

Abstract
As technology has evolved available guidelines for normal-phase flash chromatography have become less relevant. Years of experience performing chromatography with disposable columns have been condensed into simple guidelines useful for translating TLC results into either isocratic- or gradient-flash chromatography. The described studies should provide researchers with a means of selecting adequate columns and guidelines to reduce the waste of solvents, silica, time, and money.
Electronic supplementary material
The online version of this article (doi:10.1007/s11030-008-9104-x) contains supplementary material, which is available to authorized users.
Keywords: Silica gel, Flash chromatography
Introduction
Silica gel flash chromatography has become ubiquitous within organic chemistry and since its formal introduction in 1978 [1], chemists have used simple guidelines and personal experience to direct the appropriate choice of mobile phases and sample:silica ratios. As technology has evolved flash chromatography has seen an increasing use of pumps and disposable columns and preexisting guidelines have not always translated well across technologies. This has resulted in enormous waste as countless samples have needed to be further purified resulting in excessive use of silica, solvents, and time. We wish to describe some general guidelines useful for preparative separations using disposable flash columns and solvent pumps. The guidelines we suggest include column quality, sample loading, and translation of TLC results to simple isocratic or gradient use.
Many of the tenets of flash chromatography hold true regardless of the means of application. All other things being equal: (1) increasing quantities of analytes result in poorer resolution [1,2], (2) columns have an optimal flow rate determined by their geometry and silica quality (longer and narrower columns provide more theoretical plates) [3,4], (3) more homogenous stationary phases pack better and provide better resolution and more reproducible results [5], (4) stationary phases with more surface area (smaller particle sizes) generally afford better resolution [3–5]. Three of these factors are determined solely by column choice. As such, we first set out to evaluate commercially available, normal-phase, disposable chromatography columns.
Flash chromatography is not expected to provide the resolution or reproducibility of HPLC; it is a technique that can quickly improve the purity of samples to an acceptable level. Many of the equations used to describe GC and HPLC separations were developed for symmetric Gaussian peaks and application of these equations to flash chromatography requires a statistically relevant data set if such a comparison is relevant at all [6,7]. Therefore, we initially sought to identify an optimal flow rate for our studies and to determine the relevance of our data.
Initial experiments showed some variability with types of column pretreatment, methods of application, and choices of instrument. As such, all comparisons were run on the same instruments and no column pretreatments were performed. Samples were dry-loaded onto columns using 400 mg of a 1:1 mixture of acetophenone and 4-methoxyacetophenone dried onto a 4-fold, by mass, excess of 60-mesh silica gel. SINGLE StEP™(patented) columns from Thomson Instrument Company [8] were chosen for this evaluation because of the ease with which they can be dry-loaded and used. Three columns were run at each 30, 35, 40, 45, and 50 mL/min and eluent was monitored at 254 nm for elution of acetophenone and 4-methoxyacetophenone. The data from three independent runs at each flow rate provided a relevant Van Deemter plot and clearly showed that the optimal flow rate for the column was 40 mL/min [9].1 For comparison we chose commercially available columns with 230–400 mesh irregular silica gel of the same apparent quality. Columns with spherical or finer silica gel may provide better resolution; however, such columns would produce higher back pressures which may preclude use on certain systems and may afford more fine silica contamination.
Our primary focus in evaluating columns was for efficiency which could result from improved resolution (R
s), shorter elution time, or decreased solvent use. Improving any of these factors should result in a lower cost per separation. Users can choose for higher recoveries, higher purity, or the most practical balance of the two. Resolution is generally defined as the distance between band centers t
R divided by the relative separation of the bands.2 It has been shown that R
s can be related to the conditions of separation (Eq. 1) [10]. Here k is the column capacity factor (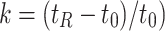 ) which is the amount of compound interacting with the stationary phase divided by the amount in the mobile phase at equilibrium.3
N is the theoretical plate number, which can be calculated [11], and \ga is the separation factor equal to k
2/k
1.
) which is the amount of compound interacting with the stationary phase divided by the amount in the mobile phase at equilibrium.3
N is the theoretical plate number, which can be calculated [11], and \ga is the separation factor equal to k
2/k
1.
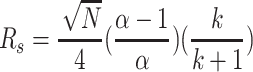 |
1 |
The utility of Eq. 1 is that it relates R s to the interdependent and separable variables k, \ga, and N. Each of these variables has a defined effect on chromatography as is shown in Fig. 1 [12].
Fig. 1.

