

Abstract
The organization of the oxidative phosphorylation (OXPHOS) system within the inner mitochondrial membrane appears to be far more complicated than previously thought. In particular, the individual protein complexes of the OXPHOS system (complexes I to V) were found to specifically interact forming defined supramolecular structures. Blue-native polyacrylamide gel electrophoresis and single particle electron microscopy proved to be especially valuable in studying the so-called “respiratory supercomplexes”? Based on these procedures, increasing evidence was presented supporting a “solid state” organization of the OXPHOS system. Here, we summarize results on the formation, organisation and function of the various types of mitochondrial OXPHOS supercomplexes.
Keywords: Supercomplexes, Respirasome, Electron microscopy, Mitochondria, Oxidative phosphorylation
Introduction
Respiration is the primary process for energy conversion in eukaryotes and is performed in mitochondria. Five membrane-embedded enzymes constitute an oxidative phosphorylation (OXPHOS) system in the inner mitochondrial membrane. Four of these protein complexes compose the “respiratory chain” and are involved in electron transfer reactions, which in three cases are coupled to proton translocation across the inner mitochondrial membrane. The resulting proton gradient is used by the ATP synthase complex for the phosphorylation ADP. Complex I or NADH dehydrogenase is the main entrance point of electrons to the respiratory chain. It uses NADH molecules generated by catabolic reactions within the mitochondrial matrix as a source of electrons and transfers them to ubiquinone within the membrane. Succinate dehydrogenase or complex II represents an alternative entrance point of electrons to the respiratory chain, which transfers electrons from succinate to ubiquinone and directly connects the Krebs cycle to the respiratory chain. This electron transfer is not coupled to proton translocation. The central component of the OXPHOS system, cytochrome c reductase or complex III, is a functional dimer. It transfers electrons from reduced ubiquinone (which is referred to as “ubiquinol”) to cytochrome c — a small mobile electron carrier associated with the outer surface of the inner membrane. Complex IV or cytochrome c oxidase represents the terminal complex of the respiratory chain and was described as a monomer upon solubilization of the inner mitochondrial membrane by mild detergent treatment but as a dimer within protein crystals. It catalyses electron transfer from cytochrome c to molecular oxygen thereby reducing the latter to water. By translocating protons across the inner mitochondrial membrane this complex makes a final contribution to the proton gradient across the inner membrane, which is used by the ATP synthase (complex V) for ATP formation. The ATP synthase is composed of two domains: a hydrophobic F0 membrane part that is connected to a water-soluble F1-headpiece by two stalks. In a nutshell, the proton gradient across the membrane domain triggers the rotation of the subunit c ring within the F0 and γ, δ and ɛ within F1, which causes the phosphorylation of ADP.
For a long time the “fluid-state” model was believed to be the best description of the organization of the OXPHOS system (Fig. 1a); it postulates that the respiratory chain complexes freely diffuse in the membrane and that electron transfer takes place on the basis of random collisions. This model is based on the finding that all individual protein complexes of the OXPHOS system can be purified in enzymatically active form and on lipid dilution experiments (reviewed in Hackenbrock et al. 1986).
Fig. 1.
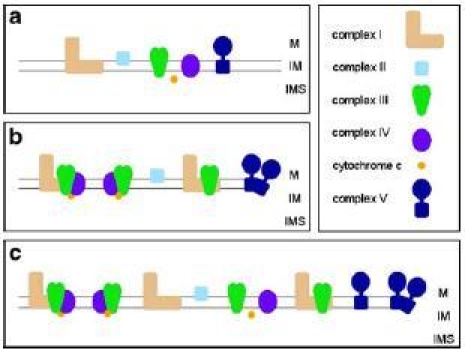
Models of the mitochondrial OXPHOS system. a The “fluid state model”. b Defined interactions of OXPHOS complexes within supercomplexes as predicted by the “solid state model”. c Integrated model of the OXPHOS system. M Matrix; IM inner mitochondrial membrane; IMS mitochondrial intermembrane space
In the last decade more and more evidences were published pointing to stable interactions of the OXPHOS complexes in the form of defined supercomplexes (Fig. 1b): (1) Flux control experiments proved that the respiratory chain is organized in one functional unit (Boumans et al. 1998; Bianchi et al. 2004). (2) Point mutations within genes encoding subunits of one OXPHOS complex affect the stability of another OXPHOS complex (Acin-Perez et al. 2004; Diaz et al. 2006). (3) Supercomplexes can be separated by blue native PAGE (Schägger and Pfeiffer 2000; Eubel et al. 2003). (4) Supercomplexes are active by in-gel activity measurements within blue native gels (Schägger and Pfeiffer 2000; Eubel et al. 2004a). (5) Oxygen uptake by isolated mitochondria of potato correlate with the abundance of supercomplexes (Eubel et al. 2004a). (6) EM structures revealed very defined interactions of OXPHOS complexes within respiratory supercomplexes (Dudkina et al. 2005a, b; Minauro-Sanmiguel et al. 2005; Schäfer et al. 2006; Heinemeyer et al. 2007).
