

Abstract
Noncovalent binding interactions between proteins are the central physicochemical phenomenon underlying biological signaling and functional control on the molecular level. Here, we perform an extensive structural analysis of a large set of bound and unbound ubiquitin conformers and study the level of residual induced fit after conformational selection in the binding process. We show that the region surrounding the binding site in ubiquitin undergoes conformational changes that are significantly more pronounced compared with the whole molecule on average. We demonstrate that these induced-fit structural adjustments are comparable in magnitude to conformational selection. Our final model of ubiquitin binding blends conformational selection with the subsequent induced fit and provides a quantitative measure of their respective contributions.
Keywords: ubiquitin binding, protein recognition, Kolmogorov–Smirnov test
The picture of protein–protein interactions has, over the decades, evolved from the early lock-and-key hypothesis (1) to the generally accepted and widely applied induced-fit model (2, 3). However, several different systems have recently been shown to follow an alternative paradigm whose central element is the idea of conformational selection (4). Within this paradigm, the conformational change in binding is thought to originate primarily from the conformational diversity of the unbound state (5–15). Simply put, the unbound protein explores the energy landscape, spending most of the time in the lowest energy conformations, but also occupying higher-energy ones, some of which are structurally similar to the bound conformations. In the course of binding, because of favorable interactions with the ligand, these conformers get preferentially selected and the population of protein microstates shifts in the direction of bound conformations (4–15). In a way, induced fit and conformational selection are two extremes of possible mechanisms underlying protein interactions (16): in the former, optimal binding is achieved by specific structural change, whereas in the latter it is brought about through selection from the already present unbound ensemble. The two mechanisms have recently been compared from the perspective of kinetics (17) and the energy landscape theory (18).
Some of the earliest-described examples of the conformational selection paradigm are the antibody–antigen interactions where an antibody can be found in different unbound conformations, exhibiting different specificity for different antigens (19–22). Binding then occurs by a simple selection of those antigens whose epitopes are already in a matching conformation for the paratope. In general, growing support for conformational selection in specific protein–protein interactions is based mainly on finding bound-like conformations of proteins in the respective unbound ensembles of structures (12, 14, 23–30). For example, Gsponer et al. (30) proposed that Ca2+-bound, ligand-free calmodulin samples the conformational space of calmodulin bound to myosin light chain kinase. Apart from such cases with bound-like conformations in the unbound state on the level of the whole molecule, there are several examples where specifically those residues that participate in binding are found in a proper conformation already before binding (31–34). Despite such successes, there are many examples of systems that cannot be explained by conformational selection. For example, Sullivan and Holyoak (35) showed that in phosphoenolpyruvate carboxykinase (PEPCK) formation of the catalytic active complex is combined with a closure of the active site. This means that, even if in the unbound state PEPCK samples bound conformations, they would simply not be available for the substrate. To overcome pitfalls in each of the above models, Grünberg et al. (11) proposed a three-step model where diffusional encounter is followed by the recognition of complementary structures within the conformational ensemble of unbound proteins and subsequent refolding, i.e., induced fit.
Advances in NMR have resulted in a high-resolution, dynamic picture of protein ensembles, allowing us to study protein–protein interactions with unprecedented resolution (36–41). In a recent tour de force study using residual dipolar couplings (RDCs), Lange and coworkers (39, 41) have demonstrated that free ubiquitin samples conformations globally similar to those in the bound state. Ubiquitin is a highly conserved, 76-residue protein that has been well studied from both structural and functional standpoints, because of its extreme importance in different key biological processes such as protein degradation, cell-cycle regulation, or transcription control (42–45). In their study, Lange and coworkers (39, 41) compared an ensemble of X-ray structures of ubiquitin, bound to different ubiquitin-binding proteins, with NMR structures of ubiquitin free in solution. The latter ensemble captured the full dynamic behavior of ubiquitin on the picosecond to microsecond time scale, extending and completing the previous picture of its dynamics (46). Surprisingly, Lange and coworkers demonstrated that for each bound ubiquitin structure there is a member of the unbound ensemble that is structurally similar to it in the rmsd sense, thus giving strong support to the conformational selection mechanism (16). Furthermore, using principal component analysis, they showed that the dominant motion in ubiquitin binding entails a pincer-like motion around the central hydrophobic patch in ubiquitin.
