

Abstract.
The glucokinase (GCK) gene was one of the first candidate genes to be identified as a human “diabetes gene". Subsequently, important advances were made in understanding the impact of GCK in the regulation of glucose metabolism. Structure elucidation by crystallography provided insight into the kinetic properties of GCK. Protein interaction partners of GCK were discovered. Gene expression studies revealed new facets of the tissue distribution of GCK, including in the brain, and its regulation by insulin in the liver. Metabolic control analysis coupled to gene overexpression and knockout experiments highlighted the unique impact of GCK as a regulator of glucose metabolism. Human GCK mutants were studied biochemically to understand disease mechanisms. Drug development programs identified small molecule activators of GCK as potential antidiabetics. These advances are summarized here, with the aim of offering an integrated view of the role of GCK in the molecular physiology and medicine of glucose homeostasis.
Keywords. Glucokinase, diabetes, glucose metabolism, insulin, hexokinase, islet of Langerhans, hepatocyte, MODY
Introduction
This review focuses on the enzyme known as mammalian glucokinase. Somewhat ironically, this enzyme is actually not a glucokinase in the strict sense, denoting a glucose phosphorylating enzyme with stringent substrate specificity for glucose, such as exists in lower organisms. Mammalian glucokinase is able to phosphorylate hexoses like mannose or fructose in addition to glucose, a property shared with three other hexokinases present at various levels and in various combinations in human, rat, and mouse tissues [1]. The four mammalian isoenzymes display extensive sequence identities, leaving no doubt about a close phylogenetic relationship [2], and arguing for the terminology hexokinases I–IV (or A–D), in which hexokinase IV (or D) designates mammalian glucokinase. In spite of these considerations, the term glucokinase has become entrenched in tradition, probably because it conveys a sense of the very unique role of this enzyme in the regulation of glucose homeostasis. This tradition is followed in the current article, with the understanding that glucokinase (GCK) stands for hexokinase IV (or D). Whether the name should be reconsidered in the future is an open question, because a gene encoding a functional ADP-dependent glucokinase reminiscent of primitive organisms was recently identified in the mouse and human genomes [3]. The level of activity and physiological role of this enzyme in mammalian tissues are currently unknown.
Glucokinase was identified as an enzyme of rat liver in 1962. In 1992, two studies using the candidate gene approach reported a genetic linkage between a form of maturity onset diabetes of the young, named MODY 2, and the GCK gene [4, 5]. This discovery came as the culmination of three decades of research in biochemistry, physiology and molecular biology, which collectively demonstrated the preferential expression of GCK in hepatocytes and β-cells of the pancreas, and suggested a key role in glucose metabolism. These investigations, which had conferred to GCK the rank of prime candidate as a “diabetes gene”, were the subject of two reviews published in 1993 [6, 7].
In recent years our perception of the pivotal role of GCK in the regulation of glucose metabolism has been reinforced by important results in a variety of experimental areas. Structural investigations by X-ray crystallography shed new light on the very special enzyme kinetics of GCK. Gene expression studies revealed novel facets of the tissue distribution of GCK, notably in brain, and of the insulin regulation of expression in the liver. Genetic manipulations in cells and mice led to major advances in our understanding of the glucose sensor function of GCK at the cellular level and its impact on whole body glucose homeostasis. This article was written to assemble knowledge from very diverse lines of investigation, with the aim of offering an integrated view of the unique place of GCK in physiology and medicine.
Three-dimensional structure of GCK: cooperative glucose kinetics explained
Glucose taken up by mammalian cells has to be converted into glucose 6-phosphate as a prelude to further utilization for glycolysis, the pentose phosphate pathway or glycogen synthesis. Although some contribution to overall glucose phosphorylation by the recently identified ADP-dependent glucokinase [3] cannot be ruled out, the bulk of glucose phosphorylation occurs at the expense of ATP in the generic reaction catalyzed by the hexokinases: hexose + ATP → hexose 6-phosphate +ADP [8].
Among the hexokinases, GCK displays unique enzyme kinetics in two respects. First, GCK has an affinity for glucose (S0.5 ≅ 6 mM) more than 20 times lower than that of the next ranking hexokinase, hexokinase II. Second, the rate of the GCK reaction displays a sigmoid rather than hyperbolic dependence on glucose concentration [1]. These two kinetic properties, low affinity and cooperativity with respect to glucose, allow GCK to operate as an ultrasensitive physiological glucose sensor in cells endowed with non-limiting glucose transport activity at the plasma membrane (see below).
The cooperativity of GCK with glucose cannot be explained by the classical mechanisms of allostery for multisubunit proteins, because GCK is catalytically active in the monomeric state [9]. Moreover, GCK harbours a single glucose binding site (the active site) [10], excluding the possibility of multisite allostery in which two molecules of glucose would bind simultaneously to the enzyme. Deviation from hyperbolic kinetics is sometimes noted for monomeric enzymes in case of random addition of substrates, but this is not the case for the GCK catalytic cycle, which involves an ordered addition of substrates in which glucose binds first and ATP second [11]. A mechanism of positive cooperativity applicable to monomeric enzymes, named the mnemonic model, was proposed for GCK on the basis of early enzymological investigations [12]. Predictions from the mnemonic model were confirmed by recent studies using pre-steady-state measurements of the intrinsic fluorescence of GCK during binding of glucose and other ligands [13, 14]. The mnemonic mechanism of cooperativity includes the following features: i) equilibrium between two conformational states of GCK with vastly different glucose affinities; ii) large predominance of the low affinity conformation in the absence of glucose; iii) slow interconversion between the conformational states for glucose-free GCK; iv) conversion from the low affinity to the high affinity conformational state strongly accelerated upon glucose binding to GCK; v) only the high affinity form of the enzyme is catalytically competent, and the rate of the catalytic step(s) is very fast compared to the conformational transition rate.
Explicit support for the model came recently from structural data on human GCK obtained by Kamata and colleagues, who solved the structure of GCK by X-ray crystallography [15]. Crystals were obtained from GCK with short truncations at the NH2-terminal end (11 or 15 amino acids) under two distinct crystallization conditions: in the presence of glucose plus a small-molecule activator of GCK (see below), or in the absence of any ligand. The overall structure of the protein was comprised of a large and a small globular domain connected by a hinge made up of three flexible loops. With glucose and the activator present, the space between the two domains assumed the shape of a narrow, deep cleft containing the glucose binding pocket. The binding site for the activator was also localized in the hinge region, at a distance from the glucose binding pocket. The structure was very similar to that of the C-terminal half of hexokinase I crystallized in the presence of glucose, called the closed form of hexokinase, and was therefore designated the “closed” form of GCK. By contrast, GCK crystals formed without ligands displayed the so-called “super-open” conformation. In this configuration, the cleft space between the two domains of GCK was much broader, as the result of two conformational changes compared to the closed form: tilt and rotation of the small domain with respect to the large domain, and extensive rearrangement of the secondary structural elements of the small domain itself [15].
The two X-ray structures of GCK could readily be fitted into the mnemonic model of enzyme mechanism. The super-open conformation represents the low glucose affinity, catalytically inactive form of enzyme. The closed conformation corresponds to the high affinity form of GCK with glucose and ATP bound at the active site during catalysis. An additional conformation of GCK, intermediary between the super-open and the closed states, was postulated for the high affinity form of the enzyme free of substrates or products. This putative conformation was designated the “open” form. The crystal structure of GCK in the open form remains to be established. However, X-ray diffraction studies of crystals of the C-terminal half of hexokinase I formed in the absence of glucose can serve as a model for the open form. Compared to the closed form, the open form displayed a wider interdomain cleft due to slight twist of the small domain relative to the large domain, without change in the internal topology of the small domain core [16]. The transition between open and closed conformations would be very fast and easily reversible, in contrast to the more complex molecular reorganization involved in the slow transition from the superopen to the open conformations. Indeed, the prediction of an intermediary open state of GCK was borne out in a recent study using targeted molecular dynamics simulation to dissect the transition from the closed to the super-open configurations of GCK [17].
Regulation of glucokinase activity by protein-protein interaction
Interaction with hepatic glucokinase regulatory protein. The best understood mechanism of rapid regulation of GCK activity relies on protein-protein interaction between GCK and a partner termed GCK regulatory protein (GCKR). The association between the two proteins is ligand-dependent and inhibitory to GCK activity. Ligands for GCKR are fructose-6-phosphate and fructose-1-phosphate, which are mutual competitors. Binding of fructose 6-phosphate to GCKR favors the GCKR-GCK interaction with a negative effect on enzyme activity, while binding of fructose-1-phosphate weakens the GCKRGCK interaction and releases active GCK. Intrahepatic fructose-1-phosphate rises postprandially after intestinal absorption of fructose and its conversion to fructose-1-phosphate by liver fructokinase. Conversely, inhibition of GCK by fructose-6-phosphate liganded to GCKR would provide a mechanism for indirect negative feedback, since fructose 6-phosphate is in equilbrium with glucose 6-phosphate (the product of the GCK reaction) through the phosphohexose isomerase step of glycolysis [18].
The conformational state of GCK preferred for binding to GCKR is the super-open form. This was suggested by experiments in a cell-free assay with purified proteins, in which the association of GCK with GCKR was estimated by co-immunoprecipitation. The amount of GCK associated with GCKR was reduced under conditions favoring the closed conformational state of GCK, that is during incubation at elevated glucose concentrations in the presence of ATP, or in the presence of glucose together with a small molecule activator of GCK. This suggested that the interface for binding to GCKR would be disrupted by the conformational transition from the super-open to the closed forms of GCK [19].
The reversible association of GCK with GCKR does more than simply regulate the catalytic activity of GCK, it also appears to underlie the intracellular trafficking of GCK between cytoplasm and nucleus. In transfected rat hepatocytes, GCK tagged with green fluorescent protein was localized in the nucleus at low glucose and was released to the cytoplasm at high glucose, but GCK mutants with reduced affinity for GCKR were predominantly cytoplasmic at low as well as high glucose [20]. In cell lines devoid of GCK and GCKR, such as COS-1, HeLa and HEK293T cells, forced expression of GCK alone resulted in cytoplasmic localization, whereas co-expression with GCKR resulted in nuclear accumulation of both proteins [20–22]. In a rat islet β-cell line with no endogenous GCKR, doxycycline-inducible expression of GCKR resulted in nuclear accumulation of GCKR together with translocation of endogenous GCK from an extranuclear localization to the nuclei [22].
A model (Fig. 1) to account for these results proposes that GCKR in the free state would shuttle between nucleus and cytoplasm. Under low glucose conditions, the assembly of GCKR-GCK complex would result in nuclear import and trapping of both proteins by molecular mechanisms not yet completely elucidated. Masking of a nuclear export sequence (NES) between amino acids 300 and 310 of GCK sequence would be part of this mechanism [21]. In metabolic states accompanied by high glucose or fructose-1-phosphate concentrations, and sufficient ATP levels [23], the GCK-GCKR complex would dissociate and the NES of GCK would become functional, allowing for the rapid export of GCK from nuclei. In the isolated perfused liver system and in primary cultures of rat hepatocytes, GCK and GCKR were indeed mainly localized to the nuclei at glucose concentrations around 5 mM. Higher glucose levels or fructose addition to the medium caused a rapid translocation of GCK to a predominant cytoplasmic localization [24, 25]. This was accompanied to a lesser degree by redistribution of GCKR to the cytoplasm [26, 27]. As predicted by this model, mice with homozygous inactivation of the GCKR gene had an extranuclear localization of hepatic GCK under all metabolic conditions [28, 29]. Interestingly, the steady state amount of GCK in the liver was reduced about 50% in the GCKR knock-out animals, suggesting the possibility that the interaction with GCKR might stabilize GCK against degradation.
