, Boreham J, Peto R, et al. Geographic association of Helicobacter pylori antibody prevalence and gastric cancer mortality in rural China. Int J Cancer. 1990;46:608–11. doi: 10.1002/ijc.2910460410. [DOI] [PubMed] [Google Scholar]
- 106.The EUROGAST Study Group An international association between Helicobacter pylori infection and gastric cancer. Lancet. 1993;341:1359–62. [PubMed] [Google Scholar]
- 107.IARC Working Group on the Evaluation of Carcinogenic Risks to Humans Schistosomes, liver flukes and Helicobacter pylori. IARC Monogr Eval Carcinog Risks Hum. 1994;61:177–240. [PMC free article] [PubMed] [Google Scholar]
- 108.Ekstrom A, Held M, Hansson L, Engstrand L, Nyren O. Helicobacter pylori in gastric cancer established by CagA immunoblot as a marker of past infection. Gastroenterology. 2001;121:784–91. doi: 10.1053/gast.2001.27999. [DOI] [PubMed] [Google Scholar]
- 109.Rieder G, Merchant JL, Haas R. Helicobacter pylori cag-type IV secretion system facilitates corpus colonization to induce precancerous conditions in Mongolian gerbils. Gastroenterology. 2005;128:1229–42. doi: 10.1053/j.gastro.2005.02.064. [DOI] [PubMed] [Google Scholar]
- 110.Kuipers EJ, Perez-Perez GI, Meuwissen SG, Blaser MJ. Helicobacter pylori and atrophic gastritis: importance of the cagA status. J Natl Cancer Inst. 1995;87:1777–80. doi: 10.1093/jnci/87.23.1777. [DOI] [PubMed] [Google Scholar]
- 111.Parsonnet J, Friedman GD, Orentreich N, Vogelman H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut. 1997;40:297–301. doi: 10.1136/gut.40.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Parkin DM. Global cancer statistics in the year 2000. Lancet Oncol. 2001;2:533–43. doi: 10.1016/S1470-2045(01)00486-7. [DOI] [PubMed] [Google Scholar]
- 113.Kuipers EJ, Nelis GF, Klinkenberg-Knol EC, Snel P, Goldfain D, Kolkman JJ, et al. Cure of Helicobacter pylori infection in patients with reflux oesophagitis treated with long term omeprazole reverses gastritis without exacerbation of reflux disease: results of a randomised controlled trial. Gut. 2004;53:12–20. doi: 10.1136/gut.53.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Leung WK, Lin SR, Ching JY, To KF, Ng EK, Chan FK, et al. Factors predicting progression of gastric intestinal metaplasia: results of a randomised trial on Helicobacter pylori eradication. Gut. 2004;53:1244–9. doi: 10.1136/gut.2003.034629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ley C, Mohar A, Guarner J, Herrera-Goepfert R, Figueroa LS, Halperin D, et al. Helicobacter pylori eradication and gastric preneoplastic conditions: a randomized, double-blind, placebo-controlled trial. Cancer Epidemiol Biomarkers Prev. 2004;13:4–10. doi: 10.1158/1055-9965.epi-03-0124. [DOI] [PubMed] [Google Scholar]
- 116.Mera R, Fontham ET, Bravo LE, Bravo JC, Piazuelo MB, Camargo MC, et al. Long term follow up of patients treated for Helicobacter pylori infection. Gut. 2004;54:1536–40. doi: 10.1136/gut.2005.072009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schenk BE, Kuipers EJ, Nelis GF, Bloemena E, Thijs JC, Snel P, et al. Effect of Helicobacter pylori eradication on chronic gastritis during omeprazole therapy. Gut. 2000;46:615–21. doi: 10.1136/gut.46.5.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wong BC, Lam SK, Wong WM, Chen JS, Zheng TT, Feng RE, et al. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: a randomized controlled trial. JAMA. 2004;291:187–94. doi: 10.1001/jama.291.2.187. [DOI] [PubMed] [Google Scholar]