Although the effort by Lange and coworkers (39, 41) provided good evidence for conformational selection in ubiquitin binding, many open questions remain. Here, we analyze the residual induced fit after the conformational selection step in ubiquitin binding and assess the possibility that induced fit and conformational selection actually coexist in the course of ubiquitin binding. Following Lange and coworkers we performed an exhaustive structural comparison between a set of ubiquitin X-ray structures bound to different partners with an RDC-refined ensemble of ubiquitin free in solution. We focus on the NMR ensemble by Lange and coworkers, but to confirm our findings, we also analyzed two other structural ensembles (Backrub ensembles from ref. 47) that were refined based on the same RDC data.
Results
Ubiquitin, when free in solution, samples a subset of structures that, in terms of their global conformation, are similar to ubiquitin in the bound state in the rmsd sense (39, 41). Global multidimensional scaling (MDS) analysis using Lange and coworkers' EROS ensemble (39, 41) supports this finding, but also adds information (Fig. 1). Calculated MDS maps inform about relative distances (here, backbone atom-positional rmsd values between conformations) of multidimensional data in a reduced dimensional space. We present our results for: (i) residues 1–70 of ubiquitin (excluding the flexible C-terminal tail) and (ii) residues 1–76 (including the tail). Although the inclusion of the C-terminal tail makes structural comparisons more difficult because of its intrinsic flexibility, our analysis indicates that this region is directly involved in binding in ≈50% of all ubiquitin complexes studied, and its contribution cannot be ignored (Fig. S1). 2D MDS maps (Fig. 1) demonstrate that, in general, there appears to be a small number of NMR structures exhibiting globally very similar conformations to bound X-ray structures, whereas the rest are significantly more different. For example, >60% of the whole X-ray ensemble is captured by only three dominant NMR unbound structures (see also Fig. S2), which is also evident from the 2D MDS maps (Fig. 1), despite the inaccuracies associated with projecting multidimensional data into two dimensions (Fig. S3). Additionally, if one focuses on all unbound structures with an rmsd in the range of 0.1 Å from the lowest rmsd value, a similar picture is obtained (Fig. S2). Altogether, this analysis supports the thesis that the unbound protein during its dynamics does not frequently adopt bound conformations. Moreover, the ensemble of unbound NMR structures is clearly separated from the bound X-ray ensemble and appears to be significantly more diffuse in a structural sense. Finally, although quantitatively different, the MDS maps with the C-terminal tail excluded or included are qualitatively fairly similar.
Fig. 1.

MDS analysis of ubiquitin binding. Global conformational analysis by MDS, using backbone atom-positional rmsd as a metric, performed on a merged ensemble consisting of X-ray (crosses) and NMR (blue and red circles) structures. (A) Residues 1–70. (B) All residues. NMR structures, which are depicted in red, represent the most similar (in rmsd sense) NMR structures to at least one X-ray structure.
More information about the high-resolution details can be obtained by looking at local conformational differences, especially close to the binding site. In Fig. 2, we see what are the average deviations of atoms between the globally most similar unbound ubiquitin structure and the corresponding bound structure, as a function of distance from the binding site. In other words, our analysis probes the residual induced fit after conformational selection. In each case (Fig. 2), the average local atom–atom rmsd (Fig. 2, red curve) is highest in the vicinity of the binding site. For example, the backbone atoms of the residues immediately surrounding the binding site deviate 31% more compared with the structure on average (Fig. 2A; 0.5-Å range, residues 1–70, δ = 31%). Even more importantly, there are structures of bound ubiquitin for which this deviation is significantly greater than the average, reaching >100% in some cases. This difference is even more pronounced if one focuses only on side-chain atoms. First, the global structural deviation between the conformationally selected structure and the corresponding bound structure is significantly greater than just for the backbone (1.4 Å versus 0.65 Å on average). Second, the local deviations around the binding site are there even more pronounced with δ = 40%. In general, the effect of the binding site proximity extends in all cases typically >5–10 Å away from it. In Fig. 2 C and D we illustrate the influence of the flexible region (residues 71–76) in ubiquitin binding. The presence of these tail residues in the binding site (Fig. 2 C and D, blue curves) increases the discrepancy of rmsd values between the binding site and the whole protein. In other words, the tail residues that participate in binding exhibit significant conformational change after conformational selection. Although the δ value for just the backbone is greater than for the side chains (78% vs. 54%), for individual cases the average local structural deviation for side chains is often higher than for the backbone (e.g., 5 Å versus 2–3 Å; Fig. 2 C and D).
Fig. 2.

