

Abstract
Screening and early identification of primary immunodeficiency disease (PID) genes is a major challenge for physicians. Many resources have catalogued molecular alterations in known PID genes along with their associated clinical and immunological phenotypes. However, these resources do not assist in identifying candidate PID genes. We have recently developed a platform designated Resource of Asian PDIs, which hosts information pertaining to molecular alterations, protein–protein interaction networks, mouse studies and microarray gene expression profiling of all known PID genes. Using this resource as a discovery tool, we describe the development of an algorithm for prediction of candidate PID genes. Using a support vector machine learning approach, we have predicted 1442 candidate PID genes using 69 binary features of 148 known PID genes and 3162 non-PID genes as a training data set. The power of this approach is illustrated by the fact that six of the predicted genes have recently been experimentally confirmed to be PID genes. The remaining genes in this predicted data set represent attractive candidates for testing in patients where the etiology cannot be ascribed to any of the known PID genes.
Keywords: RAPID, SVM, HPRD, Human Proteinpedia, NetPath
1. Introduction
Primary immunodeficiency diseases (PIDs) are a genetically heterogeneous group of disorders that affect distinct components of the innate and adaptive immune system, such as neutrophils, macrophages, dendritic cells, natural killer cells and T and B lymphocytes. The study of these diseases has provided essential insights into the functioning of our immune system. More than 120 distinct genes have been identified, whose abnormalities account for more than 150 distinct forms of PID.1 PIDs are challenging for both researchers and clinicians because they represent natural models of immunopathology, which can usually be studied effectively only in animal models, and manifest with a wide range of clinical symptoms ranging from susceptibility to infections and allergies to autoimmune and inflammatory diseases. The genetic defects that cause PIDs can affect the expression and function of proteins involved in a range of biological processes, such as immune development, effector-cell functions, signaling cascades and maintenance of immune homeostasis.2
Because genes and proteins rarely work in isolation, genes that directly or functionally interact with known PID genes could also represent additional PID genes. We have recently developed a database of PID genes designated ‘Resource of Asian PDIs (RAPID)’, which contains information pertaining to genes and proteins involved in PDIs along with other relevant information about protein–protein interactions, mouse knockout studies and microarray gene expression profiles in various cells and organs of the immune system. These significant features of PID genes, including their involvement in immune signaling pathways, were used as input binary features for the prediction of additional candidate PID genes using a support vector machine (SVM) learning approach.
SVM is a powerful machine learning technique widely used in the computational biology such as microarray data analysis,3–8 protein secondary structure prediction,9 prediction of human signal peptide cleavage sites,10 translational initiation site recognition in DNA,11 protein fold recognition,12,13 prediction of protein–protein interactions,14 prediction of protein sub-cellular localization,15–18 and peptide identification from mass-spectrometry derived data.19
SVM is a learning algorithm that can be used to generate a classifier from a set of positively and negatively labeled training data sets.20 SVM learns the classifier by mapping the input training samples into a possibly high-dimensional feature space and seeking a hyperplane in this space, which separates the two types of examples with the largest possible margin, i.e. distance to the nearest points. If the training set is not linearly separable, SVM finds a hyperplane, which optimizes a trade-off between good classification and large margin.20
For predicting a classifier between PID and non-PID genes, we have solved the above problem and obtained a linear classifier (Fig. 1). To prove generalization of the predicted classifier, we have reported leave-one-out (LOO) error for the training data set. In this approach, we have used all the known PID genes that have been described in the literature as a positive data set. The gene list for negative data sets was selected from mouse genomic informatics (MGI) database based on the criterion that mutations in mice do not result in either immune or hematopoietic system phenotypes. We trained SVM with 69 features (Supplementary Table S1) for both PID genes (positive data set) and genes that were not reported to be associated with PIDs (negative data set). The trained SVM was then used to predict candidate PID genes by testing all human genes (except those used in the training data sets) as test data set.