- 119.Take S, Mizuno M, Ishiki K, Nagahara Y, Yoshida T, Yokota K, et al. The effect of eradicating Helicobacter pylori on the development of gastric cancer in patients with peptic ulcer disease. Am J Gastroenterol. 2005;100:1037–42. doi: 10.1111/j.1572-0241.2005.41384.x. [DOI] [PubMed] [Google Scholar]
- 120.Yatsuya H, Toyoshima H, Tamakoshi A, Kikuchi S, Tamakoshi K, Kondo T, et al. Individual and joint impact of family history and Helicobacter pylori infection on the risk of stomach cancer: a nested case-control study. Br J Cancer. 2004;91:929–34. doi: 10.1038/sj.bjc.6602067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sasazuki S, Inoue M, Iwasaki M, Otani T, Yamamoto S, Ikeda S, et al. Effect of Helicobacter pylori infection combined with CagA and pepsinogen status on gastric cancer development among Japanese men and women: a nested case-control study. Cancer Epidemiol Biomarkers Prev. 2006;15:1341–7. doi: 10.1158/1055-9965.EPI-05-0901. [DOI] [PubMed] [Google Scholar]
- 122.Eidt S, Stolte M, Fischer R. Helicobacter pylori gastritis and primary gastric non-Hodgkin’s lymphomas. J Clin Pathol. 1994;47:436–9. doi: 10.1136/jcp.47.5.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Parsonnet J, Hansen S, Rodriguez L, Gelb AB, Warnke RA, Jellum E, et al. Helicobacter pylori infection and gastric lymphoma. N Engl J Med. 1994;330:1267–71. doi: 10.1056/NEJM199405053301803. [DOI] [PubMed] [Google Scholar]
- 124.Parsonnet J, Isaacson PG. Bacterial infection and MALT lymphoma. N Engl J Med. 2004;350:213–5. doi: 10.1056/NEJMp038200. [DOI] [PubMed] [Google Scholar]
- 125.Mascarel A, Ruskone-Fourmestraux A, Lavergne-Slove A, Megraud F, Dubus P, Merlio JP. Clinical, histological and molecular follow-up of 60 patients with gastric marginal zone lymphoma of mucosa-associated lymphoid tissue. Virchows Arch. 2005;446:219–24. doi: 10.1007/s00428-005-1217-3. [DOI] [PubMed] [Google Scholar]
- 126.Fischbach W, Goebeler-Kolve ME, Dragosics B, Greiner A, Stolte M. Long term outcome of patients with gastric marginal zone B cell lymphoma of mucosa associated lymphoid tissue (MALT) following exclusive Helicobacter pylori eradication therapy: experience from a large prospective series. Gut. 2004;53:34–7. doi: 10.1136/gut.53.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nakamura S, Matsumoto T, Suekane H, Matsumoto H, Esaki M, Yao T, et al. Long-term clinical outcome of Helicobacter pylori eradication for gastric mucosa-associated lymphoid tissue lymphoma with a reference to second-line treatment. Cancer. 2005;104:532–40. doi: 10.1002/cncr.21152. [DOI] [PubMed] [Google Scholar]
- 128.Wundisch T, Thiede C, Morgner A, Dempfle A, Gunther A, Liu H, et al. Long-term follow-up of gastric MALT lymphoma after Helicobacter pylori eradication. J Clin Oncol. 2005;23:8018–24. doi: 10.1200/JCO.2005.02.3903. [DOI] [PubMed] [Google Scholar]
- 129.Raderer M, Streubel B, Woehrer S, Puespoek A, Jaeger U, Formanek M, et al. High relapse rate in patients with MALT lymphoma warrants lifelong follow-up. Clin Cancer Res. 2005;11:3349–52. doi: 10.1158/1078-0432.CCR-04-2282. [DOI] [PubMed] [Google Scholar]
- 130.Inagaki H, Nakamura T, Li C, Sugiyama T, Asaka M, Kodaira J, et al. Gastric MALT lymphomas are divided into three groups based on responsiveness to Helicobacter Pylori eradication and detection of API2-MALT1 fusion. Am J Surg Pathol. 2004;28:1560–7. doi: 10.1097/00000478-200412000-00003. [DOI] [PubMed] [Google Scholar]
- 131.Liu H, Ye H, Ruskone-Fourmestraux A, Jong D, Pileri S, Thiede C, et al. T(11;18) is a marker for all stage gastric MALT lymphomas that will not respond to H. pylori eradication. Gastroenterology. 2002;122:1286–94. doi: 10.1053/gast.2002.33047. [DOI] [PubMed] [Google Scholar]