The complexes I, III and IV were found to form different types of supercomplexes in a wide range of organisms (reviewed in Dudkina et al. 2006a). Since the various OXPHOS complexes differ in abundance they are considered to not completely incorporate in supermolecular structures. Rather, OXPHOS supercomplexes and single OXPHOS complexes co-exist within the inner mitochondrial membrane (Fig. 1c). Furthermore, association of OXPHOS complexes into supercomplexes and dissociation supercomplexes into OXPHOS complexes is considered to be a dynamic process, which depends on the physiological state of a cell.
Remarkably, complex II was never observed to be included in any respiratory chain supercomplex if analyzed by blue native PAGE or single particle EM. It is noteworthy that the succinate dehydrogenase is part of the citric acid cycle and that its electron transfer function towards complex III is not a crucial function to be incorporated in a supercomplex.
Respiratory chain complexes and supercomplexes
Supercomplexes involved in the proton gradient: I, I+III2, III2+IV1, 2, I+III2 +IV
Complex I is the largest complex of the respiratory chain which catalyzes the transfer of two electrons from NADH to ubiquinone and couples it to the translocation of four protons across the inner mitochondrial membrane. The extensively studied bovine heart mitochondria complex I is made of 45 different protein subunits (Carroll et al. 2003). Fourteen homologous “core subunits” are present in all organisms including prokaryotes and chloroplast complex I and constitute the “minimal enzyme”. Other additional subunits have been depicted as “peripheral”. Structural studies describe complex I as a typical L-shaped molecule consisting of a large membrane arm and hydrophilic arm which runs perpendicular and protrudes far into the matrix (reviewed in Yagi and Matsuno-Yagi 2003). Some peripheral subunits may provide specific features to complex I in certain organisms such as an unique matrix-exposed carbonic anhydrase domain in Arabidopsis (Sunderhaus et al. 2006) and Zea mays (Peters et al. 2008) or the unknown protrusion at the tip of the membrane arm of complex I from Yarrowia lipolytica (Radermacher et al. 2006). A similar additional density at the matrix side was found at the membrane arm tip of complex I from bovine heart mitochondria (Fig. 2a), which was not previously shown in the 3D model at 18 Å deriving from cryo-EM preparation (Grigorieff 1998). The presented bovine negative stain maps are more consistent with other 2D and 3D electron microscopy data than the cryo-EM structure. A prominent feature are the overall dimensions of complex I; it is noteworthy that the height of complex I is comparable to the height of the ATP synthase monomer (Fig. 2a,d,f), which is neglected in most textbook illustrations.
Fig. 2.