Figure 1.

A schematic of SVM training strategy.
2. Materials and methods
2.1. Initial platform
RAPIDs, which is available as a worldwide web resource at http://rapid.rcai.riken.jp/21 was used as a source of information about PID genes. RAPID hosts information on sequences and expression at the mRNA and protein levels of genes reported to be involved in PID patients. The main objective of this database was to provide detailed information pertaining to genes and proteins involved in PIDs along with other relevant information about protein–protein interactions, mouse knockout studies and microarray gene expression profiles in various organs and cells of the immune system.
2.2. Features used for training the data sets
The PDIs are characterized by essential defects in the functions of the immune system, leading to increased susceptibility to infections. Although rare, these disorders cover a wide spectrum of defects, including antibody deficiencies, cellular immune deficiencies, combined immune deficiencies, phagocytic defects, complement and other innate immunity defects. On the basis of these observations for all the known PID genes, we selected 69 features (Supplementary Table S1) which not only play an important role in the development, maintenance and normal functioning of immune/hematopoietic systems but also in understanding molecular pathophysiology of PID disease causing genes. These features can be broadly classified as features for signaling pathways from NetPath and KEGG22–24 database, microarray gene expression profile from RefDIC25 database, site of expression from HPRD26 and Human Proteinpedia,27 immune/hematopoietic phenotypes from MGI28,29 and interaction with PID feature from HPRD.
2.3. Data sets
To train the SVM, two types of data sets were generated—the positive data set consists of all the known PID genes, whereas the negative data set contained genes where no immune/hematopoietic system abnormalities were described due to mouse knockouts, knockins or spontaneous mutations reported for the mouse orthologs in the MGI database.30 On the basis of these criteria, 148 PID genes were in the positive data set and 3162 genes were in the negative data set. Test data set contains 36 677 genes encoded by the human genome. Genes involved in both the training and test data sets were assigned a binary score of ‘1’ and ‘0,’ respectively, based on their presence or absence in a particular feature. The trained SVM was used to classify PID or non-PID genes from an unlabeled test data set which consists of all human genes (Fig. 2).
Figure 2.

A schematic of the algorithm for prediction of candidate PID genes.
2.4. SVM implementation
We used SVMlight (http://svmlight.joachims.org/), an implementation of Support Vector Machines in C, and also used customized functions written in MATLAB (http://www.mathtools.net/MATLAB/) for the calculation of confidence score for each predicted candidate PID gene. Absolute score also known as confidence score can be defined as 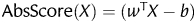 where
where 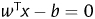 represents the separating hyperplane calculated by SVM. The score indicates how far that particular gene from the positive side of the hyperplane. In other words, higher the score more likely that a particular gene is a candidate PID gene. Using this approach, 1442 candidate PID genes were predicted which falls on the positive side of the hyperplane.
represents the separating hyperplane calculated by SVM. The score indicates how far that particular gene from the positive side of the hyperplane. In other words, higher the score more likely that a particular gene is a candidate PID gene. Using this approach, 1442 candidate PID genes were predicted which falls on the positive side of the hyperplane.
2.5. LOO error
LOO error measurement involves removing one gene from the training set, training the SVM on the remaining genes and then predicting the class label of that gene that was left out. This process is repeated until all the genes are left out exactly once. If the gene was classified correctly, the error was reported as zero, else the error was reported as one. This process was repeated by leaving out each gene once and the LOO error of the data set represent the average of individual errors.