The effect of column variables on column performance [12]. Reproduced with permission from Preston Publications
For commercial columns plate number N is largely dependant on column choice. More theoretical plates afford better resolution but N should not be considered most important when comparing columns. The mobile phase selectivity factor α has the largest effect on resolution, however, α must be optimized relative to k and this is all subject to solvent choice [13]. Increasing α results in slightly increased retention times and peak widths along with improved resolution. The column capacity factor k is thought to be best between 1 and 5 and even when resolution is possible with k < 1 results are usually unacceptable. Increasing k increases resolution but also increases retention time and peak broadening.
A comparison of four commercially available columns was undertaken: the 40 g Isco® RediSep™ (patented), 40 g Silicycle® SiliaFLASH™, 40 g Thomson Instrument Company® SINGLE StEP™, and the 50 g Biotage® SNAP™ column (Table 1). These columns were chosen because of the frequency that samples between 1 and 2 g are purified in our laboratories. All columns were compared using the same conditions at a flow rate of 40 mL/min. The SINGLE StEP™ and SNAP™ cartridges can be opened to have samples applied directly, however, all columns were run under identical conditions where the samples were dry loaded into empty 4 g columns which were attached in series. Similar experiments were performed with a loading of 2 g per column and an independent analysis was performed using 2 g of 1:1 2-nitroaniline and 4-nitroaniline on a Teledyne Isco CombiFlash® Companion. For each run either 4-methoxyacetophenone or 4-nitroanilinewas collected in order to determine sample recovery.
Table 1.
Performance of commercially available columns (n = 3)
| Column | Acetophenone—4-Methoxyacetophenone 400mg | Acetophenone—4-Methoxyacetophenone 2 g | 2-Nitroaniline—4-Nitroanilinea 2 g | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| k | α | N | R b s | k | α | N | R b s | k | α | N | R b s | |
| Biotage® SNAP™ | 8.22 | 2.81 | ≈54 | c | 7.67 | 0.59d | c | c | 7.23 | 3.34 | c | c |
| ISCO® RediSep™ | 6.55 | 3.00 | 122 | 0.29c | 6.24 | 0.71 | 43 | 0.67 | 6.28 | 3.05 | 13.7 | 1.23 |
| Silicycle® SiliaFLASH™ | 6.28 | 2.96 | 153 | 2.49c | 5.96 | 0.68 | 59 | 1.62 | 6.11 | 3.29 | 15.3 | 1.39 |
| Thomson SINGLE StEP™ | 5.29 | 3.35 | 137 | 2.39c | 5.76 | 1.03 | 53 | 1.66 | 4.44 | 2.91 | 18.7 | 1.20 |
a Estimated from printed data
b From Eq. 1
c Not calculated due to insufficient resolution
d (n = 2)
Most columns showed similar sample recovery, however, these samples were not analyzed for silica contamination. The SNAP™ columns showed slightly lower recovery than all other columns and this was attributed to peak broadening or poor separation. In our experiments values of k ranged between 4 and 7 with optimal α values between 1 and 3. Higher values of k corresponded to samples with more peak broadening, longer retention times, and more column fractions for recovery.
Larger values of R s were sometimes attributed to samples that displayed extensive peak broadening. Optimal values for R s, in our experiments, appeared to range between 1 and 2. Since purification time and solvent use are costly, the time for elution for respective columns is shown in Fig. 2. Data from
Fig. 2.
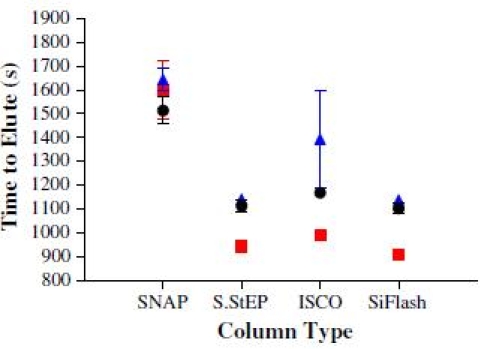
Times for samples to elute (n = 3): (▪) Acetophenones—400 mg; (≆) Acetophenones—2 g; (•) Anilines—2 g
Biotage® SNAP™ columns were difficult to analyze because peaks failed to resolve well and showed significant tailing. Overall, the SNAP™ columns showed the poorest performance in our experiments. The two best performing columns were the SiliaFLASH™ and SINGLE StEP™ columns. It is important to note that four specific columns were compared using samples that we felt relevant to standard chromatography and medicinal chemistry. Similar studies with different sized columns or other standards could provide different results. Column efficiency should be measured by more than R s as time to elute (solvent use and disposal) is also important.