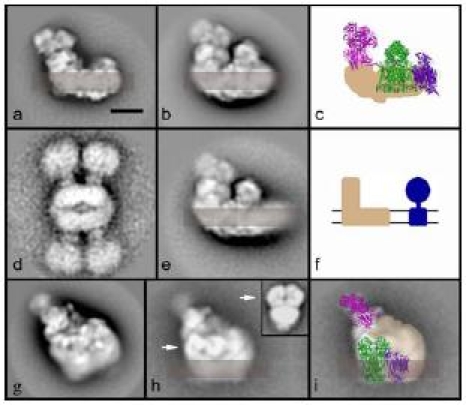
Structure of the complex I and its supercomplexes from bovine heart mitochondria. Supercomplexes were obtained by solubilization of isolated mitochondria with dodecylmaltoside (7.5 g/g) and digitonin (25 g/g) followed by purification via sucrose gradient ultracentrifugation (S. Sunderhaus and N.V. Dudkina, unpublished data) a Side-view of complex I. Position of the membrane is indicated in semi-transparent brown. b Side-view of the I + III2 supercomplex. c Modelling of the respirasome. X-ray structures of the hydrophilic domain of complex I from Thermus thermophilus (pink, Sazanov and Hinchliffe 2006), bovine cytochrome c reductase (green, Iwata et al. 1998) and bovine cytochrome c oxidase (purple, Tsukihara et al. 1996) were used. The membrane arm of complex I is given in beige. d An aggregate of two dimeric ATP synthases on the same scale as the complex I supercomplexes. e Side view of the I + III2 supercomplex with a larger membrane part, likely representing the I + III2 + IV supercomplex or respirasome. f Scheme emphasizing that ATP synthase and complex I have the same height. g Respirasome in an intermediate orientation on the carbon support film. h The same respirasome as in (e), but prepared using deep negative stain. This reveals the position of cytochrome c reductase within the I + III2 + IV supercomplex (white arrow). The insert in the upper right corner is purified cytochrome c reductase complex from Arabidopsis in a similar position (Dudkina et al. 2005a). i Model of the respirasome in the intermediate orientation. The position of complex III2 is according to (e); the hydrophilic arm of complex I was generated from the X-ray data of T. thermophilus by turning it aproximately 30° away from a side-view orientation and the remainder of the supercomplex are complex I densities (transparent beige). The location of complex IV is still uncertain. The bar of the EM frames is 10 nm
In a wide range of organisms complex I was found to form a stable association with the dimeric cytochrome c reductase complex which can be easily purified in active form from the membrane after detergent extraction. The first I + III2 supercomplex maps were described for Arabidopsis (Dudkina et al. 2005a) and for Zea mays (Peters et al. 2008). Within the supercomplex the dimeric complex III is bound at the tip of the membrane arm of complex I. The top-view projection map of the supercomplex is not completely parallel to the membrane plane although the side view map of the I+III2 supercomplex rather indicates a parallel position of the long axes of the peripheral complex I arm and complex III2 with respect to the inner membrane.
In potato (Eubel et al. 2004a, b), spinach (Krause et al. 2004) and Asparagus (Dudkina et al. 2006a) complex III2 was found to associate with one or two copies of complex IV. The structure of the mitochondrial III2 + IV1, 2 supercomplex was first studied in Saccharomyces cerevisiae because this organism lacks complex I (Heinemeyer et al. 2007). From different maps a pseudo-atomic model could be generated. It shows that two monomeric complex IV copies are attached on both sides of dimeric complex III with their convex side facing complex III2, which is the opposite to the side involved in the formation of complex IV dimers as described by X-ray crystallography. This attachment is mediated by cardiolipids (Zhang et al. 2002). Formation of the III2 + IV1, 2 supercomplex most likely has important functional implications, because the electron transport between complexes III and IV can be optimized by a contraction of cytochrome c movement between these two complexes. Moreover, in the presence of complex I it can form a more complicated structure, the “respirasome”, a supercomplex uniting complexes I, III and IV. The respirasome is able to carry out full respiration autonomously in the presence of the mobile electron carriers ubiquinone and cytochrome c.
Recent experiments with bovine heart mitochondria show that digitonin efficiently extracts complex I but mostly in combination with other respiratory chain complexes, in particular as a I + III2 + IV supercomplex (S. Sunderhaus and N.V. Dudkina, unpublished data). Projection maps were assigned to single complex I (Fig. 2a), the I + III2 supercomplex (Fig. 2b), the I + III2 + IV supercomplex or respirasome (Fig. 2e) and to tilted positions of the same particle (Fig. 2g,h), as obtained previously (Schäfer et al. 2006). Microscopy specimens were made by standard negative staining, but by using deep negative staining, which results in respirasomes fully embedded in the stain layer. Under these conditions, features of the hydrophilic parts of the core 1 and 2 subunits of complex III2 became more prominent (arrows, Fig. 2h). This leads to an unambiguous identification of the complex III2 position within the respirasome. The difference in length between the membrane arms of Fig. 2b and 2 is interpreted to be complex IV.