Induced fit in ubiquitin binding as a function of distance from the binding site. Local structural differences between the conformationally selected unbound structures and the corresponding bound structures of ubiquitin, captured as the average atomic rmsd values, and given as a function of distance from the binding site without the C-terminal tail [backbone (A) and side chains (B)] or with the C-terminal tail [backbone (C) and side chains (D) included]. The red curve represents the mean rmsd values calculated from all 19 pairs of structures, the blue curves represent the individual structural pairs, and the green curve represents the average number of residues in each distance range. The δ parameter, defined as δ = (rmsd0.5Å − rmsd25Å)/rmsd25Å) × 100%, captures the extent to which local conformational deviations close to the binding site (0.5 Å range) are greater than the global deviations (25-Å range). We use colors to illustrate the extent to which the residues in the C-terminal tail take part in forming the binding site: blue, more than ¼ of all of the tail residues are in the binding site (from 25% to 100%); yellow, <25% of tail residues are in the binding site; black, tail does not take part in the binding interaction. All error bars denote standard deviation of a given variable.
The structural deviations between a given conformationally selected structure and the corresponding bound structure appear to be more pronounced around the binding site compared with the molecule as a whole (Fig. 2). However, are these differences statistically significant? To quantitatively address this question, we have used the two-sample Kolmogorov–Smirnov (KS) P value analysis and asked whether the magnitudes of the local atomic deviations are drawn from the same distribution as the magnitudes of the global atomic deviations. In other words, we probe the statistical significance and the associated P values of the null-hypothesis that the distributions of the magnitudes of local atomic deviations between a given bound structure and the corresponding conformationally selected conformer are drawn from the same distribution as the deviations on the level of the whole molecule. We calculate this as a function of distance from the binding site (see Materials and Methods). With median values and their average deviation, we present the calculated P values in each distance range for the 19 pairs of structures (Fig. 3). Additionally, we present a fraction of structures with P < 0.1 in each distance range.
Fig. 3.

Statistical significance of induced fit motions in ubiquitin binding. The KS P value analysis of statistical significance of local conformational deviations from those of the molecule as a whole as a function of distance from the binding site. The blue curve depicts the median of P values from 19 pairs of structures, and the green curve captures the fraction of structures with P < 0.1 in each distance range, without the C-terminal tail [backbone (A) and side chains (B)] or with the C-terminal tail [backbone (C) and side chains (D)] included.
For backbone atoms, conformational differences between the local and the global, all-structure levels are statistically significant up to ≈5 Å away from the binding site, with the median P values typically <0.1. For example, if one focuses only on the backbone, >50% of structures with the C-terminal tail included exhibit P < 0.1 up to 5 Å and ≈40% without the tail included (Fig. 3 A and C). A similar situation is seen if one looks at side-chain atoms, although it appears that their fluctuations dissipate over somewhat shorter distances than those of the backbone atoms (Fig. 3 B and D). Note that the incorporation of “tail” residues typically does not change the level of statistical significance, possibly because it influences local and global rmsd values equally. A merged set, in which we pool together the magnitudes of structural deviations for all 19 structures, exhibits an even better level of significance of conformational changes, i.e., up to 10 Å (Fig. 4A). Overall, our results suggest that conformational differences close to the binding site are not representative of the global conformational changes after conformational selection, i.e., they are drawn from different distributions. Finally, to provide a structural context to our P value analysis, in Fig. 4B we map P values onto a surface of an X-ray structure of ubiquitin. Here, we use a structure whose P value curve is closest to the median curve in the rmsd sense. This approach for conformational analysis clearly shows the statistical significance of the deviation in structural changes as a function of distance from the binding site. After conformational selection, the residues close to the binding site change in ways that cannot be explained by global structural changes, hinting at induced fit optimization.
Fig. 4.

Pooled statistical significance of induced fit motions in ubiquitin binding. (A) The KS P value analysis conducted on a dataset from all 19 structural pairs pooled together into one distribution for each distance range. (B) Mapping of the calculated KS P values onto a representative ubiquitin surface [structure of ubiquitin bound to ADP-ribosylation-factor-binding protein GGA3 (gray); PDB ID code 1YD8], capturing the P value dependence on the distance from the binding site. The structure shown is closest (in the rmsd sense) in terms of its P value distribution to the median over all 19 structures examined.