Column choice
There are currently two design styles for disposable columns, those with headspace for dry-loading and those with minimal headspace which are typically sealed at the top with a 1/8” luer lock port.4 Our results have demonstrated that either of these design styles can perform well since the best performing columns tested, the SiliaFLASH™ and SINGLE StEP™ columns, showed similar resolutions and recoveries. Removable-top columns obviate the need for a secondary column and can be evenly loaded. Most samples, once adhered to silica and subjected to a mobile phase under positive pressure, do not partition back into the headspace of a column (Hill DC, unpublished research). Headspace does not appear to affect resolution.
The void volume of a column, in mL, can be approximated as 1 g SiO2 = 2 mL (CV). Since manufactured columns are similar we have recommended reasonable flow rates which should approach optimal (Table 2).
Table 2.
Recommended flow rates for commercial columns
| Column size (g) | 4 | 12 | 25 | 40 | 80 | 110 | 160 | 240 | 300 |
| Flow rate (mL/min) | 12 | 18 | 25 | 40 | 55 | 55 | 65 | 85 | 95 |
| Void volume (CV in mL) | 8 | 24 | 50 | 80 | 160 | 220 | 320 | 480 | 600 |
Solvent choice
It is most common to use TLC for solvent choice and method development with flash chromatography. The amount of material that can be adequately separated on a column is dependant on both the R f of the analyte of interest and the difference in observed R f between components (\gDR f). When translating a TLC method to a flash method column volumes (CV) are used instead of R f. CV is defined as 1/R f. and a CV is literally the volume of solvent required to elute a non-retained solute. For columns this is the same as void volume and most column manufacturers provide this information. As an example, a sample with an R f of 0.1 (CV= 10) requires 10 column volumes to elute the sample. As such, ΔCV = (1/R f2 − 1/R f1) is more directly related to resolution than is \gDR f. Since the requirements for translating from TLC to flash chromatography are different depending on the choice of elution method appropriate TLC characteristics are described with individual methods.
Substituting the polar solvent of a solvent combination typically results in a change in resolution while substituting the less polar solvent will change the R f of the components almost equally. Common binary solvent systems include ethyl acetate-hexanes, acetone-hexanes, acetonitrile-CH2Cl2, and CH2Cl2-methanol.5
The rule of half
For a given analyte R f in X% polar solvent, the R f should be about R f/2 when X/2. That is to say that an R f of about 0.4 in 40% ethyl acetate-hexanes should be about 0.2 in 20% and about 0.1 in 10% ethyl acetate-hexanes. This rule is only applicable when the weak solvent does not move the sample through the stationary phase.
Sample loading
It is noteworthy that when columns are packed by hand they are generally wetted with solvent prior to chromatography in order to better pack the stationary phase. For most separations on commercial columns pretreatment is no longer required; samples can typically be applied directly to dry columns. It is, however, practical to pretreat larger commercial columns (⩾120 g) and pretreatment can be useful when fractions are collected by automation that does not tolerate air in the solvent lines.
There are two common ways to load commercial columns. Samples are most commonly dissolved in a minimal amount of an appropriate solvent and wet-loaded. Excessive amounts of solvent can result in band broadening and loss of resolution. Samples should be applied as evenly as possible to the top of the column. When wet-loading samples which display a ΔCV larger than 2, and appropriate R′fs, we recommend sample loading of 1:20 (mass:mass). Typical solvents used for wet loading include CH2Cl2 and acetone, however, lower polarity solvents can be used when convenient. When component separation is less than ΔCV=2, or initial R f is higher than suggested, more silica should be used. With appropriate choice of mobile phase most ratios should not need to exceed 1:40. These guidelines are offered with the intent of affording a resolution between 4 and 6σ with 6σ being baseline resolution [3].
Samples can also be dried onto silica gel for loading. When this is done a 2- to 10-fold excess (by mass) of silica is generally appropriate. With some columns silica can be placed inside the column and sealed with a frit. Other columns require that dry silica be placed in an empty column, sealed into place with a frit, and attached in series. Whenever samples are dried onto silica it is important that the ratio of sample-silica to chromatographic-silica be appropriate. While a 1:20 ratio of sample to silica is appropriate for wet-loading a 1:40 ratio may be more appropriate when dry loading.
When samples are applied to columns from a solution more polar than initial separation conditions it is useful to begin elution with nonpolar solvents. In practice, we recommend eluting 1–2CV of non-polar solvent, or gradient initial conditions, prior to beginning a separation.