Figure 1.
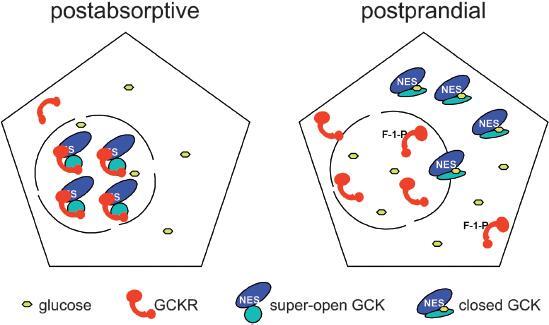
Regulation of GCK activity and subcellular localization by interaction with GCKR. The scheme depicts hepatocytes after an overnight fast (postabsorptive) and during the ingestive phase after a carbohydrate containing meal (postprandial). The nuclear compartment (circle) communicates with the cytoplasmic space by nuclear pores. The blue and cyan ovals represent the large and small lobes of GCK respectively. After carbohydrate ingestion, the glucose concentration in plasma and hepatocytes rises, as does the concentration of fructose-1-phosphate. As a result, the GCKR (red)-GCK interaction is loosened, allowing GCK to bind glucose, adopt the closed (catalytically active) conformation and exit from the nucleus to generate glucose-6-phosphate for glycogen synthesis and glycolysis.
Recently, a minor fraction of GCK and GCKR in hepatocytes was reported to be associated with the mitochondrial fraction, raising the suggestion of distinct pools of cytoplasmic GCK with specialized metabolic functions [30]. However, this notion should be considered with caution, because other authors failed to detect GCK activity or immunoreactivity in isolated mouse liver mitochondria [31].
Interaction with other proteins. The impact of the GCKR model led several investigators to search for new protein partners that might affect GCK activity or localization, notably in the islet β-cells in which GCKR is essentially absent. Alist of GCK interacting proteins with their major properties is given in Table 1. For virtually all of these proteins other than GCKR, additional investigations are necessary for definitive proof that the proposed interaction with GCK has an impact on GCK activity and/or cell function.
Table 1.
Protein interaction partners of glucokinase.
| Name | Method of identification | Biochemical effects | Cellular effects | References |
|---|---|---|---|---|
| glucokinase regulatory protein (GCKR) | protein purification from liver extract | inhibits GCK activity | regulates GCK activity and localization in hepatocytes | reviewed in [18] |
| glucokinase-associated-phosphatase | yeast two-hybrid system | increases GCK activty | unknown | [32] |
| propionyl-CoA carboxylase β-subunit | yeast two-hybrid system | increases GCK affinity and activity | unknown | [33] |
| 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase | screening of random peptide display phage library for binding to GCK | no effect in GCK activity assay | overexpression in insulinoma cells increases GCK activity | [34–36] |
| neuronal nitric oxide synthase | co-immunoprecipitation | nitrosylates GCK | anchors GCK to the periphery of insulin secretory granules | [37] |
| pentaubiquitin | identification of ubiquitin interacting motif in GCK | increases GCK affinity and activity | unknown | [39] |
| bad | peptide analysis of multiprotein complex in liver mitochondrial fraction, immunochemistry | increases GCK activity | regulates GCK activity and localization in hepatocytes and β-cells | [40, 41] |
The bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB) is responsible for the regulated formation and degradation of fructose 2,6-bisphosphate, a key allosteric activator of the enzyme 6-phosphofructo-1-kinase, and thereby of glycolysis. An interaction of the fructose-2,6-bisphosphatase domain of PFKBP with GCK was demonstrated in a yeast two-hybrid system [34]. Overexpression of PFKBP in insulinoma β-cells resulted in increased GCK specific activity without increase in the cellular content of GCK protein [35, 36]. Although provocative, the published studies did not directly demonstrate an association of GCK with PFKFB in mammalian cells, nor did they provide insight into a possible mechanism for the apparent increase in GCK activity in cells co-expressing PFKBP. These issues should be further investigated before a conclusive model of GCK regulation by association with PFKBP becomes widely accepted.
A model in which neuronal nitric oxide synthase (NOS) would serve to anchor GCK to the periphery of insulin secretory granules in the basal state in βTC3 insulinoma cells was proposed by Rizzo and Piston [37]. Autocrine activation of NOS by insulin during glucose stimulated secretion would stimulate the nitrosylation of GCK on a specific cysteine residue, and its release from the granules to the cytosol. The general validity of this model remains uncertain, however, because glucose-induced translocation of GCK from the secretory granules to the cytosol was not confirmed in MIN6 insulinoma cells [38].
Human GCK was recently found to be polyubiquitinated at multiple Lys residues in a cell-free system. Additionally, a sequence motif compatible with an ubiquitin interacting consensus was identified in the C-terminal a-helix of human GCK. This motif was the target for non-covalent binding of free pentaubiquitin chains in a cell-free system. Ubiquitin chains added to a GCK enzyme assay mixture resulted in an increased Kcat [39]. The occurrence and physiological implication of this interaction in living cells were not investigated.
Multiprotein assembly at the outer mitochondrial membrane. In investigations of the function of the BH3-only proapoptotic protein BAD, Danial and colleagues described a large multimolecular assembly comprising BAD, GCK and other proteins in a highly purified mitochondrial fraction from the mouse liver [40]. Analysis of the supramolecular complex using immunochemistry and LC-tandem MS peptide analysis revealed that the complex contained GCK and BAD, as well as the Wiskott-Aldrich family member WAVE, the catalytic subunits of protein phospatase 1 (PP1) and of protein kinase A (PKA). The essential protein in nucleating the complex appeared to be BAD, since the complex was absent in BAD knockout (BAD−/−) mice. The oxidative metabolism of glucose was reduced in hepatocytes from BAD−/− mice, suggesting a function of the complex in metabolic regulation. However, caution is warranted, because GCK associated with mitochondria was a very small fraction of the total [40], if present at all [31]. Complicating the matter further, the role and regulation of the protein kinase and phosphatase associated with the complex remain elusive.
The relevance of the Danial complex was recently extended to the β-cells of the islets of Langerhans [41]. Perifused islets from BAD−/− mice displayed glucose unresponsive insulin secretion. Total assayable GCK activity in islet homogenates from BAD−/− mice was reduced to less than 30% of the wild-type level. Glucose-sensitive insulin release was restored after treatment of islets from BAD−/− animals with cell-permeable synthetic peptides corresponding in sequence to aBH3 α-helix of BAD. Peptides harboring a phosphomimetic Ser 155 to Asp mutation were active, while a peptide with an Ala mutation was ineffective. Peptides active in the insulin secretion assay also induced strong increases in GCK activity when added to INS-1 insulinoma cells in culture. Furthermore, photoactivatable BAD peptides were shown to crosslink with GCK in homogenates of INS-1 cells. It is worth pointing out that a much larger fraction of cellular GCK appeared to be under BAD control in the islets than in the liver. Whether most or all of the GCK interacting with BAD resided at the outer mitochondrial membrane in islet cells was not experimentally demonstrated.
Regulation of glucokinase gene expression
Tissue-specific promoters. A defining feature of the GCK gene in mammalian species is the presence of two alternative promoters. The promoters are separated by approximately 30 kbp of genomic DNA and responsible for transcription initiation in a mutually exclusive manner in distinct tissues. The upstream promoter and adjacent leader exon drive the synthesis of GCK mRNA in non-hepatic tissues, that is islet cells, enteroendocrine cells, specialized glucose-sensitive neurons in the central nervous system (CNS), and additional scattered cells in various tissues [42]. This promoter is denominated the neuroendocrine promoter. By contrast, the downstream so-called liver promoter and its associated leader exon serve for transcription initiation in hepatocytes only. Each of the leader exons specifies the 5′ untranslated regions of the mRNAs as well as the first 15 codons for the GCK protein sequences, which therefore differ at their NH2-terminal ends in liver and other tissues. During splicing of the primary transcripts, the donor splice sites at the end of the neuroendocrine or liver leader exons are ligated to the acceptor site of a common exon 2 and further splicing incorporates common exons 3–10 in the two types of mRNAs. Thus, the GCK enzymes in neuroendocrine cells and hepatocytes have a common amino acid sequence overmost of their length. There is currently no known functional difference in the GCK enzymes arising from the short distinct leader sequences.
Alternative promoters allow for the control of transcription initiation by distinct sets of cis-acting regulatory elements in different tissues. In early experiments, Magnuson and colleagues showed that a 294 bp rat DNA fragment of the neuroendocrine promoter activated the expression of luciferase in insulinoma cells, but not 3T3-preadipocytes [43]. In transgenic mice, the same DNA fragment drove the expression of a human growth hormone reporter gene in pancreatic islet cells, intestinal cells and brain cells [42]. Two major functional cis-acting elements were delineated in this promoter fragment [43]. One element bound proteins which did not match known transcription factors [44], whereas the other element was identified as a binding site for Beta2/NeuroD, a basic helix-loophelix zipper (bHLHz) transcriptional activator important in the ontogeny of the endocrine pancreas [45]. Another key transcription factor of early pancreas development, Pdx-1, was proposed to activate the neuroendocrine GCK promoter [46], but this notion was subsequently dispelled [47].
There are no published investigations using transgenic mice to define the regulatory sequences responsible for the hepatocyte-specific activity of the liver GCK promoter. In transient transfection assays, a 200 bp liver promoter fragment from the rat gene was competent to stimulate the expression of a luciferase reporter in primary hepatocytes and hepatoma culture cells, but not in insulinoma cells [48, 49]. More importantly, a 300 bp enhancer active only in primary hepatocytes was mapped between nucleotides −1000 and −700 with respect to the start of transcription of the rat gene and at a similar position in the human gene. This sequence exhibited 6 DNAse I protection footprints, but the identity of the binding proteins was not elucidated [48].
Transcriptional induction of gene expression by insulin in liver. In the liver, expression of GCK is strictly dependent on the presence of insulin. Thus, GCK mRNA and protein disappear from the livers of insulino-deficient rats and are restored after insulin treatment [50]. In cultured rat hepatocytes, the effect of insulin as inducer of GCK was shown to be primarily at a transcriptional level, triggering a 15- to 30-fold increase in GCK mRNA in three hours, and taking place in glucose-free as well as glucose-containing medium without any synergy by addition of glucose [6]. Induction of GCK by insulin and repression by glucagon were also documented in hepatocytes isolated from the human liver [51].
Insulin induction of GCK in hepatocytes was shown to be suppressed by the inhibitors of phosphoinositide 3-kinase (PI3-kinase) wortmannin and LY294002, implying that activation of PI3-kinase following insulin binding to its receptor was essential. Protein kinase B (PKB, also called Akt) is a major protein kinase activated as a result of PI3-kinase stimulation. To test for a possible role of PKB in mediating GCK induction, hepatocytes were transduced for expression of an estrogen receptor-PKB chimeric protein that could be conditionally activated by tamoxifen. The transduced hepatocytes reponded to the addition of tamoxifen by a rise of GCK mRNA, mimicking the effect of insulin [52]. In separate experiments, insulin induction of GCK mRNA was abrogated by inhibitors of PKB activation [53, 54]. Additionally, GCK mRNA and protein were induced in the livers of streptozotocin diabetic mice following in vivo transduction of adenoviruses encoding kinase active versions of PFKFB, a maneuver that unexpectedly triggered the activation of PKB [55]. A role of PKB in insulin induction of GCK was questioned by Matsumoto and colleagues [56], who observed that they could inhibit insulin-mediated induction of GCK mRNA in hepatocytes by expressing a dominant negative NH2-terminal fragment of insulin receptor substrate-1 (IRS-1), while PKB activation apparently remained unaffected. An explanation for this negative result might be that GCK induction is mediated by a minor pool of PKB functionally associated with IRS-1, making a small contribution in assays of total cellular PKB activity and therefore difficult to quantify.