3. Results and discussion
Over 1500 Mendelian disorders whose molecular basis is unknown are catalogued in the online Mendelian inheritance in man (OMIM) database.31 Most of disease-gene identification efforts involve either linkage analysis or association studies.32,33 Recently, a number of in silico approaches to identify candidate disease genes have been developed that use available information reported from various studies such as functional annotation, gene expression profiles, annotated sequence features, protein–protein interactions and pathway information.34–39 Several machine learning approaches have also been employed to identify important genes for disease classification. SVM approach is generally preferred owing to its superior performance.40 In most instances, SVM is a powerful tool in dealing with high-dimensional low sample size data sets, which also performs well in various biological analyses including text categorization, evaluating microarray expression data and inferring functional annotation from protein sequence and structure data.3,4,41,42 In this study, we trained an SVM with 69 features for both positive (all known PID genes) and negative (genes with no immune/hematopoietic systems affected due to mutations from MGI) gene data sets.
As the number of genes in the positive data set is small, the LOO error was calculated for showing generalization of the algorithm. LOO error is explained in detail under the Materials and methods section. For this, we used a data set containing 148 PID genes from positive data sets along with 148 genes that were randomly selected from the negative data set. This process was repeated and from 60 such data sets, the LOO error was calculated. The average LOO error reported over 60 data sets was ∼8%. The LOO error reported by leaving out only the PID (positive) genes one by one (where training set contains same setting of 296 data points) was ∼15%.
3.1. Sensitivity and specificity
The sensitivity and specificity of the data sets was 0.85 and 0.98, respectively. On the basis of these results, we conclude that the number of genes falsely predicted to be PID genes by the trained classifier is ∼2%. We believe that availability of comprehensive and accurate biological data is a limitation that restricts the prediction accuracy and performance of this algorithm. As more data accumulates about the human genome and proteome, we expect the performance of this algorithm to improve further in the future. The complete list of predicted candidate genes is provided in Supplementary Table S2 and also available at the RAPID website http://rapid.rcai.riken.jp/. All 69 features of the predicted candidate PID genes can also be downloaded from the RAPID website.
3.2. Evaluation studies
We were able to evaluate our predictions in a limited fashion because a few studies have been published describing novel PID genes that were not included in our original list of PID genes. These experimental studies have confirmed six of the genes in our predicted list of PID genes as true PID genes. These are myeloid differentiation factor-88 (MYD88), catalytic subunit of DNA dependent serine/threonine protein kinase (PRKDC), glucose-6-phosphatase, catalytic subunit 3 (G6PC3),43–45 IL2-inducible T-cell kinase (ITK), coronin, actin binding protein 1A (CORO1A) and Interleukin 1 receptor antagonist (IL1RN).46–49 MyD88 is a key downstream adaptor protein in IL1 receptor complex and toll-like receptors signaling pathways involved in inflammatory response and host defense. In addition, MyD88 is also involved in tumorigenesis in models of hepatocarcinoma and familial associated polyposis; negative regulation of TLR3 signaling and in PKC epsilon activation.50 Patients with MyD88 deficiency are reported to be susceptible to pyogenic bacterial infections including invasive pneumococcal disease.45 Defect in PRKDC has been reported for the first time in a radiosensitive T-B-SCID patient that results in inhibition of Artemis activation and non-homologous end-joining.44 A report of mutations in G6PC3 gene has been observed among patients with severe congenital neutropenia syndrome and also shown to be susceptible to increased apoptosis that leads to disturbances in cardiac or urogenital development.43 A novel PDI, IL-2 inducible T-cell kinase (ITK) deficiency has been observed due to fatal immune dysregulation followed by EBV infection and identified homozygous mutation in the SH2 domain of ITK gene that resulted in protein destabilization and absence of NKT cells.47 A patient with T cell-deficient, B cell-sufficient and NK cell-sufficient severe combined immunodeficiency has been identified with mutation in CORO1A gene along with reduced T-cell function that was earlier demonstrated in knock-out mice of coro1a gene with similar phenotypes.49 Deficiency of the IL1-receptor antagonist, an autosomal recessive autoinflammatory disease, has been reported for the first time in children presented with clinical phenotypes of multifocal osteomyelitis, periostitis, pustulosis, thrombosis and respiratory insufficiency due to the homozygous deletion of the IL1RN gene.46,48 Further, functional analysis of these mutants confirmed diminished or lack of mRNA and protein expressions leading to cytokine abnormalities.