What is the relative magnitude of induced fit in ubiquitin interactions, when compared with conformational selection? The histograms presented in Fig. 5 compare the magnitude of conformational selection with induced fit in the first and the last distance range, representing just the binding site and the whole molecule, respectively. In all four cases, from the point of view of the whole molecule, induced fit is quantitatively of a lower magnitude compared with conformational selection (Δglobal > 0), but it is still significant, especially for side chains. When we focus on just the binding site, the magnitude of induced fit is still somewhat lower for the backbone compared with conformational selection (Δbinding-site > 0), but for side chains it is higher (Δbinding-site < 0). This practically means that side chains are more prone to undergo induced fit as opposed to backbone, especially close to the binding site, where their structural changes can even be greater than the changes caused by conformational selection. Interestingly, if one includes the tail residues (Fig. 5 C and D), the conformational selection appears to be somewhat more dominant than the global induced fit (i.e., distributions are more separated in terms of Δglobal) on average, although the local induced fit in the vicinity of the binding site can be sizable, especially in cases where the tail residues participate in binding. Further analysis (local rmsd and P values) carried out on two additional ensembles derived from the same NMR data (47) support our findings presented here. These results are further presented and discussed below and in SI Text and Table S1). Finally, we have also analyzed the correlated motions in the unbound EROS ensemble of ubiquitin. Our findings suggest that there are no major long-range correlations in the unbound state, except for those in the binding site itself (Fig. S4).
Fig. 5.

Relative magnitude of conformational selection and induced fit motions. A histogram presenting the magnitude of conformational changes and induced fit of just the binding site (dark blue) or the whole molecule (light blue), and the conformational selection part of interaction between ubiquitin and its binding partners (red), shown separately for analysis without [backbone (A) and side-chain (B)] and with the C-terminal tail [backbone (C) and side chain (D)]. The calculated Δ parameter describes the probability-weighted distance between two histograms: Δglobal − conformational selection versus induced fit of the whole molecule; Δbinding-site − conformational selection versus induced fit of the binding site.
Discussion
We have shown that apart from global conformational similarity between unbound and bound structures differences in their local conformation strongly suggest that the residual induced fit after conformational selection is a significant component underlying specificity in noncovalent interactions of ubiquitin. Local structural differences and adjustments are especially pronounced close to the binding site and are described by significantly different distributions of deviations compared with the whole molecule. Moreover, we have shown that the C-terminal tail region of ubiquitin is important in the binding interaction, and that it does undergo extensive structural rearrangements upon binding in many cases. Our results support and extend the picture of protein–protein interactions proposed by Grünberg et al. (11). In the course of a binding event, the structurally closest protein scaffold to the bound conformation is chosen by conformational selection, while subsequently the binding interface is optimized for specific interactions via induced fit (Fig. S5). In this model, the relative magnitude of conformational selection as compared with the magnitude of induced fit could vary for more dynamic proteins and could also depend on protein function. Our results suggest that the magnitude of the residual induced fit in ubiquitin binding is, on the level of the whole molecule, only marginally lower than that of conformational selection, but can locally and per atom be even greater, especially for side chains. Altogether, our study furthers our understanding of ubiquitin binding and provides a common framework for analyzing all ubiquitin interactions.
In the present study, we have chosen the rmsd measure because it is probably the most widely used measure for structural comparison of biomolecules, and it is the principal tool used in previous analyses of conformational selection (39, 41). By choosing only atom-to-atom rmsd we were able to avoid size dependence present in typical global rmsd calculations. However, we have also tested other approaches, especially for local structural comparison. We have used a nine-residue window sliding down the ubiquitin sequence and have superimposed and calculated the atom-positional rmsd inside this window. In a similar analysis, all atoms within a 6-Å radius sphere around each backbone atom were analyzed and atoms inside this sphere were superimposed and their atom-positional rmsd was calculated. Results from such analyses were in general agreement with the findings presented herein without adding new insight.
In all of our local rmsd analyses, we have chosen an unbound NMR structure with the lowest backbone global rmsd to the corresponding bound X-ray structure. Following Lange and coworkers (39, 41), this, by definition, is the conformationally selected unbound conformer associated with a given X-ray structure. The choice of this unbound structure to compare with the bound one, critically depends not only on the conformational selection model being correct, but also on its operational definition. In this sense, our analysis can be thought of as the study of the residual induced fit after conformational selection, as defined through the global backbone rmsd criteria. This fact touches on a fundamental issue concerning the very definition of conformational selection. Namely, here we have defined conformational selection in the global rmsd sense. However, one could envision a local definition of conformational selection, in which just the binding site would need to be structurally similar in the unbound and the bound state, whereas the rest of the structure could be fairly different. In this sense, the balance between conformational selection and induced fit in binding is in part a matter of definition of the two components.