- 132.Werdmuller BF, Loffeld RJ. Helicobacter pylori infection has no role in the pathogenesis of reflux esophagitis. Dig Dis Sci. 1997;42:103–5. doi: 10.1023/a:1018841222856. [DOI] [PubMed] [Google Scholar]
- 133.Fallone CA, Barkun AN, Gottke MU, Best LM, Loo VG, Veldhuyzen van Zanten S, et al. Association of Helicobacter pylori genotype with gastroesophageal reflux disease and other upper gastrointestinal diseases. Am J Gastroenterol. 2000;95:659–69. doi: 10.1111/j.1572-0241.2000.01970.x. [DOI] [PubMed] [Google Scholar]
- 134.Labenz J, Blum AL, Bayerdorffer E. Curing Helicobacter pylori infection in patients with duodenal ulcer may provoke reflux esophagitis. Gastroenterology. 1997;112:1442–7. doi: 10.1016/s0016-5085(97)70024-6. [DOI] [PubMed] [Google Scholar]
- 135.Vakil N, Hahn B, McSorley D. Recurrent symptoms and gastro-oesophageal reflux disease in patients with duodenal ulcer treated for Helicobacter pylori infection. Aliment Pharmacol Ther. 2000;14:45–51. doi: 10.1046/j.1365-2036.2000.00677.x. [DOI] [PubMed] [Google Scholar]
- 136.Schwizer W, Thumshirn M, Dent J, Guldenschuh I, Menne D, Cathomas G, et al. Helicobacter pylori and symptomatic relapse of gastro-oesophageal reflux disease: a randomised controlled trial. Lancet. 2001;357:1738–42. doi: 10.1016/S0140-6736(00)04894-7. [DOI] [PubMed] [Google Scholar]
- 137.Suzuki H, Masaoka T, Nomura S, Hoshino Y, Kurabayashi K, Minegishi Y, et al. Current consensus on the diagnosis and treatment of H. pylori-associated gastroduodenal disease. Keio J Med. 2003;52:163–73. doi: 10.2302/kjm.52.163. [DOI] [PubMed] [Google Scholar]
- 138.Warren JR, Marshall BJ. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet. 1983;1(8336):1273–5. [PubMed] [Google Scholar]
- 139.Marshall BJ, Warren JR, Francis GJ, Langton SR, Goodwin CS, Chir B, et al. Rapid urease test in the management of Campylobacter pyloridis-associated gastritis. Am J Gastroenterol. 1987;82:200–10. [PubMed] [Google Scholar]
- 140.Klein PD, Malaty HM, Martin RF, Graham KS, Genta RM, Graham DY. Noninvasive detection of Helicobacter pylori infection in clinical practice: the 13C urea breath test. Am J Gastroenterol. 1996;91:690–4. [PubMed] [Google Scholar]
- 141.Wesjnen C, Hendriks H, Hoes A, Verweij W, Verheij T, Wit N. New immunoassay for the detection of Helicobacter pylori infection compared with urease test, 13C test and histology: validation in the primary care setting. J Microbiol Methods. 2001;46:235–40. doi: 10.1016/s0167-7012(01)00275-5. [DOI] [PubMed] [Google Scholar]
- 142.Narinesingh D, Ngo T. Immunochromatographic flow-injection method for detection of human serum immunoglobulin G antibodies to Helicobacter pylori. Ann Chim Acta. 2002;453:53–61. [Google Scholar]
- 143.Fujioka N, Fahey M, Hamada G, Nishimoto I, Kowalski L, Iriya K, et al. Serological Immunoglobulin G antibody titers to Helicobacter pylori in Japanese Brazilian and Non-Japanese Brazilian gastric cancer patients and controls in Sao Paulo. Jpn J Cancer Res. 2001;92:829–35. doi: 10.1111/j.1349-7006.2001.tb01168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wu D, Kuo C, Lu C, Su Y, Yu F, Lee Y, et al. Evaluation of an office-based urine test for detecting Helicobacter pylori: a prospective pilot study. Hepatogastroenterology. 2001;48:614–7. [PubMed] [Google Scholar]
- 145.Graham D, Reddy S. Rapid detection of anti-Helicobacter pylori IgG in urine using immunochromatography. Aliment Pharmacol Ther. 2001;15:699–702. doi: 10.1046/j.1365-2036.2001.00968.x. [DOI] [PubMed] [Google Scholar]