There are two recent independent reports51,52 on the identification and prioritization of candidate disease genes in general as well as specific to primary immunodeficiencies by integrating functional annotations from gene ontology and compilation of protein interaction network data sets from BIND,53 BioGRID54 and HPRD.26 In the latter studies, 24 candidate genes were reported that are likely to be involved in PID have been identified using these parameters, out of which, over 80% of these genes are already listed as candidates in our SVM analysis, thereby, paving the way for successful implementation of this approach in the future.
We have also summarized reports of genome-wide association studies and other related studies for newly identified candidate PID genes and the associated immunological disorder (Table 1). Because the candidate PID gene list is still large, this approach of integrating data from high-throughput studies would allow further prioritization of genes for confirmation in patients with PID where the exact gene is not yet identified. We hope that such integrated approaches should assist PID physicians and researchers to gain insights into the pathophysiology of these diseases at a faster pace, which could be translated to improve the diagnosis and/or treatment of PIDs.
Table 1.
A list of predicted PID genes whose association with immunological disorders has been reported recently
| Gene symbol | Molecule class | Immunological disorder(s) | Reference(s) |
|---|---|---|---|
| ITGAM | Cell surface receptor | Systemic lupus erythematosus | Harley et al., Nat. Genet., 2008 (PubMed ID: 18204446);55 Nath et al., Nature Genetics, 2008 (PubMed ID: 18204448)56 |
| BANK1 | Chaperone | Systemic lupus erythematosus | Kozyrev et al., Nat. Genet., 2008 (PubMed ID: 18204447)57 |
| MST1 | Growth factor | Inflammatory bowel disease | Goyette et al., Mucosal Immunol., 2008 (PubMed ID: 19079170)58 |
| CYLD | Ubiquitin–proteasome system protein | Crohn's disease | Johnson and O'Donnell et al., BMC Med. Genet., 2009 (PubMed ID: 19161620)59 |
| PTPN2 | Tyrosine phosphatase | Crohn's disease | Wellcome Trust Case Control Consortium, 2007;60 Todd et al., Nat. Genet., 2007 (PubMed ID: 17554260)61 |
| PTPN22 | Tyrosine phosphatase | Systemic lupus erythematosus | Wellcome Trust Case Control Consortium, 2007;60 Harley et al., Nat. Genet., 2008 (PubMed ID: 18204446)55 |
| TNFAIP3 | Transcription regulatory protein | Rheumatoid arthritis | Plenge et al., Nat. Genet., 2007 (PubMed ID: 17982456)62 |
| STAT4 | Transcription factor | Systemic lupus erythematosus | Remmers et al., N Engl J Med., 2007 (PubMed ID: 17804842)63 |
| TNFSF4 | Ligand | Systemic lupus erythematosus | Graham et al., Nat. Genet., 2008 (PubMed ID: 18059267)64 |
| CTLA4 | Adhesion molecule | Autoimmune thyroid diseases | Ueda et al., Nature, 2003 (PubMed ID: 12724780);65 Ikegami et al., J. Clin. Endocrinol. Metab., 2006 (PubMed ID: 16352685)66 |
3.3. Availability
The list of predicted PID genes is available as Supplementary Table S2 and at the RAPID website http://rapid.rcai.riken.jp/.
Supplementary Data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
We thank the Department of Biotechnology of the Government of India for research support to the Institute of Bioinformatics, Bangalore.
Supplementary Material
Acknowledgements
The authors thank Shigeaki Nonoyama, Hirokazu Kanegane, Toshio Miyawaki, Koichi Oshima and Atsushi Hijikata for their valuable input and suggestions.