By using the X-ray structures of complexes III, IV and the peripheral arm of complex I we could determine the positions of these single complexes within the 2D maps of the respirasome (Fig. 2c,i). Although the hydrophilic arm of Thermus thermophius complex I is built up of less subunits than in bovine it resembles its overall shape in side-view orientation (Fig. 2c). The position of complex III2 was determined exactly due to the well recognizable features of core 1 and 2 subunits of this complex. The smaller complex IV is placed at the tip of the membrane arm of complex I, because there is a striking difference in the length of the membrane part of supercomplex (Fig. 2b,e). The exact rotational orientation, however, is unclear because the complex IV is mostly embedded into the membrane and because there are only two different views of the respirasome. The exact position is, however, different between yeast and bovine, as we will explain here: In the some specific side view maps on which the yeast model was based (Heinemeyer et al. 2007), the core 1 and 2 subunits of complex III2 and the complex IV monomers are in nonoverlapping positions. The side view map of the bovine I+III2+IV supercomplex, however, shows the core 1 and 2 subunits in an overlap position, which is only possible by rotating the complex III dimer about 90° in the membrane plane. Because the complex IV monomer is mostly if not all in a non-overlapping position (Fig. 2c) this implies that it binds at a different position. Complex IV is placed next to the complex III2 within the bovine respirasome model from Fig. 2c and the remainder densities belong to complex I.
Finally, both monomers of the complex III dimer can be an attachment site for the complex I, because they are identical. But remarkably, in the Arabidopsis I + III2 supercomplex only one of the sites is occupied. This could be an effect of a non-stoichiometric presence of complexes I and III, because it appears that in potato indeed two copies of complex I can bind to complex III2 (J.B. Bultema, R. Kouřil, unpublished data).
Previous inhibitor titration experiments showed that cytochrome c does not exhibit pool behavior in yeast and that the whole respiratory chain of yeast behaves like a single functional unit (Boumans et al. 1998). Wittig et al. 2006 proposed that the respirasome is the unit of a higher level organization of the respiratory chain into long “string”-like megastructures. Our data on bovine supercomplexes, where high numbers of respirasomes were found after mild detergent solubilization, are in line with this suggestion. Such an organization makes the electron transfer rate through the OXPHOS system more efficient and, thus, provides a basis for most effective respiration.
Supercomplexes using the proton gradient: dimeric ATP synthase
The composition of dimeric F0F1 ATP synthase from bovine and yeast has been studied in detail (reviewed in Wittig and Schägger 2008). The water-soluble F1-part is mostly constituted of three α and three β subunits. It is connected to the ring-like subunit c oligomer of the F0-part by a central stalk consisting of γ, δ and g3 subunits and by a peripheral stalk made from subunits OSCP (Su 5), b, d, F6 (h) (different names of the yeast counterparts in brackets). In addition, the F0-part is composed of subunits a (Su 6), A6L (Su 8), e, f, g. The yeast protein has two specific subunits i and k.
The first biochemical evidence about a dimeric organization of the ATP synthases complex in yeast came from the BN-PAGE work of Arnold et al. 1998. The supercomplex includes dimer specific subunits named e, g and k, which were not detected in monomers. Later, dimers were described in beef, Arabidopsis and several other organisms by the same technique (Schägger and Pfeiffer 2000; Eubel et al. 2003). Recently, low-resolution 2D structures of dimeric ATP synthase were solved in bovine (Minauro-Sanmiguel et al. 2005), the colorless green alga Polytomella (Dudkina et al. 2005b) and S. cerevisiae (Dudkina et al. 2006b). In all organisms studied until now two monomers associate via the membrane F0 parts and make an angle of 35–90°. Dimeric ATP synthase from green algae (Fig. 3e) seems to be the most stable one and is represented exclusively by molecules with an angle of 70°. These data, together with BN-PAGE evidences of trimeric and tetrameric organizations of mammalian ATP synthases (Krause et al. 2005), give support for an oligomeric arrangement of ATP synthases in the membrane. The presence of rows of ATP synthase dimers was previously demonstrated in mitochondria of Paramecium by rapid-freeze deep-etch electron microscopy (Allen et al. 1989) and in yeast mitochondria by ultrasectioning and negative staining of osmotically shocked Polytomella mitochondria (Dudkina et al. 2006b). Recently, new supporting data based on electron cryo-tomography were presented which show that the ATP synthases of mammalian mitochondria are arranged in long rows of dimers located at the apex of cristae membranes (Strauss et al. 2008).