The transcriptional regulators which respond to the insulin activation of PKB (and perhaps of accessory signaling cascades) and stimulate transcription at the liver GCK promoter remain elusive. The sterol-regulatory element binding protein SREBP1c, a master regulator of lipogenic enzymes such as fatty acid synthase (FAS) and acetyl-CoA carboxylase (ACC), was proposed to be a mediator of insulin induction of GCK. Briefly, SREBP1c itself was shown to be induced at the transcriptional level by insulin in hepatocytes [57, 58]. Overexpression of SREBP1c in primary hepatocytes was accompanied by an elevation of GCK mRNA and, conversely, a dominant negative mutant of SREBP1c inhibited insulin-dependent induction of GCK mRNA [59]. More recently, investigators identified a tandem DNA element around position −200 in the rat liver GCK promoter, which could bind bacterially expressed mature SREBP1c in DNAse protection assay and electrophoretic mobility shift assay (EMSA) [60]. However, in other investigations, inducible expression of mature SREBP1c in hepatocytes failed to produce a significant increase of GCK mRNA [61]. In SREBP1c ko mice, GCK mRNA was induced normally during the fasting-refeeding transition [62]. Moreover, liver transduction of adenovirus encoding active PKB (i.e. mimicking insulin signaling) resulted in strong induction of hepatic GCK mRNA in normal as well as SREBP1 ko mice [63]. In our own experiments in insulin stimulated hepatocytes, SREBP1c failed to bind to the liver GCK promoter, while its binding to the FAS promoter was readily discerned by chromatin immunoprecipitation [64]. Also, insulin produced amore than 10-fold augmentation of GCK mRNA amount in a time frame in which there was strictly no increase in transcriptionally mature SREBP1c protein. Vice-versa, activation of the nuclear receptor liver X receptor (LXR) with a synthetic ligand produced a strong induction of SREBP1c and its target gene FAS without any rise of GCK mRNA [64]. Together, these results seem to virtually rule out a determinant role of SREBP1c in the rapid induction of liver GCK by insulin in rat hepatocytes.
Binding sites for the transcriptional activators hepatocyte nuclear factor-4 (HNF-4) and hypoxia-inducible factor-1a (HIF-1α) were identified within the first 100 bp of the liver GCK promoter [65, 66]. Coexpression of HNF-4 and HIF-1α with GCK reporter plasmids in primary rat hepatocytes resulted in additive transactivation of the GCK promoter, which was further augmented by expression of the co-activator p300. More importantly, co-transfection of plasmids encoding constitutively active PI3K or PKB stimulated GCK promoter activity, as long as an intact binding site for HIF-1α was present in the promoter DNA fragment. Together, these findings suggested that co-operation between HNF-4, HIF-1α and p300 at the liver GCK promoter might be important for induction of the gene in response to acute insulin signaling [66]. The proposal that HIF-1α might be a mediator in insulin signaling is significant, because hypoxia and insulin were previously shown to synergize in inducing GCK gene expression in cultured hepatocytes [67]. This type of synergy might in part account for the enrichment of insulin inducible enzymes such as GCK in the underoxygenated perivenous region of the liver lobule, compared to the periportal region [68]. Based on findings in another system, insulin might hypothetically induce HIF-1α target genes by relieving HIF-1a from corepression by a forkhead box class O (FOXO) factor, through a mechanism involving PKB-mediated phosphorylation and functional inactivation of FOXO [69]. However, arguing against this possibility, levels of hepatic GCK mRNA in adult mice with liver specific knockout of FOXO1 were similar to those of control mice in both the fasted and fed states [70].
Hormone and nutrient regulation of glucokinase gene expression in islets of Langerhans. Glucose refeeding after a fast is an experimental paradigm often used to study acute hyperinsulinemia in the intact animal. Consistent with data in cultured hepatocytes, GCK mRNA was virtually absent from the livers of fasted rats and accumulated massively within hours of feeding a glucose-enriched diet [71]. By contrast, GCK mRNA and protein levels in islets of Langerhans were not significantly depressed in fasted rats compared to freely fed animals and, more importantly, were not acutely induced during the fasting-refeeding transition [72]. Thus, constitutive expression of GCK in the pancreatic islets contrasted with inducible GCK gene expression in the liver.
In the MIN6 insulinoma cell line, GCK mRNA was higher during culture at 30 mM than at 3 mM glucose concentration, or following the addition of subnanomolar concentrations of insulin to the medium [73]. However, luciferase reporter plasmids driven by the neuroendocrine GCK promoter were unaffected after a rise in glucose or insulin concentrations [74], suggesting that, in insulinoma cells, the major effects of glucose or insulin might be to stabilize GCK mRNA against degradation rather than stimulate transcription from the neuroendocrine promoter. In rat INS-1 insulinoma cells, GCK mRNA and protein levels were increased about 50% after 24 hours of culture in the presence of insulin-like growth factor-1 (IGF-1), consistent with a modest transcriptional effect of IGF-1. An inhibitory cis-acting DNA element targeted by FOXO1 was identified at position −500 in the neuroendocrine promoter. Thus, FOXO1 might act in this system as a repressor, itself negatively regulated by IGF-1 [75].
Metabolic role of hepatic glucokinase
Regulatory role of glucokinase at the hepatocellular level. The liver is equipped with an insulin-independent facilitative glucose transport system (now known to be mostly by GLUT2) with apparent affinity constant of 17 mM and high transport rate, allowing extremely rapid equilibration of the glucose concentration across the hepatocyte plasma membrane [76]. Unlimiting glucose transport, and the unique glucose affinity of GCK(which is the predominant hexokinase isoenzyme in the liver), ensure that fluctuations of the plasma glucose concentration are rapidly translated into changes in the rate of glucose phosphorylation inside the hepatocyte. If, in turn, the GCK reaction controls the substrate flux along the entire length of the pathways from glucose to final products, the overall metabolic rate in these pathways will be regulated autonomously in response to changes in the plasma glucose concentration.
In hepatocytes with adenovirus mediated overexpression of GCK, compared to hepatocytes transduced with control vectors, rates of glycogen synthesis were augmented in proportion to GCK activity [77, 78]. This effect was attributed to an increase in the cellular content of glucose-6-phosphate, a product of the GCK reaction and an allosteric activator of glycogen synthase [79]. Overexpression of GCK also resulted in proportionate stimulation of glycolysis and glucose oxidation [80, 81]. As stated above, the fact that GCK exerts dominant control on glycogen synthesis and glycolysis has a crucial physiological implication: it confers to hepatocytes the ability for cell autonomous regulation of glucose metabolism in response to fluctuations of the plasma glucose.
Regulatory role of liver glucokinase at the organismal level. The impact of altering liver glucokinase activity on whole body glucose homeostasis was illustrated in transgenic mice with an extracopy of the GCK gene locus. These animals had an approximately 50% increase in hepatic GCK activity over the wild-type level and a 25% reduction in plasma glucose in the postabsorptive state, reflecting increased glucose clearance. During an hyperglycemic clamp, overall glucose utilization was similar in control and transgenic mice, in spite of plasma insulin levels that were 40% lower in the transgenics. Liver glycogen synthesis over the period of hyperglycemia was increased about 3.5 times in transgenic mice compared to wild-type [82]. The level of immunodetectable GCK was lower in the pancreatic islets of transgenic mice with additional GCK gene copies than in wild-type animals, perhaps owing to destabilization of the GCK protein in islets at low glycemic levels.
Another mouse model with overexpression of liver GCK was produced by transgenesis of GCK cDNA fused to the phosphoenolpyruvate carboxykinase (PEPCK) promoter. This construct allowed for high GCK activity to be maintained in the livers of transgenic mice made insulino-deficient by streptozotocin treatment, while virtual suppression of hepatic GCK was noted in nontransgenic littermates [83]. Transgenic mice were largely protected against streptozotocin-induced hyperglycemia. They maintained normal levels of liver glycogen and lactate, compared to severe depletion of these metabolites in the livers of nontransgenic diabetic mice, consistent with a determinant role of GCK in hepatic glycogen synthesis and glycolysis.
The effects of loss of hepatic GCK function on glucose homeostasis were investigated using a gene targeting system allowing for liver-specific inactivation of the GCK gene [84]. Mice with homozygous GCK gene deletion in the hepatocytes survived to adult age with virtually complete absence of GCK enzyme activity in liver tissue. In the freely fed state, the mice exhibited modest hyperglycemia with relative hyperinsulinemia compared to controls, indicative of an insulin resistant state. Liver glycogen in the freely fed state was normal, implying that gluconeogenesis (and perhaps high affinity hexokinases) produced sufficient glucose-6-phosphate in the basal state to ensure glycogen formation and compensate for the ablation of GCK. However, during a hyperglycemic clamp, the rate of liver glycogen synthesis failed to rise and whole body glucose turnover was lower than in control animals. Interestingly, glucose-induced insulin secretion during hyperglycemia was markedly diminished in liver knockout mice, possibly reflecting functional damage to the β-cells of the islets of Langerhans due to chronic hyperglycemia and secretory overstimulation [84].
The role of liver GCK in restraining hepatic glucose output in response to hyperglycemia was also highlighted in mice with global heterozygous inactivation of the GCK gene [85]. These mice exhibited a 50% reduction of GCK activity in the livers, and presumably in islets and other neuroendocrine cells as well. Glucose metabolism was investigated by the glucose clamp technique combined with pancreatic clamping with somatostatin, in order to maintain circulating insulin concentrations at the same fixed level in knockout and wild-type mice. Hyperglycemia caused a 10% reduction of the rate of hepatic glucose production in knockout mice, compared to 40% in controls, demonstrating that liver GCK is instrumental in insulin-independent inhibition of hepatic glucose production in response to high glucose [85]. Since GCK gene inactivation was ubiquitous in these mice, it cannot be excluded that decreased GCK activity at extrahepatic sites, for example in the hypothalamus (see below), might have compromised a neurallymediated mechanism contributing to the suppression of hepatic glucose production during hyperglycemia.
Metabolic role of islet β-cell glucokinase
Regulatory role of glucokinase in the β-cell. Insulin secretion elicited by hyperglycemia requires stimulation of glucose metabolism in the β-cells. Coupling between metabolism and secretion relies on the generation of metabolic products, most prominently ATP generated by glycolysis and glucose oxidation, which act as upstream signals in cascades leading to the stimulation of the exocytic machinery [86, 87]. For instance, a rise in the cellular ATP/AMP ratio causes the inhibition of ATP-sensitive K+ channels in the plasma membrane, leading to membrane depolarization and activation of voltage-sensitive Ca++ channels. The influx of extracellular Ca++ ions then somehow promotes the exocytosis of insulin secretory granules. The ability of the β-cells to increase their rate of glucose metabolism in response to a rise in the extracellular glucose concentration is basic to the regulation of insulin secretion. The biochemical properties which allow glucose to regulate its own metabolism, and thereby insulin secretion, are the same in the β-cells and in hepatocytes. The first property is unrestricted glucose transport to ensure rapid glucose equilibration across the plasma membrane [88, 89]. The second aspect is that glucose phosphorylation is predominantly catalyzed by GCK [90], which provides for concentration-dependent adjustment of the rate of the initial reaction of glucose metabolism. The third property is a metabolic organization in which flux along the entire pathways of glycolysis and glucose oxidation is mostly controlled at the initial reaction, that catalyzed by GCK.