References
- 1.Geha R.S., Notarangelo L.D., Casanova J.L., et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J. Allergy Clin. Immunol. 2007;120:776–94. doi: 10.1016/j.jaci.2007.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marodi L., Notarangelo L.D. Immunological and genetic bases of new primary immunodeficiencies. Nat. Rev. Immunol. 2007;7:851–61. doi: 10.1038/nri2195. [DOI] [PubMed] [Google Scholar]
- 3.Brown M.P., Grundy W.N., Lin D., et al. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc. Natl Acad. Sci. USA. 2000;97:262–7. doi: 10.1073/pnas.97.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furey T.S., Cristianini N., Duffy N., Bednarski D.W., Schummer M., Haussler D. Support vector machine classification and validation of cancer tissue samples using microarray expression data. Bioinformatics. 2000;16:906–14. doi: 10.1093/bioinformatics/16.10.906. [DOI] [PubMed] [Google Scholar]
- 5.Pirooznia M., Yang J.Y., Yang M.Q., Deng Y. A comparative study of different machine learning methods on microarray gene expression data. BMC Genomics. 2008;9(Suppl 1):S13. doi: 10.1186/1471-2164-9-S1-S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang L., Zhu J., Zou H. Hybrid huberized support vector machines for microarray classification and gene selection. Bioinformatics. 2008;24:412–9. doi: 10.1093/bioinformatics/btm579. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y., Tetko I.V., Hall M.A., et al. Gene selection from microarray data for cancer classification—a machine learning approach. Comput. Biol. Chem. 2005;29:37–46. doi: 10.1016/j.compbiolchem.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Yeang C.H., Ramaswamy S., Tamayo P., et al. Molecular classification of multiple tumor types. Bioinformatics. 2001;17(Suppl 1):S316–22. doi: 10.1093/bioinformatics/17.suppl_1.s316. [DOI] [PubMed] [Google Scholar]
- 9.Hua S., Sun Z. A novel method of protein secondary structure prediction with high segment overlap measure: support vector machine approach. J. Mol. Biol. 2001;308:397–407. doi: 10.1006/jmbi.2001.4580. [DOI] [PubMed] [Google Scholar]
- 10.Jagla B., Schuchhardt J. Adaptive encoding neural networks for the recognition of human signal peptide cleavage sites. Bioinformatics. 2000;16:245–50. doi: 10.1093/bioinformatics/16.3.245. [DOI] [PubMed] [Google Scholar]
- 11.Zien A., Ratsch G., Mika S., Scholkopf B., Lengauer T., Muller K.R. Engineering support vector machine kernels that recognize translation initiation sites. Bioinformatics. 2000;16:799–807. doi: 10.1093/bioinformatics/16.9.799. [DOI] [PubMed] [Google Scholar]
- 12.Cai Y.D., Liu X.J., Xu X., Zhou G.P. Support vector machines for predicting protein structural class. BMC Bioinformatics. 2001;2:3. doi: 10.1186/1471-2105-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding C.H., Dubchak I. Multi-class protein fold recognition using support vector machines and neural networks. Bioinformatics. 2001;17:349–58. doi: 10.1093/bioinformatics/17.4.349. [DOI] [PubMed] [Google Scholar]
- 14.Bock J.R., Gough D.A. Predicting protein–protein interactions from primary structure. Bioinformatics. 2001;17:455–60. doi: 10.1093/bioinformatics/17.5.455. [DOI] [PubMed] [Google Scholar]
- 15.Bhasin M., Raghava G.P. ESLpred: SVM-based method for subcellular localization of eukaryotic proteins using dipeptide composition and PSI-BLAST. Nucleic Acids Res. 2004;32:W414–9. doi: 10.1093/nar/gkh350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garg A., Bhasin M., Raghava G.P. Support vector machine-based method for subcellular localization of human proteins using amino acid compositions, their order, and similarity search. J. Biol. Chem. 2005;280:14427–32. doi: 10.1074/jbc.M411789200. [DOI] [PubMed] [Google Scholar]