Fig. 3.
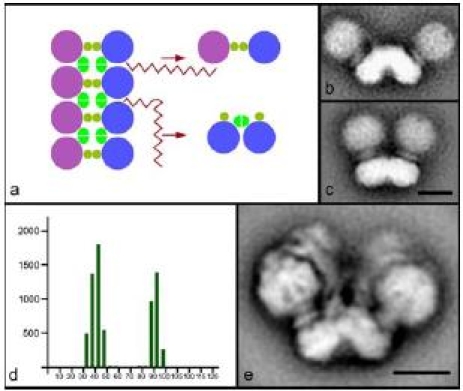
Dimers and multimers of ATP synthase. a Scheme of the multimeric chain of ATP synthases in mitochondria, which can be disrupted by detergent in two ways giving small and large angle dimers. Purple and blue circles symbolize monomeric ATP synthase complexes and ochre and bright green represent dimer-specific subunits. In mitochondria of S. cerevisiae the large angle dimers of 90° (b) and small angle dimers of 35° (c) were found in similar proportions. d Distribution of angles between the monomers of dimeric ATP synthase from S. cerevisiae. y-axis: number of particles; x-axis: size of the angle in degrees. e Dominant projection map of dimeric ATP synthase from Polytomella which corresponds to a stable dimer in which the monomers exclusively make an angle of 70°. The bars are 10 nm
In our scheme of the multimeric chain of ATP synthase complexes in mitochondria (Fig. 3a) we try to explain the difference in angle between two types of monomers. The two types are products of detergent disruption that may happen in two different ways. Chains can be disrupted to give a large angle dimer (70–90°) or a small angle dimer (35–40°) that differ in the interface. In mitochondria of S. cerevisiae the large angle dimers of 90° (Fig. 3b) and small angle dimers of 35° (Fig. 3c) are represented in similar numbers (Fig. 3d), which indicates the interactions between monomers within both types of dimers have an equal stability. In the case of Polytomella the presence of only 70° dimers suggests that the interaction between two neighboring monomers of the dimers is very weak and can be easily broken by detergent, likely due to the presence of unique dimer specific subunits in this organism (Vázquez-Acevedo et al. 2006). Although originally only dimers of 40° were reported in bovine (Minauro-Sanmiguel et al. 2005) we could observe small amounts of dimers with an angle of 90° (N.V. Dudkina, unpublished data), which also was reported in the recent study on intact bovine mitochondria (Strauss et al. 2008). The large angle dimers were also found in mitochondria of Z. mays (N.V. Dudkina, unpublished data).
Though the EM studies provide the first look on the structural organization of dimeric ATP synthase, they do not give a clear statement about the dimer interface. Wittig and Schägger 2008 summarize the most recent data about the monomer-monomer and dimer-dimer interaction. The dimer specific subunits e and g, as well as the subunits a, b, h and i of the monomer, stabilize the monomer-monomer interface. These data are in agreement with electron microscopy results showing that the dimerization occurs via the membrane F0- and peripheral stalk parts (Dudkina et al. 2005b, 2006b). The dimer-dimer interface is less well understood. Subunits e, f, g, Su 8 (A6L), transmembrane helixes of the a-subunit not involved in monomer-monomer interaction, as well as carriers for inorganic phosphate and for ADP/ATP are candidates for the oligomerization of ATP synthases.