Isocratic elution
For a two component mixture a solvent system that affords ΔCV ≥ 2 is typically appropriate for translation to isocratic flash chromatography. For ΔR f = 0.1, this requires that samples have an R f between 0.1 and 0.3. For a ΔR f of 0.2 samples must have an R f between 0.45 and 0.1. For a two component mixture with a ΔR f = 0.1 and an upper R f of 0.5 in a solvent system that is X% polar solvent, the rule of half suggests that an appropriate solvent choice for isocratic flash chromatography should be between 1/2X and 1/5X. Since the weak solvent may move components across the stationary phase we recommend testing the system by TLC at 1/4X and/or 1/5X in order to demonstrate the utility of a mobile phase for translation to flash chromatography.
Linear gradients
The widespread use of technology for flash chromatography has led many users to opt for gradient elution. Some of the many reasons for running gradients include: (1) reduced separation time, (2) reduced solvent waste, (3) improved separation of analytes (both polar and nonpolar samples can be resolved) [14], and (4) regardless of gradient choice, samples are likely to be eluted in a state of higher purity. Linear gradients are simple to program and samples are often eluted in fewer fractions than observed for similar isocratic methods. The initial R f of samples can vary more for gradient elution and in practice we recommend an R f between 0.1 and 0.5. When analytes show a ΔR f of ⩾0.1 (ΔCV ≥ 2) and the components of interest lie in the preferred TLC zone, with X% polar solvent, the following steps should provide reasonable sample separation [15].
Divide X/4 for initial gradient conditions or set them to zero if X/4 approaches zero.
Wash the column with 1 CV of weak solvent or initial mixture.
Program a gradient of 10 CV from X/4 to X×2
Hold at X×2 for 1 CV
Results
For acetophenone and 4-methoxyacetophenone samples displayed appropriate R f values for isocratic elution) in 10% ethyl acetate and appropriate values for gradient elution in 20%ethyl acetate-hexanes [16].6 Sample mixtures (1:1) were run under recommended isocratic and gradient conditions with a column loading of 1:20; 1 g of each component was dissolved and applied to a 40 g column and the results are shown in Fig. 3.
Fig. 3.
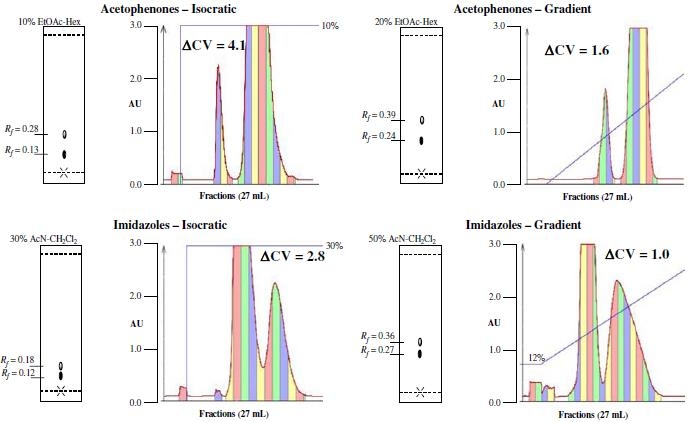
Representative separations. Each fraction is 27 mL
 A pair of polar imidazole containing compounds was used to further demonstrate the utility of our guidelines. These samples run quite close on TLC using acetonitrile-CH2Cl2 however we were able to obtain adequate separations using our guidelines. The poor solubility of the sulfone containing compound necessitated dry loading and the samples were run at 1:20 (sample:column silica) loading, after drying a 2 g mixture onto 4 g of 60-mesh silica. The silica-sample mixture was then sealed onto the top of a dry 40 g SINGLE StEP™ column using a head-space frit.
A pair of polar imidazole containing compounds was used to further demonstrate the utility of our guidelines. These samples run quite close on TLC using acetonitrile-CH2Cl2 however we were able to obtain adequate separations using our guidelines. The poor solubility of the sulfone containing compound necessitated dry loading and the samples were run at 1:20 (sample:column silica) loading, after drying a 2 g mixture onto 4 g of 60-mesh silica. The silica-sample mixture was then sealed onto the top of a dry 40 g SINGLE StEP™ column using a head-space frit.
Summary
In an attempt to help researchers choose suitable commercial columns we have described a method for comparing available (silica) flash chromatography columns. General methods for TLC development and translation to either isocratic or gradient elution have been described and recommendations for dry-loading and solvent loading have also been described. These recommendations should prove useful for translating TLC results to either isocratic or gradient flash chromatography methods. Taken together this information should help researchers reduce waste of solvent (use and disposal), silica, time, and money.