The extent to which glucose phosphorylation does control the rate of glycolysis in insulin secreting cells was investigated in INS-1 derived insulinoma cells that had been engineered for doxycycline inducible GCK expression. Moderate increases in GCK activity elicited by small doses of doxycycline were accompanied by quantitatively comparable fractional increases in glycolytic rate at physiologically relevant extracellular glucose concentration. The fact that the ratio of fractional increase in glycolytic flux on fractional increase in GCK activity was close to unity showed that GCK exerted overwhelming control over the glycolytic rate [91]. Not surprisingly, at very high levels of GCK overexpression and especially at high glucose levels, a limit to the increase in glycolytic flux was attained, indicating that one or several steps in the lower part of the glycolytic pathway became limiting [91, 92]. The glyceraldehyde-phosphate dehydrogenase reaction was incriminated as such a step, due to NAD+ depletion [93].
Regulatory role of β-cell glucokinase at the organismal level. A strategy targeting the neuroendocrine promoter region of the GCK gene was used to generate mice with GCK deficiency in the β-cells of the islets of Langerhans (and other cell-types utilizing the same promoter) [94]. Mice homozygous for the deleted allele developed severe diabetes and died during the first week after birth. Glucose-stimulated insulin secretion was completely absent in islets from newborn null mice. Mice heterozygous for the deleted allele survived to adulthood with slight hyperglycemia, intolerance to a glucose load and reduced glucose-stimulated hyperinsulinemia. As expected, GCK activity in islets was reduced to about 50% of the wild-type level in the heterozygotes. Isolated islets form heterozygous mice exhibited impaired glucose stimulated insulin release, most apparent at a glucose concentration around 10 mM [94]. Similar findings were reported in mice made deficient in islet GCK activity by a LoxP strategy using Cre recombinase expressed from the rat insulin II gene promoter. The mice with heterozygous inactivation of β-cell GCK were markedly impaired in ability to raise plasma insulin during a hyperglycemic clamp in comparison to controls [84]. Collectively, these data underlined the role of islet GCK in glucose-induced insulin secretion and maintenance of glucose homeostasis in the whole animal. Whether the ablation of GCK in other neuroendocrine or insulin expressing cells might have contributed to the observed phenotype in the above experiments remains to be determined.
Glucokinase in the brain
Transgenic mice harboring a chimeric human growth hormone (hGH) structural gene transcribed from the rat neuroendocrine GCK promoter had abundant hGH in their pancreatic islets as expected, and more surprisingly, also displayed strong immunoreactivity to hGH in isolated hypothalamic cells [42]. Following this discovery, Jetton and colleagues detected GCK mRNA by reverse transcription-polymerase chain reaction (RT-PCR) in several regions of the rat brain and localized GCK transcripts as well as immunoreactive protein in situ in the hypothalamus [42]. Subsequently, the neuroendocrine form of GCK mRNA was selectively identified by RT-PCR assay as well as in situ hybridization in the ventromedial and arcuate nuclei of the rat hypothalamus [95]. At the protein level, immunoblotting and enzyme assays confirmed the presence of GCK in rat hypothalamic extracts [96].
In the ventromedial hypothalamus, GCK mRNA was shown to be expressed in neurons synthesizing pre-proopiomelanocortin (POMC) and neuropeptide Y (NPY)/agouti-related peptide (AgRP), which play critical roles in neural pathways involved in the regulation of food intake and energy expenditure. Experiments using primary cultures of rat hypothalamic neurons incubated with metabolic inhibitors such as mannoheptulose or alloxan suggested that GCK activity was instrumental for the glucose response of glucose-excited and glucose-inhibited neurons as well [97]. Transcript profiling at the single cell level confirmed the presence of GCK mRNA in approximately 50%of glucose-excited and glucose-inhibited neurons in the ventromedial area of the hypothalamus. Some of these neurons also contained mRNAs for subunits of ATP-sensitive K+ channels [98]. In primary cultures of neurons from the rat ventromedial hypothalamus, treatment with specific small interfering RNA (siRNA) to knock-down GCK resulted in the virtual disappearance of excitatory as well as inhibitory responses to glucose elevation [99]. Collectively, these findings supported the idea that the glucose sensor of at least a fraction of ventromedial hypothalamic neurons was GCK, functioning as the rate determining enzyme of glucose metabolism and controlling neuronal excitability. However, this proposal raises puzzling questions. First, the interstitial glucose concentration in brain in the postabsorptive state is around 1.5 mM, well below the optimal range of concentrations where GCK displays ultrasensitive kinetics with respect to glucose concentration. The second key issue is that of coupling between glucose metabolism and neuronal activity. Many neurons are equipped with ATP-sensitive K+ channels, and the excitability of such neurons might then be controlled by the rate of glucose metabolism and the attending fluctuations in cellular ATP content, in ways reminiscent of the islet β-cells [100]. Other glucose-derived metabolites with a signaling role might be intermediates of the lipogenic pathway [101].
Molecular medicine of glucokinase
Maturity onset diabetes of the young (MODY2). Approximately 200 mutations of the human GCK gene, including missense, non-sense, and splice site mutations, were described in the literature. The majority of these mutations cause a form of familial hyperglycemia or mild diabetes, transmitted as an autosomal dominant trait and often diagnosed before 25 years of age. Based on these characteristics, diabetes associated with GCK mutations was included in the group of maturity onset diabetes of the young (MODY), and was named MODY2. Clinically, MODY2 was defined as a syndrome of impaired fasting glucose level or mild diabetes, with hyperglycemia maintained at stable level for many years, little propensity for late neurovascular complications and often adequate glycemic control with diet alone [102, 103]. Several cases of MODY2 were diagnosed in women with a history of gestational diabetes [104]. Recently, a new nomenclature was proposed for the MODY syndromes, MODY2 being replaced by the descriptive name “familial mild fasting hyperglycemia” [105]. Rare cases of infants inheriting GCK mutations from two heterozygous parents were described in the literature. They presented an autosomal recessive form of permanent neonatal diabetes mellitus (PNDM) [106].
Clinical investigations in patients with MODY2 revealed disorders of liver metabolism and pancreatic islet function predicted from basic studies in cells and animals. Net synthesis of glycogen in the liver after each of three daily meals was decreased by about 50% in patients compared to controls [107]. Additionally, the suppression of hepatic glucose output by hyperglycemia was deficient in patients with a GCK mutation [108]. Patients with a variety of missense or nonsense GCK mutations exhibited postprandial plasma insulin levels comparable to controls, although glucose concentrations were higher in the patients, a finding compatible with a loss of sensitivity of the β-cells to glucose. More specifically, during a ramping glucose infusion, the glucose dose-response curve for insulin secretion was found to be shifted to the right in the MODY2 patients [109].
The biochemical properties of many GCK mutants identified in MODY2 individuals were investigated using bacterially produced protein. A wide range of abnormalities affecting the turn-over number of the enzyme, affinity for the substrates, and cooperativity with respect to glucose were described [110–112]. Mathematical models based on the kinetics properties were used to predict the threshold of glucose-stimulated insulin secretion by β-cells harboring a given heterozygous mutation. On average, prediction for a number of mutants was in agreement with the level of fasting hyperglycemia noted in cohorts of MODY2 patients [111]. Some mutants were found to have unaltered kinetic properties compared to the wild-type enzyme, but to display accelerated temperature-dependent loss of activity in the test tube, in one case correlating with decreased protein half-life in transduced cells [111, 113].
Several mouse lines with heritable hyperglycemia were produced through phenotype-driven mutagenesis programs using the potent mutagen N-ethyl-N-nitrosourea (ENU). Interestingly, the majority of hyperglycemic lines that were genetically characterized so far harbored mutations in the GCK gene [114, 115], several of which corresponded to mutations that had been described in pedigrees with MODY2 or in one case in PNDM [116]. These mice displayed spontaneous hyperglycemia in the freely fed state, and glucose intolerance accompanied by relative hypoinsulinemia after an oral glucose load, similar to abnormalities found in human MODY2 patients [116].
Persistent hyperinsulinemic hypoglycemia of infancy (PHHI). A small number of heterozygous missense GCK mutations were identified as probable cause of hyperinsulinism in infancy, a rare syndrome characterized by chronic low fasting plasma glucose and bouts of symptomatic hypoglycemia [117]. Kinetic studies of the mutant enzymes revealed an increase in glucose affinity with or without increased turnover number (kcat) [118–120]. A gene knock-in strategy was used to produce mice with a GCK allele encoding a hyperactive mutant GCK with increased affinity for glucose. The mice were hyperinsulinemic relative to their plasma glucose levels. However, the severity of hypoglycemia was less than expected, suggesting a compensatory mechanism in the liver. Paradoxically, liver GCK activity and protein content in these mice were lower than normal, suggesting the possibility of a shorter half-life of the mutant enzyme due to impaired interaction with GCKR [121].
Small-molecule activators of glucokinase as potential antidiabetic drugs
Type 2 diabetesmelltus (T2D) is a common disease of multifactorial etiology, in which both a genetic predisposition determined bymultiple genes, and lifestyle factors such as excess calorie intake and low physical activity, play causative roles. The increasing prevalence of T2D, and its serious consequences on health at both the individual and public levels, create incentives for the development of novel antidiabetic drugs. Physiopathologically, T2D consists of defective insulin action in target tissue, including reduced insulin suppression of liver glucose production, and defective insulin secretion and other functions in the β-cells of the islets of Langerhans [122]. The central role of GCK in facilitating glucose disposal by the liver on the one hand, and insulin secretion by the islets of Langerhans on the other hand, provided a strong rationale for the search of small molecule activators of GCK in drug discovery programs aimed at developing a new class of antidiabetic drugs. Such programs were implemented by several major drug companies, and a limited number of molecules acting as GCK activators are currently in early stages of clinical trials.
Small molecule activators of GCK emerged from a high throughput screening strategy based on the capacity of chemicals to increase the activity of recombinant GCK in an enzyme assay mixture containing GCKR. The first promising compound was described in 2003 by Grippo and colleagues [123] and shown actually to be a direct activator of GCK in the absence of GCKR. This compound, designated RO-28-1675, had the dual effects of increasing both the affinity for glucose and the turnover number of GCK, that is, it shifted the glucose S0.5 of GCK from 8.5 mM to 0.8 mM and increased the kcat by approximately 80% at maximal concentration. When added to isolated rat islets of Langerhans, RO-28-1675 stimulated the rate of glycolysis, lowered the threshold for glucose-stimulated insulin secretion and increased the maximal secretory response. A single oral administration of RO-28-1675 to normal or obese mice had a clear-cut hypoglycemic effect accompanied by hyperinsulinemia. Treated mice displayed improved glucose tolerance after an oral glucose load. Endogenous glucose production was reduced during a hyperglycemic hyperinsulinemic clamp in RO-28-1675-treated rats compared to controls, consistent with an effect of the drug on hepatic GCK activity [123]. A variety of chemically distinct small molecule activators of GCK were subsequently discovered in several laboratories. The biochemical and cellular effects, as well as biological actions in experimental animals, were essentially similar to those of the prototype RO-28-1675 [19, 124–126]. An expert review of the current status of GCK activators in drug development was recently published [127].