The angle between two monomers in a dimer provides direct evidence that oligomerization of ATP synthases leads to the curvature of the inner mitochondrial membrane. Ultractructural studies on yeast null mutants of dimer specific e and g subunits which lack the dimeric state of ATP synthases indicated that their mitochondria exhibit unusual onion-shape morphology without any membrane foldings (Paumard et al. 2002; Giraud et al. 2002). This strongly suggests that dimerization of ATP synthases is essential for cristae morphology. The recent study on bovine and rat mitochondria proved that the ATP synthases in mammalian mitochondrial membranes are organized in long ribbons of dimers and, furthermore, the mitochondrial cristae act as proton traps and, as a result, ATP synthases may optimize their own performance under proton-limited conditions (Strauss et al. 2008).
Perspectives and open questions
Membrane proteins are difficult objects for any high-resolution structural investigation but the first low-resolution data are a good initial point on the way to high-resolution 3D structures. It is relatively easy to obtain medium-resolution maps of the dimeric ATP synthase. Unfortunately, only a low resolution 3D structure of a respirasome is available up to now (Schäfer et al. 2007). At a resolution around 30 Å, as obtained in the 3D structure, it is impossible to fit unambiguously the X-ray structures of single complexes. At least a resolution of about 15 Å or better is necessary for the precise modeling, as was obtained in some of our beef 2D maps. 2D maps, however, have the disadvantage that the complexes are often only present in a very limited number of orientations, with the exception of the yeast supercomplex III2 + IV1, 2 (Heinemeyer et al. 2007). Hence, higher 2D and 3D resolution is needed to solve the single complexes interfaces within the supercomplex. Currently there are two methods to obtain higher resolution structures of such complexes: crystallization of the rather stable supercomplexes and merging of EM and X-ray data. In dimeric ATP synthase specific subunits must be responsible for the tilt of the membrane domain. There are now several proteins known in which some of the membrane spanning α-helices make large angles within proteins, e.g. LHCII (Liu et al. 2004). A next step in dimeric ATP synthase research would be to reveal such α-helices that make large angles between different subunits and thereby contribute in membrane bending.
Acknowledgement
We thank Dr. Roman Kouřil for discussion. We also gratefully acknowledge funding by the Netherlands organization of scientific research (NWO) and by the Deutsche Forschungsgemeinschaft (grants Br1829-7/3 and Br1829-8/1).
References
- Acín-Pérez R, Bayona-Bafaluy M, Fernández-Silva P, Moreno-Loshuertos R, Pérez-Martos A, Bruno C, Moraes C, Enríquez J (2004) Mol Cell 13:805–815 [DOI] [PMC free article] [PubMed]
- Allen RD, Schroeder CC, Fok AK (1989) J Cell Biol 108:2233–2240 [DOI] [PMC free article] [PubMed]
- Arnold I, Pfeiffer K, Neupert W, Stuart RA, Schägger H (1998) EMBO J 17:7170–7178 [DOI] [PMC free article] [PubMed]
- Bianchi C, Genova ML, Castelli GP, Lenaz G (2004) J Biol Chem 279:36562–36569 [DOI] [PubMed]
- Boumans H, Grivell LA, Berden JA (1998) J Biol Chem 273:4872–4877 [DOI] [PubMed]
- Carroll J, Fearnley IM, Shannon RJ, Hirst J, Walker JE (2003) Mol Cel Proteomics 2:117–126 [DOI] [PubMed]