We have carried out a detailed analysis based on an NMR ensemble (39, 41) that is, to the best of our knowledge, the most detailed available structural description of ubiquitin dynamics, covering a dynamical range from ps to μs. However, it is known that different structural ensembles can reproduce the NMR data to a similar extent, including ensembles that were generated through refinement (39, 41, 47) or even represent different structural models obtained by other methods such as X-ray crystallography (48). For this reason, we have repeated our complete analysis on two additional NMR ensembles generated from the same RDC data as the EROS ensemble [Backrub ensembles generated via Monte Carlo simulations (47)]. All of the principal results obtained with the EROS ensemble are reproduced with these ensembles (see SI Text). For example, 47% of the structures in the Backrub 1.2 ensemble (Table S1) without the tail, and 58% with the tail included, exhibit significantly greater fluctuations (P < 0.1) in the region <1.5 Å away from the binding site compared with the molecule as a whole. However, here one should emphasize that the original EROS ensemble still slightly better reproduces the original RDC data (47) and may potentially be somewhat more realistic as it was generated by using time-dependent molecular dynamics simulations.
Finally, our findings could be of use for further development of computational docking methods, which have recently started to incorporate the conformational selection model for ligand–receptor interactions (49). One particularly exciting future development would entail using molecular dynamics simulations to sample bound and unbound ensembles of different interacting partners. The principal advantage of such an approach is that one would obtain information about populations of each conformational state. Altogether, the methods developed herein provide a quantitative basis for comparing induced fit and conformational selection in general and open up ways for similar efforts in different systems.
Materials and Methods
Structural dataset used in our analysis is based on the one used by Lange and coworkers (39, 41). It consists of two ensembles: (i) an ensemble of X-ray structures containing a number of ubiquitin conformers bound to different partners and (ii) an NMR ensemble of ubiquitin structures free in solution, the EROS ensemble refined from RDCs (39, 41). The first ensemble consists of 19 high-resolution X-ray structures of ubiquitin bound to different binding partners (Table S2), chosen from the Protein Data Bank (PDB). The selection criteria together with the exact PDB codes are given in SI Text. The NMR EROS ensemble includes 116 conformations of unbound ubiquitin. For additional analysis, we have used two Backrub ensembles described in ref. 47 (two ensembles of 50 structures with maximum segment length 3 and 12 and kT = 2.4 and 1.2, respectively) that also reproduce the RDC data well. However, if not further specified, our analysis refers to the EROS ensemble. In addition to the analysis of ubiquitin without the flexible C-terminal region (residues 71–76 comprising ubiquitin's tail), as done by Lange and coworkers (39, 41), we have performed analysis on the complete structure of ubiquitin (including all 76 residues, when available, or the maximum number of residues, when the structure of the complete molecule was not available, such as in a number of X-ray structures). The reason for including the tail residues is that, in fact, they often directly participate in ubiquitin binding and are in many cases an integral part of ubiquitin's binding site (Fig. S1).
Structural rmsd Analysis.
Our structural analysis focused on: (i) ubiquitin's global structural features, encompassing the whole molecule and (ii) local structural features and their dependence on the distance from the binding site. For the global analysis we have used nonmetric MDS [the Sammon mapping algorithm as implemented in R (50) and modified for our purposes] and applied it to a distance matrix produced from backbone atom-positional rmsd values calculated for all pairs from the merged set (X-ray with NMR ensemble). The exact procedure for generating MDS maps is given in SI Text.
Our local structural analysis is based on first finding, for each X-ray structure, one unbound NMR structure with the most similar conformation in the backbone atom-positional rmsd sense. As suggested before, this structure is the one that is conformationally selected for a given binding partner (39, 41). Subsequently, we analyze individual atom-to-atom rmsd (separately for backbone and side-chain atoms) between the atoms in the ubiquitin X-ray structures and the matching atoms from the corresponding conformationally selected NMR structures, as a function of distance from the binding site (Fig. 2). Importantly, these local deviations are analyzed upon global superposition of the two structures by using all backbone atoms for superposition. To acquire the dependence of the deviations on the distance from the binding site, we have grouped all ubiquitin atoms in each pair into nine distance ranges: from 0 to 0.5, 1.5, 3, 5, 7.5, 10, 15, 20, and 25 Å, depending on the distance from the binding site. A given atom is assigned to one of the above ranges if its distance to the closest nonhydrogen atom from the binding partner is less than the sum of the upper bound of a given range and the van der Waals radii of the two atoms. The binding site itself is defined as all of the atoms in the 0.5-Å distance range (51). Note that distance ranges are cumulative, meaning that if an atom belongs to one group it also belongs to all other groups with larger upper bounds.