An implicit hope in developing activators of the physiological glucose sensor was the idea of restoring “physiological” glucose-sensing, particularly in the islet β-cells. However, the strong increase in the glucose affinity of GCK induced by all activators known to date might drastically lower the threshold for insulin release and cause maximal insulin secretion at low plasma glucose levels. Whether the design of drug preparations without liability of inducing serious hypoglycemia is possible remains to be seen. Another concern with GCK activators is related to the activation of hepatic lipogenesis leading to hepatosteatosis (and associated insulin resistance), as was noted in transgenic mice with hepatic overexpression of GCK [128, 129]. Finally, in a more general vein, the long term benefit of using insulinotropic agents in the treatment of T2D (at least early in the disease) was recently questioned, because of concerns about the possibility of precipitating islet failure through hypersecretory activity [130].
Conclusions and perspectives
The evidence presented in this review should leave little doubt about the status of GCK as an eminent player in the regulation of glucose metabolism and homeostasis. This status relies on the mechanism whereby GCK operates as an ultrasensitive glucose sensor, recently illuminated by structural data. The localization of such a sensor inside, rather than on the surface, of cells is striking. It is increasingly clear that the traffic of GCK between distinct subcellular sites, regulated by reversible interactions with diverse scaffold proteins, might contribute to shape the metabolic activity of cells, but by which exact molecular mechanisms remains to be explained, notably in the pancreatic β-cells. In hypothalamic nuclei and other brain areas, it will be important to resolve the apparent paradox of GCK operating under less than optimal glucose concentrations for substrate regulation of the enzyme reaction. Some of the most intriguing unanswered questions pertain to the regulation of GCK gene expression in hepatocytes. The transcriptional induction of GCK is a dramatic effect of insulin in the liver, perhaps the best example of induction of any gene in any cell-type by insulin. However, details about the regulatory proteins of transcription which mediate this effect are still a black box. The burstlike pattern of regulation of GCK transcription in rat liver with very large, rapid and transient inductive effects of insulin for a protein with relatively slow turnover raises puzzling questions with respect to the biological advantage that might arise from this type of regulation. In the diabetes field, the development of GCK activators through the preclinical phase has generated exciting results, but also illustrated some liabilities. If these can be overcome, drugs with a novel mechanism of action will become available as an additional option in the panoply of oral antidiabetics.
Acknowledgement
The long term support of research in the author’s laboratory by the Swiss National Science Foundation is gratefully acknowledged.
Footnotes
Received 11 June 2008; received after revision 18 July 2008; accepted 30 July 2008
References
- 1.Cardenas M. L., Cornish-Bowden A., Ureta T. Evolution and regulatory role of the hexokinases. Biochim. Biophys. Acta. 1998;1401:242–264. doi: 10.1016/S0167-4889(97)00150-X. [DOI] [PubMed] [Google Scholar]
- 2.Kawai S., Mukai T., Mori S., Mikami B., Murata K. Hypothesis: structures, evolution, and ancestor of glucose kinases in the hexokinase family. J. Biosci. Bioeng. 2005;99:320–330. doi: 10.1263/jbb.99.320. [DOI] [PubMed] [Google Scholar]
- 3.Ronimus R. S., Morgan H.W. Cloning and biochemical characterization of a novel mouse ADP-dependent glucokinase. Biochem. Biophys. Res. Commun. 2004;315:652–658. doi: 10.1016/j.bbrc.2004.01.103. [DOI] [PubMed] [Google Scholar]
- 4.Froguel P., Vaxillaire M., Sun F., Velho G., Zouali H., Butel M. O., Lesage S., Vionnet N., Clement K., Fougerousse F., et al. Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent diabetes mellitus. Nature. 1992;356:162–164. doi: 10.1038/356162a0. [DOI] [PubMed] [Google Scholar]
- 5.Hattersley A. T., Turner R. C., Permutt M. A., Patel P., Tanizawa Y., Chiu K. C., O’Rahilly S., Watkins P. J., Wainscoat J. S. Linkage of type 2 diabetes to the glucokinase gene. Lancet. 1992;339:1307–1310. doi: 10.1016/0140-6736(92)91958-B. [DOI] [PubMed] [Google Scholar]
- 6.Iynedjian P. B. Mammalian glucokinase and its gene. Biochem. J. 1993;293(Pt1):1–13. doi: 10.1042/bj2930001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Printz R. L., Magnuson M. A., Granner D. K. Mammalian glucokinase. Annu. Rev. Nutr. 1993;13:463–496. doi: 10.1146/annurev.nu.13.070193.002335. [DOI] [PubMed] [Google Scholar]
- 8.Wilson J. E. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003;206:2049–2057. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- 9.Cardenas M. L., Rabajille E., Niemeyer H. Maintenance of the monomeric structure of glucokinase under reacting conditions. Arch. Biochem. Biophys. 1978;190:142–148. doi: 10.1016/0003-9861(78)90261-8. [DOI] [PubMed] [Google Scholar]
- 10.Moukil M. A., Van Schaftingen E. Analysis of the cooperativity of human beta-cell glucokinase through the stimulatory effect of glucose on fructose phosphorylation. J.Biol. Chem. 2001;276:3872–3878. doi: 10.1074/jbc.M008722200. [DOI] [PubMed] [Google Scholar]
- 11.Monasterio O., Cardenas M. L. Kinetic studies of rat liver hexokinase D (‘glucokinase’) in non-co-operative conditions show an ordered mechanism with MgADP as the last product to be released. Biochem. J. 2003;371:29–38. doi: 10.1042/BJ20020728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Storer A. C., Cornish-Bowden A. Kinetic evidence for a ‘mnemonical’ mechanism for rat liver glucokinase. Biochem. J. 1977;165:61–69. doi: 10.1042/bj1650061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heredia V. V., Thomson J., Nettleton D., Sun S. Glucose-induced conformational changes in glucokinase mediate allosteric regulation: transient kinetic analysis. Biochemistry. 2006;45:7553–7562. doi: 10.1021/bi060253q. [DOI] [PubMed] [Google Scholar]
- 14.Kim Y. B., Kalinowski S. S., Marcinkeviciene J. A pre-steady state analysis of ligand binding to human glucokinase: evidence for a preexisting equilibrium. Biochemistry. 2007;46:1423–1431. doi: 10.1021/bi0617308. [DOI] [PubMed] [Google Scholar]
- 15.Kamata K., Mitsuya M., Nishimura T., Eiki J., Nagata Y. Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase. Structure. 2004;12:429–438. doi: 10.1016/j.str.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Aleshin A. E., Zeng C., Bartunik H. D., Fromm H. J., Honzatko R. B. Regulation of hexokinase I: crystal structure of recombinant human brain hexokinase complexed with glucose and phosphate. J. Mol. Biol. 1998;282:345–357. doi: 10.1006/jmbi.1998.2017. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J., Li C., Chen K., Zhu W., Shen X., Jiang H. Conformational transition pathway in the allosteric process of human glucokinase. Proc. Natl. Acad. Sci. USA. 2006;103:13368–13373. doi: 10.1073/pnas.0605738103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Schaftingen E., Detheux M., Veiga da Cunha M. Short-term control of glucokinase activity: role of a regulatory protein. Faseb. J. 1994;8:414–419. doi: 10.1096/fasebj.8.6.8168691. [DOI] [PubMed] [Google Scholar]
- 19.Futamura M., Hosaka H., Kadotani A., Shimazaki H., Sasaki K., Ohyama S., Nishimura T., Eiki J., Nagata Y. An allosteric activator of glucokinase impairs the interaction of glucokinase and glucokinase regulatory protein and regulates glucose metabolism. J. Biol. Chem. 2006;281:37668–37674. doi: 10.1074/jbc.M605186200. [DOI] [PubMed] [Google Scholar]
- 20.de la Iglesia N., Veiga-da-Cunha M., Van Schaftingen E., Guinovart J. J., Ferrer J. C. Glucokinase regulatory protein is essential for the proper subcellular localisation of liver glucokinase. FEBS Lett. 1999;456:332–338. doi: 10.1016/S0014-5793(99)00971-0. [DOI] [PubMed] [Google Scholar]
- 21.Shiota C., Coffey J., Grimsby J., Grippo J. F., Magnuson M. A. Nuclear import of hepatic glucokinase depends upon glucokinase regulatory protein, whereas export is due to a nuclear export signal sequence in glucokinase. J. Biol. Chem. 1999;274:37125–37130. doi: 10.1074/jbc.274.52.37125. [DOI] [PubMed] [Google Scholar]
- 22.Bosco D., Meda P., Iynedjian P. B. Glucokinase and glucokinase regulatory protein: mutual dependence for nuclear localization. Biochem. J. 2000;348(Pt1):215–222. doi: 10.1042/0264-6021:3480215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guigas B., Bertrand L., Taleux N., Foretz M., Wiernsperger N., Vertommen D., Andreelli F., Viollet B., Hue L. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside and metformin inhibit hepatic glucose phosphorylation by an AMP-activated protein kinase-independent effect on glucokinase translocation. Diabetes. 2006;55:865–874. doi: 10.2337/diabetes.55.04.06.db05-1178. [DOI] [PubMed] [Google Scholar]
- 24.Toyoda Y., Miwa I., Kamiya M., Ogiso S., Nonogaki T., Aoki S., Okuda J. Evidence for glucokinase translocation by glucose in rat hepatocytes. Biochem. Biophys. Res. Commun. 1994;204:252–256. doi: 10.1006/bbrc.1994.2452. [DOI] [PubMed] [Google Scholar]
- 25.Brown K. S., Kalinowski S. S., Megill J. R., Durham S. K., Mookhtiar K. A. Glucokinase regulatory protein may interact with glucokinase in the hepatocyte nucleus. Diabetes. 1997;46:179–186. doi: 10.2337/diabetes.46.2.179. [DOI] [PubMed] [Google Scholar]
- 26.Toyoda Y., Miwa I., Satake S., Anai M., Oka Y. Nuclear location of the regulatory protein of glucokinase in rat liver and translocation of the regulator to the cytoplasm in response to high glucose. Biochem. Biophys. Res. Commun. 1995;215:467–473. doi: 10.1006/bbrc.1995.2488. [DOI] [PubMed] [Google Scholar]
- 27.Mukhtar M., Stubbs M., Agius L. Evidence for glucose and sorbitol-induced nuclear export of glucokinase regulatory protein in hepatocytes. FEBS Lett. 1999;462:453–458. doi: 10.1016/S0014-5793(99)01580-X. [DOI] [PubMed] [Google Scholar]