- Diaz F, Fukui H, Garcia S, Moraes CT (2006) Mol Cell Biol 26:4872–4881 [DOI] [PMC free article] [PubMed]
- Dudkina NV, Eubel H, Keegstra W, Boekema EJ, Braun HP (2005a) Proc Natl Acad Sci U S A 102:3225–3229 [DOI] [PMC free article] [PubMed]
- Dudkina NV, Heinemeyer J, Keegstra W, Boekema EJ, Braun HP (2005b) FEBS Lett 579:5769–5772 [DOI] [PubMed]
- Dudkina NV, Heinemeyer J, Sunderhaus S, Boekema EJ, Braun HP (2006a) Trends Plant Sci 11:232–240 [DOI] [PubMed]
- Dudkina NV, Sunderhaus S, Braun HP, Boekema EJ (2006b) FEBS Lett 580:3427–3432 [DOI] [PubMed]
- Eubel H, Jänsch L, Braun HP (2003) Plant Physiol 133:274–286 [DOI] [PMC free article] [PubMed]
- Eubel H, Heinemeyer J, Braun HP (2004a) Plant Physiol 134:1450–1459 [DOI] [PMC free article] [PubMed]
- Eubel H, Heinemeyer J, Sunderhaus S, Braun HP (2004b) Plant Physiol Biochem 42:937–942 [DOI] [PubMed]
- Giraud MF, Paumard P, Soubannier V, Vaillier J, Arselin G, Salin B, Schaeffer J, Brethes D, di Rago P, Velours J (2002) Biochim Biophys Acta 1555:174–180 [DOI] [PubMed]
- Grigorieff N (1998) J Mol Biol 277:1033–1046 [DOI] [PubMed]
- Hackenbrock CR, Chazotte B, Gupte SS (1986) J Bioenerg Biomembr 18:331–368 [DOI] [PubMed]
- Heinemeyer J, Braun HP, Boekema EJ, Kouřil R (2007) J Biol Chem 282:12240–12248 [DOI] [PubMed]
- Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S, Jap BK (1998) Science 281:64–71 [DOI] [PubMed]
- Krause F, Reifschneider NH, Vocke D, Seelert H, Rexroth S, Dencher NA (2004) J Biol Chem 279:48369–48375 [DOI] [PubMed]
- Krause F, Reifschneider NH, Goto S, Dencher NA (2005) Biochem Biophys Res Commun 329:583–590 [DOI] [PubMed]
- Liu Z, Yan H, Wang K, Kuang T, Zhang J, Gui L, An X, Chang W (2004) Nature 428:287–292 [DOI] [PubMed]
- Minauro-Sanmiguel F, Wilkens S, García JJ (2005) Proc Natl Acad Sci U S A 102:12356–12358 [DOI] [PMC free article] [PubMed]
- Paumard P, Vaillier J, Coulary B, Schaeffer J, Soubannier V, Mueller DM, Brethes D, di Rago JP, Velours J (2002) EMBO J 21:221–230 [DOI] [PMC free article] [PubMed]
- Peters K, Dudkina NV, Jänsch L, Braun HP, Boekema EJ (2008) Biochim Biophys Acta 1777:84–93 [DOI] [PubMed]
- Radermacher M, Ruiz T, Clason T, Benjamin S, Brandt U, Zickermann V (2006) J Struct Biol 154:269–279 [DOI] [PMC free article] [PubMed]
- Sazanov LA, Hinchliffe P (2006) Science 311:1430–1436 [DOI] [PubMed]
- Schäfer E, Seelert H, Reifschneider NH, Krause F, Dencher NA, Vonck J (2006) J Biol Chem 281:15370–15375 [DOI] [PubMed]
- Schäfer E, Dencher NA, Vonck J, Parcej DN (2007) Biochemistry 46:12579–12585 [DOI] [PubMed]
- Schägger H, Pfeiffer K (2000) EMBO J 19:1777–1783 [DOI] [PMC free article] [PubMed]
- Strauss M, Hofhaus G, Schröder RR, Kühlbrandt W (2008) EMBO J 27:1154–1160 [DOI] [PMC free article] [PubMed]
- Sunderhaus S, Dudkina NV, Jänsch L, Klodmann J, Perales M, Zabaleta E, Boekema E, Braun HP (2006) J Biol Chem 281:6482–6488 [DOI] [PubMed]
- Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S (1996) Science 272:1136–1144 [DOI] [PubMed]
- Vázquez-Acevedo M, Cardol P, Cano-Estrada A, Lapaille M, Remacle C, González-Halphen D (2006) J Bioenerg Biomembr 38:271–282 [DOI] [PubMed]
- Wittig I, Schägger H (2008) Biochim Biophys Acta 1777:592–598 [DOI] [PubMed]
- Wittig I, Carrozzo R, Santorelli FM, Schägger H (2006) Biochim Biophys Acta 1757:1066–1072 [DOI] [PubMed]
- Yagi T, Matsuno-Yagi A (2003) Biochemistry 42:2266–2274 [DOI] [PubMed]
- Zhang M, Mileykovskaya E, Dowhan W (2002) J Biol Chem 277:43553–43556 [DOI] [PubMed]