Statistical Significance of Structural Differences.
We have used the standard two-sample KS test to compare, for each pair of structures, the distribution of atom-to-atom rmsd values in each distance range with the distribution of atom-to-atom rmsd values in the last distance range (25 Å) representing the whole molecule. We have chosen this nonparametric test, because the distributions of magnitudes of atomic deviations did not conform well to any distribution for which a parametric test might be used. In addition to applying the above test to individual structural pairs, we have applied our P value analysis to a merged dataset where data from all 19 structural pairs was pooled into one distribution for each distance range. The details about the KS test are given in SI Text.
Quantitative Comparison of Conformational Selection and Induced Fit.
One can quantify the magnitude of conformational selection as the average backbone atom-positional rmsd value between all unbound structures and the one chosen to be the most similar to the bound state. Similarly, the calculated local atom-positional rmsd values (for pairs of X-ray and NMR structures) can be taken as a measure of the magnitude of induced fit. The differences in magnitude between conformational selection and induced fit in ubiquitin binding is analyzed here via: (i) histograms of average backbone atom-positional rmsd values between all unbound structures and the one chosen to be the most similar to the bound state, capturing the magnitude of conformational selection for all 19 structures, and (ii) histograms of structural atom-to-atom deviations from the first and the last distance range (see above), capturing the induced fit of the binding site and the whole molecule, respectively. Additionally, to quantitatively describe relative differences between induced fit and conformational selection, we have calculated probability-weighted “distance” between distributions:
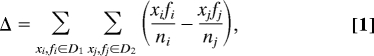 |
where D1 and D2 are distributions of magnitudes of conformational selection and induced fit, respectively, x are rmsd values, f are frequencies, and n is a number of points in each distribution.
Supplementary Material
Acknowledgments.
We thank I. Dikic, V. S. Pande, S. Bacallado, and the members of the Laboratory of Computational Biophysics at the Mediterranean Institute for Life Sciences for useful comments on the manuscript and T. Kortemme (University of California, San Francisco, CA) for providing the Backrub-refined ensembles of ubiquitin. This work was supported in part by the National Foundation for Science, Higher Education, and Technological Development of Croatia, a European Molecular Biology Organization installation grant (to B.Z.), Unity Through Knowledge Fund Grant UKF 1A (to B.Z.), and a Federation of European Biochemical Societies Summer Research Fellowship (to T.W.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0906966106/DCSupplemental.
References
- 1.Fisher E. Einfluss der Konfiguration auf die Wirkung der Enzyme. Ber Dt Chem Ges. 1894;27:2985–2993. [Google Scholar]
- 2.Koshland DE. Application of a theory of enzyme specificity to protein synthesis. Proc Natl Acad Sci USA. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bosshard HR. Molecular recognition by induced fit: How fit is the concept? News Physiol Sci. 2001;16:171–173. doi: 10.1152/physiologyonline.2001.16.4.171. [DOI] [PubMed] [Google Scholar]
- 4.Ma B, Kumar S, Tsai CJ, Nussinov R. Folding funnels and binding mechanism. Protein Eng. 1999;12:713–720. doi: 10.1093/protein/12.9.713. [DOI] [PubMed] [Google Scholar]
- 5.Tsai CJ, Kumar S, Ma B, Nussinov R. Folding funnels, binding funnels, and protein function. Protein Sci. 1999;8:1181–1190. doi: 10.1110/ps.8.6.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsai CJ, Ma B, Nussinov R. Folding and binding cascades: Shifts in energy landscapes. Proc Natl Acad Sci USA. 1999;96:9970–9972. doi: 10.1073/pnas.96.18.9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar S, Ma B, Tsai CJ, Sinha N, Nussinov R. Folding and binding cascades: Dynamic landscapes and population shifts. Protein Sci. 2000;9:10–19. doi: 10.1110/ps.9.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai CJ, Ma B, Sham YY, Kumar S, Nussinov R. Structured disorder and conformational selection. Proteins. 2001;44:418–427. doi: 10.1002/prot.1107. [DOI] [PubMed] [Google Scholar]