Electronic supplementary material
The Below is the Electronic Supplementary Material.
Acknowledgments
Deepest thanks to Samuel A. Ellis of Thomson Instrument Company (TIC) for encouraging this project and for donating time, materials, and expertise. We thank Ken Bordeau (Sanofi- Aventis), Melissa Ellis (TIC), Prof. Erik Sorensen (Princeton University), and Douglas McLeod (Princeton University and Bristol-Myers Squibb) for support, assistance, and critical reviews. This work was supported by Exelixis, Inc. and AstraZeneca Pharmaceuticals LP.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
The equation used for the van Deemter plot, shown in the supplementary material, squares the data and errors making it difficult to obtain a relevant data set.
R s = 2(t R2 − t R1)/(w 1 + w 2)
N = 16t 2R/w 2.
We have encountered many problems using columns that have luer ports, especially when samples precipitate on the column during loading.
With CH2Cl2-methanol we have had mixed results. Washing commercial columns with 10% methanol in CH2Cl2 to remove water and polar contaminants then washing with CH2Cl2 to remove methanol has occasionally afforded columns which provide adequate resolution.
TLC results are highly variable relative to individual techniques.
References
- 1.Still CW, Kahn M, Mitra AJ. Rapid chromatographic technique for preparative separations with moderate resolution. J Org Chem. 1978;43:2923–2925. doi: 10.1021/jo00408a041. [DOI] [Google Scholar]
- 2.Cox GB, Snyder LR. Preparative high-performance liquid chromatography under isocratic conditions. II. The consequences of two adjacent bands having unequal column capacities. J Chromatogr. 1989;483:95–110. doi: 10.1016/S0021-9673(01)93113-9. [DOI] [Google Scholar]
- 3.McGuffin VL. Chromatography. 6. Oxford: Elsevier; 2004. [Google Scholar]
- 4.Snyder LR. A rapid approach to selecting the best experimental conditions for high-speed liquid column chromatography. III. Small-particle columns. J Chromatogr. 1977;15:441–449. [Google Scholar]
- 5.Wellings DA. Preparative HPLC. Oxford: Elsevier; 2006. [Google Scholar]
- 6.Stout RW, DeStefano JJ, Snyder LR. High-performance liquid chromatographic column efficiency as a function of particle composition and geometry and capacity factor. J Chromatogr. 1983;282:263–286. doi: 10.1016/S0021-9673(00)91607-8. [DOI] [Google Scholar]
- 7.Snyder LR. Rapid approach to selecting the best experimental conditions for high-speed liquid column chromatography. I. Estimating initial sample resolution and the final resolution required by a given problem. J Chromatogr Sci. 1972;10:200–212. [Google Scholar]
- 8.Thomson Instrument Company: http://www.htslabs.com
- 9.van Deemter JJ, Zuiderweg FJ, Klinkenberg A. Longitudinal diffusion and resistance to mass transfer as a cause of nonideality in chromatography. Chem Eng Sci. 1956;5:271–289. doi: 10.1016/0009-2509(56)80003-1. [DOI] [Google Scholar]
- 10.Purnell H. Gas chromatography. New York: Wiley; 1962. [Google Scholar]
- 11.McNaught AD, Wilkinson A, editors. IUPAC compendium of chemical technology. 2. Oxford: Blackwell Science; 1997. [Google Scholar]
- 12.Snyder LR. Rapid approach to selecting the best experimental conditions for high-speed liquid column chromatography. II. Estimating column length, operating pressure, and separation time for some required sample resolution. J Chromatogr Sci. 1972;10:369–379. [Google Scholar]
- 13.Snyder LR, Glajch JL, Kirkland JJ. Practical HPLC method development. New York: Wiley-Interscience; 1988. [Google Scholar]
- 14.McCaffery P, Evans J, Koul O, Volpert A, Reid K, Ullmann MD. Retinoid quantification by HPLC/MSn. J Lipid Res. 2002;43:1143–1149. doi: 10.1194/jlr.D200012-JLR200. [DOI] [PubMed] [Google Scholar]
- 15.Horsman JA, Bickler JR (2006) Control system and method for flash separation. US Patent application 0219633 A1
- 16.Snyder LR. Advanced concepts in thin-layer chromatography—a summary. J Chromatogr. 1971;63:7–8. doi: 10.1016/S0021-9673(01)85610-7. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.