- 146.Manes G, Balzano A, Iaquinto G, Ricci C, Piccirillo M, Giardullo N, et al. Accuracy of stool antigen test in posteradication assessment of Helicobacter pylori infection. Dig Dis Sci. 2001;46:2440–4. doi: 10.1023/a:1012319718848. [DOI] [PubMed] [Google Scholar]
- 147.Leodolter A, Agha-Amiri K, Peitz U, Gerards C, Ebert M, Malfertheiner P. Validity of a Helicobacter pylori stool antigen assay for the assessment of H. pylori status following eradication therapy. Eur J Gastroenterol Hepatol. 2001;13:673–6. doi: 10.1097/00042737-200106000-00010. [DOI] [PubMed] [Google Scholar]
- 148.Vaira D, Vakil N, Menegatti M, Hoff B, Ricci C, Gatta L, et al. The stool antigen test for detection of Helicobacter pylori after eradication therapy. Ann Intern Med. 2002;136:280–7. doi: 10.7326/0003-4819-136-4-200202190-00007. [DOI] [PubMed] [Google Scholar]
- 149.Gisbert J, Pajares J. Diagnosis of Helicobacter pylori infection by stool antigen determination: a systematic review. Am J Gastroenterol. 2001;96:2829–38. doi: 10.1111/j.1572-0241.2001.04235.x. [DOI] [PubMed] [Google Scholar]
- 150.Lind T, Veldhuyzen van Zanten S, Unge P, Spiller R, Bayerdorffer E, O’Morain C, et al. Eradication of Helicobacter pylori using one-week triple therapies combining omeprazole with two antimicrobials: the MACH I Study. Helicobacter. 1996;1:138–44. doi: 10.1111/j.1523-5378.1996.tb00027.x. [DOI] [PubMed] [Google Scholar]
- 151.Matsuhisa T, Kawai T, Masaoka T, Suzuki H, Ito M, Kawamura Y, et al. Efficacy of metronidazole as second-line drug for the treatment of Helicobacter pylori infection in the Japanese population: a multicenter study in the Tokyo Metropolitan Area. Helicobacter. 2006;11:152–8. doi: 10.1111/j.1523-5378.2006.00394.x. [DOI] [PubMed] [Google Scholar]
- 152.Gisbert JP, Castro-Fernandez M, Bermejo F, Perez-Aisa A, Ducons J, Fernandez-Bermejo M, et al. Third-line rescue therapy with levofloxacin after two H. pylori treatment failures. Am J Gastroenterol. 2006;101:243–7. doi: 10.1111/j.1572-0241.2006.00457.x. [DOI] [PubMed] [Google Scholar]
- 153.Nishizawa T, Suzuki H, Kurabayashi K, Masaoka T, Muraoka H, Mori M, et al. Gatifloxacin resistance and mutations in gyra after unsuccessful Helicobacter pylori eradication in Japan. Antimicrob Agents Chemother. 2006;50:1538–40. doi: 10.1128/AAC.50.4.1538-1540.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nishizawa T, Suzuki H, Umezawa A, Muraoka H, Iwasaki E, Masaoka T, et al. Rapid detection of point mutations conferring resistance to fluoroquinolone in gyr A of Helicobacter pylori by allele-specific polymerase chain reaction. J Clin Microbiol 2006; Nov.22; [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 155.Boer WA, Tytgat GN. The best therapy for Helicobacter pylori infection: should efficacy or side-effect profile determine our choice? Scand J Gastroenterol. 1995;30:401–7. doi: 10.3109/00365529509093298. [DOI] [PubMed] [Google Scholar]
- 156.Fischbach LA, Zanten S, Dickason J. Meta-analysis: the efficacy, adverse events, and adherence related to first-line anti-Helicobacter pylori quadruple therapies. Aliment Pharmacol Ther. 2004;20:1071–82. doi: 10.1111/j.1365-2036.2004.02248.x. [DOI] [PubMed] [Google Scholar]
- 157.Sakaki N. Problems occurring after Helicobacter pylori eradication and their countermeasures. Nippon Rinsho. 2003;61:119–24. [PubMed] [Google Scholar]