- 28.Farrelly D., Brown K. S., Tieman A., Ren J., Lira S. A., Hagan D., Gregg R., Mookhtiar K. A., Hariharan N. Mice mutant for glucokinase regulatory protein exhibit decreased liver glucokinase: a sequestration mechanism in metabolic regulation. Proc. Natl. Acad. Sci. USA. 1999;96:14511–14516. doi: 10.1073/pnas.96.25.14511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grimsby J., Coffey J. W., Dvorozniak M. T., Magram J., Li G., Matschinsky F. M., Shiota C., Kaur S., Magnuson M. A., Grippo J. F. Characterization of glucokinase regulatory protein-deficient mice. J. Biol. Chem. 2000;275:7826–7831. doi: 10.1074/jbc.275.11.7826. [DOI] [PubMed] [Google Scholar]
- 30.Arden C., Baltrusch S., Agius L. Glucokinase regulatory protein is associated with mitochondria in hepatocytes. FEBS Lett. 2006;580:2065–2070. doi: 10.1016/j.febslet.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 31.Bustamante E., Pediaditakis P., He L., Lemasters J. J. Isolated mouse liver mitochondria are devoid of glucokinase. Biochem. Biophys. Res. Commun. 2005;334:907–910. doi: 10.1016/j.bbrc.2005.06.174. [DOI] [PubMed] [Google Scholar]
- 32.Munoz-Alonso M. J., Guillemain G., Kassis N., Girard J., Burnol A. F., Leturque A. A novel cytosolic dual specificity phosphatase, interacting with glucokinase, increases glucose phosphorylation rate. J. Biol. Chem. 2000;275:32406–32412. doi: 10.1074/jbc.M000841200. [DOI] [PubMed] [Google Scholar]
- 33.Shiraishi A., Yamada Y., Tsuura Y., Fijimoto S., Tsukiyama K., Mukai E., Toyoda Y., Miwa I., Seino Y. A novel glucokinase regulator in pancreatic beta cells: precursor of propionyl-CoA carboxylase beta subunit interacts with glucokinase and augments its activity. J. Biol. Chem. 2001;276:2325–2328. doi: 10.1074/jbc.C000530200. [DOI] [PubMed] [Google Scholar]
- 34.Baltrusch S., Lenzen S., Okar D. A., Lange A. J., Tiedge M. Characterization of glucokinase-binding protein epitopes by a phage-displayed peptide library. Identification of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase as a novel interaction partner. J Biol Chem. 2001;276:43915–43923. doi: 10.1074/jbc.M105470200. [DOI] [PubMed] [Google Scholar]
- 35.Massa L., Baltrusch S., Okar D. A., Lange A. J., Lenzen S., Tiedge M. Interaction of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK-2/FBPase-2) with glucokinase activates glucose phosphorylation and glucose metabolism in insulin-producing cells. Diabetes. 2004;53:1020–1029. doi: 10.2337/diabetes.53.4.1020. [DOI] [PubMed] [Google Scholar]
- 36.Baltrusch S., Langer S., Massa L., Tiedge M., Lenzen S. Improved metabolic stimulus for glucose-induced insulin secretion through GK and PFK-2/FBPase-2 coexpression in insulin-producing RINm5F cells. Endocrinology. 2006;147:5768–5776. doi: 10.1210/en.2006-0694. [DOI] [PubMed] [Google Scholar]
- 37.Rizzo M. A., Piston D. W. Regulation of beta cell glucokinase by S-nitrosylation and association with nitric oxide synthase. J. Cell. Biol. 2003;161:243–248. doi: 10.1083/jcb.200301063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arden C., Harbottle A., Baltrusch S., Tiedge M., Agius L. Glucokinase is an integral component of the insulin granules in glucose-responsive insulin secretory cells and does not translocate during glucose stimulation. Diabetes. 2004;53:2346–2352. doi: 10.2337/diabetes.53.9.2346. [DOI] [PubMed] [Google Scholar]
- 39.Bjorkhaug L., Molnes J., Sovik O., Njolstad P. R., Flatmark T. Allosteric activation of human glucokinase by free polyubiquitin chains and its ubiquitin-dependent cotranslational proteasomal degradation. J. Biol. Chem. 2007;282:22757–22764. doi: 10.1074/jbc.M700517200. [DOI] [PubMed] [Google Scholar]
- 40.Danial N. N., Gramm C. F., Scorrano L., Zhang C. Y., Krauss S., Ranger A. M., Datta S. R., Greenberg M. E., Licklider L. J., Lowell B. B., Gygi S. P., Korsmeyer S. J. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 2003;424:952–956. doi: 10.1038/nature01825. [DOI] [PubMed] [Google Scholar]
- 41.Danial N. N., Walensky L. D., Zhang C. Y., Choi C. S., Fisher J. K., Molina A. J., Datta S. R., Pitter K. L., Bird G. H., Wikstrom J. D., Deeney J. T., Robertson K., Morash J., Kulkarni A., Neschen S., Kim S., Greenberg M. E., Corkey B. E., Shirihai O. S., Shulman G. I., Lowell B. B., Korsmeyer S. J. Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nat.Med. 2008;14:144–153. doi: 10.1038/nm1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jetton T. L., Liang Y., Pettepher C. C., Zimmerman E. C., Cox F. G., Horvath K., Matschinsky F. M., Magnuson M. A. Analysis of upstream glucokinase promoter activity in transgenic mice and identification of glucokinase in rare neuroendocrine cells in the brain and gut. J. Biol. Chem. 1994;269:3641–3654. [PubMed] [Google Scholar]
- 43.Shelton K. D., Franklin A. J., Khoor A., Beechem J., Magnuson M. A. Multiple elements in the upstream glucokinase promoter contribute to transcription in insulinoma cells. Mol. Cell. Biol. 1992;12:4578–4589. doi: 10.1128/mcb.12.10.4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moates J. M., Magnuson M. A. The Pal elements in the upstream glucokinase promoter exhibit dyad symmetry and display cell-specific enhancer activity when multimerised. Diabetologia. 2004;47:1632–1640. doi: 10.1007/s00125-004-1497-1. [DOI] [PubMed] [Google Scholar]
- 45.Moates J. M., Nanda S., Cissell M. A., Tsai M. J., Stein R. BETA2 activates transcription from the upstream glucokinase gene promoter in islet beta-cells and gut endocrine cells. Diabetes. 2003;52:403–408. doi: 10.2337/diabetes.52.2.403. [DOI] [PubMed] [Google Scholar]
- 46.Watada H., Kajimoto Y., Umayahara Y., Matsuoka T., Kaneto H., Fujitani Y., Kamada T., Kawamori R., Yamasaki Y. The human glucokinase gene beta-cell-type promoter: an essential role of insulin promoter factor 1/PDX-1 in its activation in HIT-T15 cells. Diabetes. 1996;45:1478–1488. doi: 10.2337/diabetes.45.11.1478. [DOI] [PubMed] [Google Scholar]
- 47.Chakrabarti S. K., James J. C., Mirmira R. G. Quantitative assessment of gene targeting in vitro and in vivo by the pancreatic transcription factor, Pdx1. Importance of chromatin structure in directing promoter binding. J. Biol. Chem. 2002;277:13286–13293. doi: 10.1074/jbc.M111857200. [DOI] [PubMed] [Google Scholar]
- 48.Iynedjian P. B., Marie S., Wang H., Gjinovci A., Nazaryan K. Liver-specific enhancer of the glucokinase gene. J. Biol. Chem. 1996;271:29113–29120. doi: 10.1074/jbc.271.46.29113. [DOI] [PubMed] [Google Scholar]
- 49.Iynedjian P. B. Identification of upstream stimulatory factor as transcriptional activator of the liver promoter of the glucokinase gene. Biochem. J. 1998;333(Pt3):705–712. doi: 10.1042/bj3330705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iynedjian P. B., Gjinovci A., Renold A. E. Stimulation by insulin of glucokinase gene transcription in liver of diabetic rats. J. Biol. Chem. 1988;263:740–744. [PubMed] [Google Scholar]
- 51.Iynedjian P. B., Marie S., Gjinovci A., Genin B., Deng S. P., Buhler L., Morel P., Mentha G. Glucokinase and cytosolic phosphoenolpyruvate carboxykinase (GTP) in the human liver. Regulation of gene expression in cultured hepatocytes. J. Clin. Invest. 1995;95:1966–1973. doi: 10.1172/JCI117880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Iynedjian P. B., Roth R. A., Fleischmann M., Gjinovci A. Activation of protein kinase B/cAkt in hepatocytes is sufficient for the induction of expression of the gene encoding glucokinase. Biochem. J. 2000;351(Pt3):621–627. doi: 10.1042/0264-6021:3510621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ribaux P. G., Iynedjian P. B. Analysis of the role of protein kinase B (cAKT) in insulin-dependent induction of glucokinase and sterol regulatory element-binding protein 1 (SREBP1)mRNAs in hepatocytes. Biochem. J. 2003;376:697–705. doi: 10.1042/BJ20031287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iynedjian P. B. Lack of evidence for a role of TRB3/NIPK as an inhibitor of PKB-mediated insulin signalling in primary hepatocytes. Biochem. J. 2005;386:113–118. doi: 10.1042/BJ20041425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu C., Okar D. A., Stoeckman A. K., Peng L. J., Herrera A. H., Herrera J. E., Towle H. C., Lange A. J. A potential role for fructose-2,6-bisphosphate in the stimulation of hepatic glucokinase gene expression. Endocrinology. 2004;145:650–658. doi: 10.1210/en.2003-1290. [DOI] [PubMed] [Google Scholar]
- 56.Matsumoto M., Ogawa W., Teshigawara K., Inoue H., Miyake K., Sakaue H., Kasuga M. Role of the insulin receptor substrate 1 and phosphatidylinositol 3-kinase signaling pathway in insulin-induced expression of sterol regulatory element binding protein 1c and glucokinase genes in rat hepatocytes. Diabetes. 2002;51:1672–1680. doi: 10.2337/diabetes.51.6.1672. [DOI] [PubMed] [Google Scholar]
- 57.Foretz M., Pacot C., Dugail I., Lemarchand P., Guichard C., Le Liepvre X., Berthelier-Lubrano C., Spiegelman B., Kim J. B., Ferre P., Foufelle F. ADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucose. Mol. Cell. Biol. 1999;19:3760–3768. doi: 10.1128/mcb.19.5.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fleischmann M., Iynedjian P. B. Regulation of sterol regulatory-element binding protein 1 gene expression in liver: role of insulin and protein kinase B/cAkt. Biochem. J. 2000;349:13–17. doi: 10.1042/0264-6021:3490013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Foretz M., Guichard C., Ferre P., Foufelle F. Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of glucokinase and lipogenesis-related genes. Proc. Natl. Acad. Sci. USA. 1999;96:12737–12742. doi: 10.1073/pnas.96.22.12737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim S. Y., Kim H. I., Kim T. H., Im S. S., Park S. K., Lee I. K., Kim K. S., Ahn Y. H. SREBP-1c mediates the insulin-dependent hepatic glucokinase expression. J. Biol. Chem. 2004;279:30823–30829. doi: 10.1074/jbc.M313223200. [DOI] [PubMed] [Google Scholar]
- 61.Stoeckman A. K., Towle H. C. The role of SREBP-1c in nutritional regulation of lipogenic enzyme gene expression. J. Biol. Chem. 2002;277:27029–27035. doi: 10.1074/jbc.M202638200. [DOI] [PubMed] [Google Scholar]