- 9.Ma B, Shatsky M, Wolfson HJ, Nussinov R. Multiple diverse ligands binding at a single protein site: A matter of pre-existing populations. Protein Sci. 2002;11:184–197. doi: 10.1110/ps.21302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.James LC, Tawfik DS. Conformational diversity and protein evolution: A 60-year-old hypothesis revisited. Trends Biochem Sci. 2003;28:361–368. doi: 10.1016/S0968-0004(03)00135-X. [DOI] [PubMed] [Google Scholar]
- 11.Grünberg R, Leckner J, Nilges M. Complementarity of structure ensembles in protein–protein binding. Structure (London) 2004;12:2125–2136. doi: 10.1016/j.str.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 12.Eisenmesser EZ, et al. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- 13.Tobi D, Bahar I. Structural changes involved in protein binding correlate with intrinsic motions of proteins in the unbound state. Proc Natl Acad Sci USA. 2005;102:18908–18913. doi: 10.1073/pnas.0507603102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313:1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- 15.Kapur S, Khosla C. Fit for an enzyme. Nature. 2008;454:832–833. doi: 10.1038/454832a. [DOI] [PubMed] [Google Scholar]
- 16.Boehr DD, Wright PE. How do proteins interact? Science. 2008;320:1429–1430. doi: 10.1126/science.1158818. [DOI] [PubMed] [Google Scholar]
- 17.Weikl TR, von Deuster C. Selected-fit versus induced-fit protein binding: Kinetic differences and mutational analysis. Proteins. 2009;75:104–110. doi: 10.1002/prot.22223. [DOI] [PubMed] [Google Scholar]
- 18.Okazaki K, Takada S. Dynamic energy landscape view of coupled binding and protein conformational change: Induced-fit versus population-shift mechanisms. Proc Natl Acad Sci USA. 2008;105:11182–11187. doi: 10.1073/pnas.0802524105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foote J, Milstein C. Conformational isomerism and the diversity of antibodies. Proc Natl Acad Sci USA. 1994;91:10370–10374. doi: 10.1073/pnas.91.22.10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berger C, et al. Antigen recognition by conformational selection. FEBS Lett. 1999;450:149–153. doi: 10.1016/s0014-5793(99)00458-5. [DOI] [PubMed] [Google Scholar]
- 21.James LC, Roversi P, Tawfik DS. Antibody multispecificity mediated by conformational diversity. Science. 2003;299:1362–1367. doi: 10.1126/science.1079731. [DOI] [PubMed] [Google Scholar]
- 22.Jimenez R, Salazar G, Baldridge KK, Romesberg FE. Flexibility and molecular recognition in the immune system. Proc Natl Acad Sci USA. 2003;100:92–97. doi: 10.1073/pnas.262411399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kovrigin EL, Loria JP. Enzyme dynamics along the reaction coordinate: Critical role of a conserved residue. Biochemistry. 2006;45:2636–2647. doi: 10.1021/bi0525066. [DOI] [PubMed] [Google Scholar]
- 24.Koglin A, et al. Structural basis for the selectivity of the external thioesterase of the surfactin synthetase. Nature. 2008;454:907–911. doi: 10.1038/nature07161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osborne MJ, Schnell J, Benkovic SJ, Dyson HJ, Wright PE. Backbone dynamics in dihydrofolate reductase complexes: Role of loop flexibility in the catalytic mechanism. Biochemistry. 2001;40:9846–9859. doi: 10.1021/bi010621k. [DOI] [PubMed] [Google Scholar]
- 26.Pontiggia F, Zen A, Micheletti C. Small- and large-scale conformational changes of adenylate kinase: A molecular dynamics study of the subdomain motion and mechanics. Biophys J. 2008;95:5901–5912. doi: 10.1529/biophysj.108.135467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang C, Schwieters CD, Clore GM. Open-to-closed transition in apo maltose-binding protein observed by paramagnetic nmr. Nature. 2007;449:1078–1082. doi: 10.1038/nature06232. [DOI] [PubMed] [Google Scholar]
- 28.Xu Y, et al. Induced-fit or preexisting equilibrium dynamics? Lessons from protein crystallography and MD simulations on acetylcholinesterase and implications for structure-based drug design. Protein Sci. 2008;17:601–605. doi: 10.1110/ps.083453808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang L, et al. Insights into equilibrium dynamics of proteins from comparison of NMR and X-ray data with computational predictions. Structure (London) 2007;15:741–749. doi: 10.1016/j.str.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gsponer J, et al. A coupled equilibrium shift mechanism in calmodulin-mediated signal transduction. Structure (London) 2008;16:736–746. doi: 10.1016/j.str.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Najmanovich R, Kuttner J, Sobolev V, Edelman M. Side-chain flexibility in proteins upon ligand binding. Proteins. 2000;39:261–268. doi: 10.1002/(sici)1097-0134(20000515)39:3<261::aid-prot90>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 32.Li X, Keskin O, Ma B, Nussinov R, Liang J. Protein–protein interactions: Hot spots and structurally conserved residues often locate in complemented pockets that preorganized in the unbound states: Implications for docking. J Mol Biol. 2004;344:781–795. doi: 10.1016/j.jmb.2004.09.051. [DOI] [PubMed] [Google Scholar]