- 158.Raghunath A, Hungin AP, Wooff D, Childs S. Prevalence of Helicobacter pylori in patients with gastro-oesophageal reflux disease: systematic review. BMJ. 2003;326:737. doi: 10.1136/bmj.326.7392.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Raghunath AS, Hungin AP, Wooff D, Childs S. Systematic review: the effect of Helicobacter pylori and its eradication on gastro-oesophageal reflux disease in patients with duodenal ulcers or reflux oesophagitis. Aliment Pharmacol Ther. 2004;20:733–44. doi: 10.1111/j.1365-2036.2004.02172.x. [DOI] [PubMed] [Google Scholar]
- 160.Moayyedi P, Feltbower R, Brown J, Mason S, Mason J, Nathan J, et al. Effect of population screening and treatment for Helicobacter pylori on dyspepsia and quality of life in the community: a randomised controlled trial. Leeds HELP Study Group. Lancet. 2000;355:1665–9. doi: 10.1016/s0140-6736(00)02236-4. [DOI] [PubMed] [Google Scholar]
- 161.Harvey RF, Lane JA, Murray LJ, Harvey IM, Donovan JL, Nair P. Randomised controlled trial of effects of Helicobacter pylori infection and its eradication on heartburn and gastro-oesophageal reflux: Bristol Helicobacter project. BMJ. 2004;328:1417. doi: 10.1136/bmj.38082.626725.EE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Nomura S, Esumi H, Job C, Tan SS. Lineage and clonal development of gastric glands. Dev Biol. 1998;204:124–35. doi: 10.1006/dbio.1998.9055. [DOI] [PubMed] [Google Scholar]
- 163.Mills JC, Andersson N, Hong CV, Stappenbeck TS, Gordon JI. Molecular characterization of mouse gastric epithelial progenitor cells. Proc Natl Acad Sci U S A. 2002;99:14819–24. doi: 10.1073/pnas.192574799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mahdavi J, Sonden B, Hurtig M, Olfat FO, Forsberg L, Roche N, et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science. 2002;297:573–8. doi: 10.1126/science.1069076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Evans DG, Karjalainen TK, Evans DJ, Jr, Graham DY, Lee CH. Cloning, nucleotide sequence, and expression of a gene encoding an adhesin subunit protein of Helicobacter pylori. J Bacteriol. 1993;175:674–83. doi: 10.1128/jb.175.3.674-683.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Bennett HJ, Roberts IS. Identification of a new sialic acid-binding protein in Helicobacter pylori. FEMS Immunol Med Microbiol. 2005;44:163–9. doi: 10.1016/j.femsim.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 167.Bury-Mone S, Thiberge JM, Contreras M, Maitournam A, Labigne A, Reuse H. Responsiveness to acidity via metal ion regulators mediates virulence in the gastric pathogen Helicobacter pylori. Mol Microbiol. 2004;53:623–38. doi: 10.1111/j.1365-2958.2004.04137.x. [DOI] [PubMed] [Google Scholar]
- 168.Oh JD, Kling-Backhed H, Giannakis M, Engstrand LG, Gordon JI. Interactions between gastric epithelial stem cells and Helicobacter pylori in the setting of chronic atrophic gastritis. Curr Opin Microbiol. 2006;9:21–7. doi: 10.1016/j.mib.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 169.Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, et al. Gastric cancer originating from bone marrow-derived cells. Science. 2004;306:1568–71. doi: 10.1126/science.1099513. [DOI] [PubMed] [Google Scholar]
- 170.Suzuki H, Marshall BJ, Hibi T. Overview: Helicobacter pylori and extragastric disease. Int J Hematol. 2006;84:291–300. doi: 10.1532/IJH97.06180. [DOI] [PubMed] [Google Scholar]
- 171.Veneri D, Krampera M, Franchini M. High prevalence of sustained remission of idiopathic thrombocytopenic purpura after Helicobacter pylori eradication: a long-term follow-up study. Platelets. 2005;16:117–9. doi: 10.1080/09537100400015153. [DOI] [PubMed] [Google Scholar]