- 62.Liang G., Yang J., Horton J. D., Hammer R. E., Goldstein J. L., Brown M. S. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J. Biol. Chem. 2002;277:9520–9528. doi: 10.1074/jbc.M111421200. [DOI] [PubMed] [Google Scholar]
- 63.Ono H., Shimano H., Katagiri H., Yahagi N., Sakoda H., Onishi Y., Anai M., Ogihara T., Fujishiro M., Viana A. Y., Fukushima Y., Abe M., Shojima N., Kikuchi M., Yamada N., Oka Y., Asano T. Hepatic Akt activation induces marked hypoglycemia, hepatomegaly, and hypertriglyceridemia with sterol regulatory element binding protein involvement. Diabetes. 2003;52:2905–2913. doi: 10.2337/diabetes.52.12.2905. [DOI] [PubMed] [Google Scholar]
- 64.Hansmannel F., Mordier S., Iynedjian P. B. Insulin induction of glucokinase and fatty acid synthase in hepatocytes: analysis of the roles of sterol-regulatory-element-binding protein-1c and liver X receptor. Biochem. J. 2006;399:275–283. doi: 10.1042/BJ20060811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roth U., Jungermann K., Kietzmann T. Activation of glucokinase gene expression by hepatic nuclear factor 4alpha in primary hepatocytes. Biochem. J. 2002;365:223–228. doi: 10.1042/BJ20020340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roth U., Curth K., Unterman T. G., Kietzmann T. The transcription factors HIF-1 and HNF-4 and the coactivator p300 are involved in insulin-regulated glucokinase gene expression via the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2004;279:2623–2631. doi: 10.1074/jbc.M308391200. [DOI] [PubMed] [Google Scholar]
- 67.Kietzmann T., Roth U., Freimann S., Jungermann K. Arterial oxygen partial pressures reduce the insulin-dependent induction of the perivenously located glucokinase in rat hepatocyte cultures: mimicry of arterial oxygen pressures by H2O2. Biochem. J. 1997;321(Pt1):17–20. doi: 10.1042/bj3210017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jungermann K., Kietzmann T. Oxygen: modulator of metabolic zonation and disease of the liver. Hepatology. 2000;31:255–260. doi: 10.1002/hep.510310201. [DOI] [PubMed] [Google Scholar]
- 69.Emerling B. M., Weinberg F., Liu J. L., Mak T. W., Chandel N. S. PTEN regulates p300-dependent hypoxia-inducible factor 1 transcriptional activity through Forkhead transcription factor 3a (FOXO3a) Proc.Natl. Acad. Sci. USA. 2008;105:2622–2627. doi: 10.1073/pnas.0706790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matsumoto M., Pocai A., Rossetti L., Depinho R. A., Accili D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor foxo1 in liver. Cell Metab. 2007;6:208–216. doi: 10.1016/j.cmet.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 71.Iynedjian P. B., Ucla C., Mach B. Molecular cloning of glucokinase cDNA. Developmental and dietary regulation of glucokinase mRNA in rat liver. J. Biol. Chem. 1987;262:6032–6038. [PubMed] [Google Scholar]
- 72.Iynedjian P. B., Pilot P. R., Nouspikel T., Milburn J. L., Quaade C., Hughes S., Ucla C., Newgard C. B. Differential expression and regulation of the glucokinase gene in liver and islets of Langerhans. Proc. Natl. Acad. Sci. USA. 1989;86:7838–7842. doi: 10.1073/pnas.86.20.7838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Da Silva Xavier G., Qian Q., Cullen P. J., Rutter G. A. Distinct roles for insulin and insulin-like growth factor-1 receptors in pancreatic beta-cell glucose sensing revealed by RNA silencing. Biochem. J. 2004;377:149–158. doi: 10.1042/BJ20031260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.da Silva Xavier G., Varadi A., Ainscow E. K., Rutter G. A. Regulation of gene expression by glucose in pancreatic beta -cells (MIN6) via insulin secretion and activation of phosphatidylinositol 3′-kinase. J. Biol. Chem. 2000;275:36269–36277. doi: 10.1074/jbc.M006597200. [DOI] [PubMed] [Google Scholar]
- 75.Yoshida K., Murao K., Imachi H., Cao W. M., Yu X., Li J., Ahmed R. A., Kitanaka N., Wong N. C., Unterman T. G., Magnuson M. A., Ishida T. Pancreatic glucokinase is activated by insulin-like growth factor-I. Endocrinology. 2007;148:2904–2913. doi: 10.1210/en.2006-1149. [DOI] [PubMed] [Google Scholar]
- 76.Williams T. F., Exton J. H., Park C. R., Regen D. M. Stereospecific transport of glucose in the perfused rat liver. Am. J. Physiol. 1968;215:1200–1209. doi: 10.1152/ajplegacy.1968.215.5.1200. [DOI] [PubMed] [Google Scholar]
- 77.Seoane J., Gomez-Foix A. M., O’Doherty R. M., Gomez-Ara C., Newgard C. B., Guinovart J. J. Glucose 6-phosphate produced by glucokinase, but not hexokinase I, promotes the activation of hepatic glycogen synthase. J. Biol. Chem. 1996;271:23756–23760. doi: 10.1074/jbc.271.39.23756. [DOI] [PubMed] [Google Scholar]
- 78.Agius L., Peak M., Newgard C. B., Gomez-Foix A. M., Guinovart J. J. Evidence for a role of glucose-induced translocation of glucokinase in the control of hepatic glycogen synthesis. J. Biol. Chem. 1996;271:30479–30486. doi: 10.1074/jbc.271.48.30479. [DOI] [PubMed] [Google Scholar]
- 79.Gomis R. R., Ferrer J. C., Guinovart J. J. Shared control of hepatic glycogen synthesis by glycogen synthase and glucokinase. Biochem. J. 2000;351(Pt3):811–816. doi: 10.1042/0264-6021:3510811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Takeuchi H., Inoue Y., Ishihara H., Oka Y. Overexpression of either liver type or pancreatic beta cell type glucokinase via recombinant adenovirus enhances glucose oxidation in isolated rat hepatocytes. FEBS Lett. 1996;393:60–64. doi: 10.1016/0014-5793(96)00833-2. [DOI] [PubMed] [Google Scholar]
- 81.Seoane J., Barbera A., Telemaque-Potts S., Newgard C. B., Guinovart J. J. Glucokinase overexpression restores glucose utilization and storage in cultured hepatocytes from male Zucker diabetic fatty rats. J. Biol. Chem. 1999;274:31833–31838. doi: 10.1074/jbc.274.45.31833. [DOI] [PubMed] [Google Scholar]
- 82.Niswender K. D., Shiota M., Postic C., Cherrington A. D., Magnuson M. A. Effects of increased glucokinase gene copy number on glucose homeostasis and hepatic glucose metabolism. J. Biol. Chem. 1997;272:22570–22575. doi: 10.1074/jbc.272.36.22570. [DOI] [PubMed] [Google Scholar]
- 83.Ferre T., Pujol A., Riu E., Bosch F., Valera A. Correction of diabetic alterations by glucokinase. Proc. Natl. Acad. Sci. USA. 1996;93:7225–7230. doi: 10.1073/pnas.93.14.7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Postic C., Shiota M., Niswender K. D., Jetton T. L., Chen Y., Moates J. M., Shelton K. D., Lindner J., Cherrington A. D., Magnuson M. A. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem. 1999;274:305–315. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- 85.Rossetti L., Chen W., Hu M., Hawkins M., Barzilai N., Efrat S. Abnormal regulation of HGP by hyperglycemia in mice with a disrupted glucokinase allele. Am. J. Physiol. 1997;273:E743–750. doi: 10.1152/ajpendo.1997.273.4.E743. [DOI] [PubMed] [Google Scholar]
- 86.Henquin J. C., Ravier M. A., Nenquin M., Jonas J. C., Gilon P. Hierarchy of the beta-cell signals controlling insulin secretion. Eur. J. Clin. Invest. 2003;33:742–750. doi: 10.1046/j.1365-2362.2003.01207.x. [DOI] [PubMed] [Google Scholar]
- 87.MacDonald M. J., Fahien L. A., Brown L. J., Hasan N. M., Buss J. D., Kendrick M. A. Perspective: emerging evidence for signaling roles of mitochondrial anaplerotic products in insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2005;288:E1–15. doi: 10.1152/ajpendo.00218.2004. [DOI] [PubMed] [Google Scholar]
- 88.Whitesell R. R., Powers A. C., Regen D. M., Abumrad N. A. Transport and metabolism of glucose in an insulin-secreting cell line, beta TC-1. Biochemistry. 1991;30:11560–11566. doi: 10.1021/bi00113a011. [DOI] [PubMed] [Google Scholar]
- 89.Tal M., Liang Y., Najafi H., Lodish H. F., Matschinsky F. M. Expression and function of GLUT-1 and GLUT-2 glucose transporter isoforms in cells of cultured rat pancreatic islets. J. Biol. Chem. 1992;267:17241–17247. [PubMed] [Google Scholar]
- 90.Schuit F., Moens K., Heimberg H., Pipeleers D. Cellular origin of hexokinase in pancreatic islets. J. Biol. Chem. 1999;274:32803–32809. doi: 10.1074/jbc.274.46.32803. [DOI] [PubMed] [Google Scholar]
- 91.Wang H., Iynedjian P. B. Modulation of glucose responsiveness of insulinoma beta-cells by graded overexpression of glucokinase. Proc. Natl. Acad. Sci. USA. 1997;94:4372–4377. doi: 10.1073/pnas.94.9.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang H., Iynedjian P. B. Acute glucose intolerance in insulinoma cells with unbalanced overexpression of glucokinase. J. Biol. Chem. 1997;272:25731–25736. doi: 10.1074/jbc.272.41.25731. [DOI] [PubMed] [Google Scholar]
- 93.Berman H. K., Newgard C. B. Fundamental metabolic differences between hepatocytes and islet betacells revealed by glucokinase overexpression. Biochemistry. 1998;37:4543–4552. doi: 10.1021/bi9726133. [DOI] [PubMed] [Google Scholar]
- 94.Terauchi Y., Sakura H., Yasuda K., Iwamoto K., Takahashi N., Ito K., Kasai H., Suzuki H., Ueda O., Kamada N., et al. Pancreatic beta-cell-specific targeted disruption of glucokinase gene. Diabetes mellitus due to defective insulin secretion to glucose. J. Biol. Chem. 1995;270:30253–30256. doi: 10.1074/jbc.270.51.30253. [DOI] [PubMed] [Google Scholar]
- 95.Yang X. J., Kow L. M., Funabashi T., Mobbs C. V. Hypothalamic glucose sensor: similarities to and differences from pancreatic beta-cell mechanisms. Diabetes. 1999;48:1763–1772. doi: 10.2337/diabetes.48.9.1763. [DOI] [PubMed] [Google Scholar]
- 96.Roncero I., Alvarez E., Vazquez P., Blazquez E. Functional glucokinase isoforms are expressed in rat brain. J. Neurochem. 2000;74:1848–1857. doi: 10.1046/j.1471-4159.2000.0741848.x. [DOI] [PubMed] [Google Scholar]
- 97.Dunn-Meynell A. A., Routh V. H., Kang L., Gaspers L., Levin B. E. Glucokinase is the likely mediator of glucosensing in both glucose-excited and glucose-inhibited central neurons. Diabetes. 2002;51:2056–2065. doi: 10.2337/diabetes.51.7.2056. [DOI] [PubMed] [Google Scholar]
- 98.Kang L., Routh V. H., Kuzhikandathil E. V., Gaspers L. D., Levin B. E. Physiological and molecular characteristics of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes. 2004;53:549–559. doi: 10.2337/diabetes.53.3.549. [DOI] [PubMed] [Google Scholar]
- 99.Kang L., Dunn-Meynell A. A., Routh V. H., Gaspers L. D., Nagata Y., Nishimura T., Eiki J., Zhang B. B., Levin B. E. Glucokinase is a critical regulator of ventromedial hypothalamic neuronal glucosensing. Diabetes. 2006;55:412–420. doi: 10.2337/diabetes.55.02.06.db05-1229. [DOI] [PubMed] [Google Scholar]