- 33.Rajamani D, Thiel S, Vajda S, Camacho CJ. Anchor residues in protein–protein interactions. Proc Natl Acad Sci USA. 2004;101:11287–11292. doi: 10.1073/pnas.0401942101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yogurtcu ON, Erdemli SB, Nussinov R, Turkay M, Keskin O. Restricted mobility of conserved residues in protein–protein interfaces in molecular simulations. Biophys J. 2008;94:3475–3485. doi: 10.1529/biophysj.107.114835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sullivan SM, Holyoak T. Enzymes with lid-gated active sites must operate by an induced fit mechanism instead of conformational selection. Proc Natl Acad Sci USA. 2008;105:13829–13834. doi: 10.1073/pnas.0805364105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. J Am Chem Soc. 1982;104:4546–4570. [Google Scholar]
- 37.Akke M, Palmer AG. Monitoring macromolecular motions on microsecond to millisecond time scales by R1ρ-R1 constant relaxation time NMR spectroscopy. J Am Chem Soc. 1996;118:911–912. [Google Scholar]
- 38.Meiler J, Prompers JJ, Peti W, Griesinger C, Brüschweiler R. Model-free approach to the dynamic interpretation of residual dipolar couplings in globular proteins. J Am Chem Soc. 2001;123:6098–6107. doi: 10.1021/ja010002z. [DOI] [PubMed] [Google Scholar]
- 39.Lange OF, et al. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science. 2008;320:1471–1475. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- 40.Markwick PRL, Malliavin T, Nilges M. Structural biology by NMR: Structure, dynamics, and interactions. PLoS Comput Biol. 2008;4:e1000168. doi: 10.1371/journal.pcbi.1000168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lakomek N, et al. Residual dipolar couplings as a tool to study molecular recognition of ubiquitin. Biochem Soc Trans. 2008;36:1433–1437. doi: 10.1042/BST0361433. [DOI] [PubMed] [Google Scholar]
- 42.Hicke L, Schubert HL, Hill CP. Ubiquitin-binding domains. Nat Rev Mol Cell Biol. 2005;6:610–621. doi: 10.1038/nrm1701. [DOI] [PubMed] [Google Scholar]
- 43.Harper JW, Schulman BA. Structural complexity in ubiquitin recognition. Cell. 2006;124:1133–1136. doi: 10.1016/j.cell.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 44.Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. 2007;315:201–205. doi: 10.1126/science.1127085. [DOI] [PubMed] [Google Scholar]
- 45.Ikeda F, Dikic I. Atypical ubiquitin chains: New molecular signals. EMBO Rep. 2008;9:536–542. doi: 10.1038/embor.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lindorff-Larsen K, Best RB, Depristo MA, Dobson CM, Vendruscolo M. Simultaneous determination of protein structure and dynamics. Nature. 2005;433:128–132. doi: 10.1038/nature03199. [DOI] [PubMed] [Google Scholar]
- 47.Friedland GD, Lakomek N, Griesinger C, Meiler J, Kortemme T. A correspondence between solution-state dynamics of an individual protein and the sequence and conformational diversity of its family. PLoS Comput Biol. 2009;5:e1000393. doi: 10.1371/journal.pcbi.1000393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Best RB, Lindorff-Larsen K, DePristo MA, Vendruscolo M. Relation between native ensembles and experimental structures of proteins. Proc Natl Acad Sci USA. 2006;103:10901–10906. doi: 10.1073/pnas.0511156103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chaudhury S, Gray JJ. Conformer selection and induced fit in flexible backbone protein–protein docking using computational and NMR ensembles. J Mol Biol. 2008;381:1068–1087. doi: 10.1016/j.jmb.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.R Development Core Team. A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2008. [Google Scholar]
- 51.Tsai CJ, Lin SL, Wolfson HJ, Nussinov R. A dataset of protein–protein interfaces generated with a sequence-order-independent comparison technique. J Mol Biol. 1996;260:604–620. doi: 10.1006/jmbi.1996.0424. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.