- 172.Veneri D, Franchini M, Krampera M, Matteis G, Solero P, Pizzolo G. Analysis of HFE and TFR2 gene mutations in patients with acute leukemia. Leuk Res. 2005;29:661–4. doi: 10.1016/j.leukres.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 173.Inaba T, Mizuno M, Take S, Suwaki K, Honda T, Kawai K, et al. Eradication of Helicobacter pylori increases platelet count in patients with idiopathic thrombocytopenic purpura in Japan. Eur J Clin Invest. 2005;35:214–9. doi: 10.1111/j.1365-2362.2005.01471.x. [DOI] [PubMed] [Google Scholar]
- 174.Konno M, Muraoka S, Takahashi M, Imai T. Iron-deficiency anemia associated with Helicobacter pylori gastritis. J Pediatr Gastroenterol Nutr. 2000;31:52–6. doi: 10.1097/00005176-200007000-00012. [DOI] [PubMed] [Google Scholar]
- 175.Baysoy G, Ertem D, Ademoglu E, Kotiloglu E, Keskin S, Pehlivanoglu E. Gastric histopathology, iron status and iron deficiency anemia in children with Helicobacter pylori infection. J Pediatr Gastroenterol Nutr. 2004;38:146–51. doi: 10.1097/00005176-200402000-00008. [DOI] [PubMed] [Google Scholar]
- 176.Nahon S, Lahmek P, Massard J, Lesgourgues B, Mariaud de Serre N, Traissac L, et al. Helicobacter pylori-associated chronic gastritis and unexplained iron deficiency anemia: a reliable association? Helicobacter. 2003;8:573–7. doi: 10.1111/j.1523-5378.2003.00184.x. [DOI] [PubMed] [Google Scholar]
- 177.Choe YH, Kim SK, Hong YC. The relationship between Helicobacter pylori infection and iron deficiency: seroprevalence study in 937 pubescent children. Arch Dis Child. 2003;88:178. doi: 10.1136/adc.88.2.178-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Annibale B, Marignani M, Monarca B, Antonelli G, Marcheggiano A, Martino G, et al. Reversal of iron deficiency anemia after Helicobacter pylori eradication in patients with asymptomatic gastritis. Ann Intern Med. 1999;131:668–72. doi: 10.7326/0003-4819-131-9-199911020-00006. [DOI] [PubMed] [Google Scholar]
- 179.Marignani M, Angeletti S, Bordi C, Malagnino F, Mancino C, Delle Fave G, et al. Reversal of long-standing iron deficiency anaemia after eradication of Helicobacter pylori infection. Scand J Gastroenterol. 1997;32:617–22. doi: 10.3109/00365529709025109. [DOI] [PubMed] [Google Scholar]
- 180.Kostaki M, Fessatou S, Karpathios T. Refractory iron-deficiency anaemia due to silent Helicobacter pylori gastritis in children. Eur J Pediatr. 2003;162:177–9. doi: 10.1007/s00431-002-1139-x. [DOI] [PubMed] [Google Scholar]
- 181.Russo-Mancuso G, Branciforte F, Licciardello M, La Spina M. Iron deficiency anemia as the only sign of infection with Helicobacter pylori: a report of 9 pediatric cases. Int J Hematol. 2003;78:429–31. doi: 10.1007/BF02983815. [DOI] [PubMed] [Google Scholar]
- 182.Shiotani A, Okada K, Yanaoka K, Itoh H, Nishioka S, Sakurane M, et al. Beneficial effect of Helicobacter pylori eradication in dermatologic diseases. Helicobacter. 2001;6:60–5. doi: 10.1046/j.1523-5378.2001.00009.x. [DOI] [PubMed] [Google Scholar]
- 183.Fukuda S, Shimoyama T, Umegaki N, Mikami T, Nakano H, Munakata A. Effect of Helicobacter pylori eradication in the treatment of Japanese patients with chronic idiopathic urticaria. J Gastroenterol. 2004;39:827–30. doi: 10.1007/s00535-004-1397-7. [DOI] [PubMed] [Google Scholar]
- 184.Federman DG, Kirsner RS, Moriarty JP, Concato J. The effect of antibiotic therapy for patients infected with Helicobacter pylori who have chronic urticaria. J Am Acad Dermatol. 2003;49:861–4. doi: 10.1016/s0190-9622(03)00846-6. [DOI] [PubMed] [Google Scholar]