- 100.Parton L. E., Ye C. P., Coppari R., Enriori P. J., Choi B., Zhang C. Y., Xu C., Vianna C. R., Balthasar N., Lee C. E., Elmquist J. K., Cowley M. A., Lowell B. B. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature. 2007;449:228–232. doi: 10.1038/nature06098. [DOI] [PubMed] [Google Scholar]
- 101.Wolfgang M. J., Lane M. D. The role of hypothalamic malonyl-CoA in energy homeostasis. J. Biol. Chem. 2006;281:37265–37269. doi: 10.1074/jbc.R600016200. [DOI] [PubMed] [Google Scholar]
- 102.Velho G., Blanche H., Vaxillaire M., Bellanne-Chantelot C., Pardini V. C., Timsit J., Passa P., Deschamps I., Robert J. J., Weber I. T., Marotta D., Pilkis S. J., Lipkind G. M., Bell G. I., Froguel P. Identification of 14 new glucokinase mutations and description of the clinical profile of 42 MODY-2 families. Diabetologia. 1997;40:217–224. doi: 10.1007/s001250050666. [DOI] [PubMed] [Google Scholar]
- 103.Fajans S. S., Bell G. I., Polonsky K. S. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N. Engl. J. Med. 2001;345:971–980. doi: 10.1056/NEJMra002168. [DOI] [PubMed] [Google Scholar]
- 104.Ellard S., Beards F., Allen L. I., Shepherd M., Ballantyne E., Harvey R., Hattersley A. T. A high prevalence of glucokinase mutations in gestational diabetic subjects selected by clinical criteria. Diabetologia. 2000;43:250–253. doi: 10.1007/s001250050038. [DOI] [PubMed] [Google Scholar]
- 105.Murphy R., Ellard S., Hattersley A. T. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol. Metab. 2008;4:200–213. doi: 10.1038/ncpendmet0778. [DOI] [PubMed] [Google Scholar]
- 106.Njolstad P. R., Sagen J. V., Bjorkhaug L., Odili S., Shehadeh N., Bakry D., Sarici S. U., Alpay F., Molnes J., Molven A., Sovik O., Matschinsky F. M. Permanent neonatal diabetes caused by glucokinase deficiency: inborn error of the glucose-insulin signaling pathway. Diabetes. 2003;52:2854–2860. doi: 10.2337/diabetes.52.11.2854. [DOI] [PubMed] [Google Scholar]
- 107.Velho G., Petersen K. F., Perseghin G., Hwang J. H., Rothman D. L., Pueyo M. E., Cline G. W., Froguel P., Shulman G. I. Impaired hepatic glycogen synthesis in glucokinase-deficient (MODY-2) subjects. J. Clin. Invest. 1996;98:1755–1761. doi: 10.1172/JCI118974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tappy L., Dussoix P., Iynedjian P., Henry S., Schneiter P., Zahnd G., Jequier E., Philippe J. Abnormal regulation of hepatic glucose output in maturity-onset diabetes of the young caused by a specific mutation of the glucokinase gene. Diabetes. 1997;46:204–208. doi: 10.2337/diabetes.46.2.204. [DOI] [PubMed] [Google Scholar]
- 109.Byrne M. M., Sturis J., Clement K., Vionnet N., Pueyo M. E., Stoffel M., Takeda J., Passa P., Cohen D., Bell G. I., et al. Insulin secretory abnormalities in subjects with hyperglycemia due to glucokinase mutations. J. Clin. Invest. 1994;93:1120–1130. doi: 10.1172/JCI117064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Miller S. P., Anand G. R., Karschnia E. J., Bell G. I., LaPorte D. C., Lange A. J. Characterization of glucokinase mutations associated with maturity-onset diabetes of the young type 2 (MODY-2): different glucokinase defects lead to a common phenotype. Diabetes. 1999;48:1645–1651. doi: 10.2337/diabetes.48.8.1645. [DOI] [PubMed] [Google Scholar]
- 111.Davis E. A., Cuesta-Munoz A., Raoul M., Buettger C., Sweet I., Moates M., Magnuson M. A., Matschinsky F. M. Mutants of glucokinase cause hypoglycaemiaand hyperglycaemia syndromes and their analysis illuminates fundamental quantitative concepts of glucose homeostasis. Diabetologia. 1999;42:1175–1186. doi: 10.1007/s001250051289. [DOI] [PubMed] [Google Scholar]
- 112.Galan M., Vincent O., Roncero I., Azriel S., Boix-Pallares P., Delgado-Alvarez E., Diaz-Cadorniga F., Blazquez E., Navas M. A. Effects of novel maturity-onset diabetes of the young (MODY)-associated mutations on glucokinase activity and protein stability. Biochem. J. 2006;393:389–396. doi: 10.1042/BJ20051137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Burke C. V., Buettger C. W., Davis E. A., McClane S. J., Matschinsky F. M., Raper S. E. Cell-biological assessment of human glucokinase mutants causing maturity-onset diabetes of the young type 2 (MODY-2) or glucokinase-linked hyperinsulinaemia (GK-HI) Biochem. J. 1999;342(Pt2):345–352. doi: 10.1042/0264-6021:3420345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Toye A. A., Moir L., Hugill A., Bentley L., Quarterman J., Mijat V., Hough T., Goldsworthy M., Haynes A., Hunter A. J., Browne M., Spurr N., Cox R. D. A new mouse model of type 2 diabetes, produced by N-ethylin the glucokinase gene. Diabetes. 2004;53:1577–1583. doi: 10.2337/diabetes.53.6.1577. [DOI] [PubMed] [Google Scholar]
- 115.Aigner B., Rathkolb B., Herbach N., Hrabe de Angelis M., Wanke R., Wolf E. Diabetes models by screen for hyperglycemia in phenotype-driven ENU mouse mutagenesis projects. Am. J. Physiol. Endocrinol. Metab. 2008;294:E232–240. doi: 10.1152/ajpendo.00592.2007. [DOI] [PubMed] [Google Scholar]
- 116.Inoue M., Sakuraba Y., Motegi H., Kubota N., Toki H., Matsui J., Toyoda Y., Miwa I., Terauchi Y., Kadowaki T., Shigeyama Y., Kasuga M., Adachi T., Fujimoto N., Matsumoto R., Tsuchihashi K., Kagami T., Inoue A., Kaneda H., Ishijima J., Masuya H., Suzuki T., Wakana S., Gondo Y., Minowa O., Shiroishi T., Noda T. A series of maturity onset diabetes of the young, type 2 (MODY2) mouse models generated by a large-scale ENU mutagenesis program. Hum. Mol. Genet. 2004;13:1147–1157. doi: 10.1093/hmg/ddh133. [DOI] [PubMed] [Google Scholar]
- 117.Glaser B., Kesavan P., Heyman M., Davis E., Cuesta A., Buchs A., Stanley C. A., Thornton P. S., Permutt M. A., Matschinsky F. M., Herold K. C. Familial hyperinsulinism caused by an activating glucokinase mutation. N. Engl. J. Med. 1998;338:226–230. doi: 10.1056/NEJM199801223380404. [DOI] [PubMed] [Google Scholar]
- 118.Gloyn A. L., Noordam K., Willemsen M. A., Ellard S., Lam W. W., Campbell I. W., Midgley P., Shiota C., Buettger C., Magnuson M. A., Matschinsky F. M., Hattersley A. T. Insights into the biochemical and genetic basis of glucokinase activation from naturally occurring hypoglycemia mutations. Diabetes. 2003;52:2433–2440. doi: 10.2337/diabetes.52.9.2433. [DOI] [PubMed] [Google Scholar]
- 119.Cuesta-Munoz A. L., Huopio H., Otonkoski T., Gomez-Zumaquero J. M., Nanto-Salonen K., Rahier J., Lopez-Enriquez S., Garcia-Gimeno M. A., Sanz P., Soriguer F. C., Laakso M. Severe persistent hyperinsulinemic hypoglycemia due to a de novo glucokinase mutation. Diabetes. 2004;53:2164–2168. doi: 10.2337/diabetes.53.8.2164. [DOI] [PubMed] [Google Scholar]
- 120.Wabitsch M., Lahr G., Van de Bunt M., Marchant C., Lindner M., von Puttkamer J., Fenneberg A., Debatin K. M., Klein R., Ellard S., Clark A., Gloyn A. L. Heterogeneity in disease severity in a family with a novel G68V GCK activating mutation causing persistent hyperinsulinaemic hypoglycaemia of infancy. Diabet. Med. 2007;24:1393–1399. doi: 10.1111/j.1464-5491.2007.02285.x. [DOI] [PubMed] [Google Scholar]
- 121.Pino M. F., Kim K. A., Shelton K. D., Lindner J., Odili S., Li C., Collins H.W., Shiota M., Matschinsky F. M., Magnuson M. A. Glucokinase thermolability and hepatic regulatory protein binding are essential factors for predicting the blood glucose phenotype of missense mutations. J. Biol. Chem. 2007;282:13906–13916. doi: 10.1074/jbc.M610094200. [DOI] [PubMed] [Google Scholar]
- 122.Muoio D. M., Newgard C. B. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat. Rev. Mol. Cell. Biol. 2008;9:193–205. doi: 10.1038/nrm2327. [DOI] [PubMed] [Google Scholar]
- 123.Grimsby J., Sarabu R., Corbett W. L., Haynes N. E., Bizzarro F. T., Coffey J. W., Guertin K. R., Hilliard D.W., Kester R. F., Mahaney P. E., Marcus L., Qi L., Spence C. L., Tengi J., Magnuson M. A., Chu C. A., Dvorozniak M. T., Matschinsky F. M., Grippo J. F. Allosteric activators of glucokinase: potential role in diabetes therapy. Science. 2003;301:370–373. doi: 10.1126/science.1084073. [DOI] [PubMed] [Google Scholar]
- 124.Brocklehurst K. J., Payne V. A., Davies R. A., Carroll D., Vertigan H. L., Wightman H. J., Aiston S., Waddell I. D., Leighton B., Coghlan M. P., Agius L. Stimulation of hepatocyte glucose metabolism by novel small molecule glucokinase activators. Diabetes. 2004;53:535–541. doi: 10.2337/diabetes.53.3.535. [DOI] [PubMed] [Google Scholar]
- 125.Efanov A. M., Barrett D. G., Brenner M. B., Briggs S. L., Delaunois A., Durbin J. D., Giese U., Guo H., Radloff M., Gil G. S., Sewing S., Wang Y., Weichert A., Zaliani A., Gromada J. A novel glucokinase activator modulates pancreatic islet and hepatocyte function. Endocrinology. 2005;146:3696–3701. doi: 10.1210/en.2005-0377. [DOI] [PubMed] [Google Scholar]
- 126.Fyfe M. C., White J. R., Taylor A., Chatfield R., Wargent E., Printz R. L., Sulpice T., McCormack J. G., Procter M. J., Reynet C., Widdowson P. S., Wong-Kai-In P. Glucokinase activator PSN-GK1 displays enhanced antihyperglycaemic and insulinotropic actions. Diabetologia. 2007;50:1277–1287. doi: 10.1007/s00125-007-0646-8. [DOI] [PubMed] [Google Scholar]
- 127.Coghlan M., Leighton B. Glucokinase activators in diabetes management. Expert Opin. Investig. Drugs. 2008;17:145–167. doi: 10.1517/13543784.17.2.145. [DOI] [PubMed] [Google Scholar]
- 128.O’Doherty R. M., Lehman D. L., Telemaque-Potts S., Newgard C. B. Metabolic impact of glucokinase overexpression in liver: lowering of blood glucose in fed rats is accompanied by hyperlipidemia. Diabetes. 1999;48:2022–2027. doi: 10.2337/diabetes.48.10.2022. [DOI] [PubMed] [Google Scholar]
- 129.Ferre T., Riu E., Franckhauser S., Agudo J., Bosch F. Long-term overexpression of glucokinase in the liver of transgenic mice leads to insulin resistance. Diabetologia. 2003;46:1662–1668. doi: 10.1007/s00125-003-1244-z. [DOI] [PubMed] [Google Scholar]
- 130.Aston-Mourney K., Proietto J., Morahan G., Andrikopoulos S. Too much of a good thing: why it is bad to stimulate the beta cell to secrete insulin. Diabetologia. 2008;51:540–545. doi: 10.1007/s00125-008-0930-2. [DOI] [PubMed] [Google Scholar]