- 17.Hua S., Sun Z. Support vector machine approach for protein subcellular localization prediction. Bioinformatics. 2001;17:721–8. doi: 10.1093/bioinformatics/17.8.721. [DOI] [PubMed] [Google Scholar]
- 18.Shi J.Y., Zhang S.W., Pan Q., Cheng Y.M., Xie J. Prediction of protein subcellular localization by support vector machines using multi-scale energy and pseudo amino acid composition. Amino Acids. 2007;33:69–74. doi: 10.1007/s00726-006-0475-y. [DOI] [PubMed] [Google Scholar]
- 19.Anderson D.C., Li W., Payan D.G., Noble W.S. A new algorithm for the evaluation of shotgun peptide sequencing in proteomics: support vector machine classification of peptide MS/MS spectra and SEQUEST scores. J. Proteome. Res. 2003;2:137–46. doi: 10.1021/pr0255654. [DOI] [PubMed] [Google Scholar]
- 20.Park K.J., Gromiha M.M., Horton P., Suwa M. Discrimination of outer membrane proteins using support vector machines. Bioinformatics. 2005;21:4223–9. doi: 10.1093/bioinformatics/bti697. [DOI] [PubMed] [Google Scholar]
- 21.Keerthikumar S., Raju R., Kandasamy K., et al. RAPID: resource of Asian primary immunodeficiency diseases. Nucleic Acids Res. 2009;37:D863–7. doi: 10.1093/nar/gkn682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanehisa M., Araki M., Goto S., et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–4. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanehisa M., Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanehisa M., Goto S., Hattori M., et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–7. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hijikata A., Kitamura H., Kimura Y., et al. Construction of an open-access database that integrates cross-reference information from the transcriptome and proteome of immune cells. Bioinformatics. 2007;23:2934–41. doi: 10.1093/bioinformatics/btm430. [DOI] [PubMed] [Google Scholar]
- 26.Keshava Prasad T.S., Goel R., Kandasamy K., et al. Human protein reference database—2009 update. Nucleic Acids Res. 2009;37:D767–72. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kandasamy K., Keerthikumar S., Goel R., et al. Human Proteinpedia: a unified discovery resource for proteomics research. Nucleic Acids Res. 2009;37:D773–81. doi: 10.1093/nar/gkn701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blake J.A., Bult C.J., Eppig J.T., Kadin J.A., Richardson J.E. The mouse genome database genotypes:phenotypes. Nucleic Acids Res. 2009;37:D712–9. doi: 10.1093/nar/gkn886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bult C.J., Eppig J.T., Kadin J.A., Richardson J.E., Blake J.A. The mouse genome database (MGD): mouse biology and model systems. Nucleic Acids Res. 2008;36:D724–8. doi: 10.1093/nar/gkm961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eppig J.T., Blake J.A., Bult C.J., Kadin J.A., Richardson J.E. The mouse genome database (MGD): new features facilitating a model system. Nucleic Acids Res. 2007;35:D630–7. doi: 10.1093/nar/gkl940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amberger J., Bocchini C.A., Scott A.F., Hamosh A. McKusick's online Mendelian inheritance in man (OMIM) Nucleic Acids Res. 2009;37:D793–6. doi: 10.1093/nar/gkn665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Botstein D., Risch N. Discovering genotypes underlying human phenotypes: past successes for Mendelian disease, future approaches for complex disease. Nat. Genet. 2003;33(Suppl):228–37. doi: 10.1038/ng1090. [DOI] [PubMed] [Google Scholar]
- 33.Glazier A.M., Nadeau J.H., Aitman T.J. Finding genes that underlie complex traits. Science. 2002;298:2345–9. doi: 10.1126/science.1076641. [DOI] [PubMed] [Google Scholar]
- 34.Freudenberg J., Propping P. A similarity-based method for genome-wide prediction of disease-relevant human genes. Bioinformatics. 2002;18(Suppl 2):S110–5. doi: 10.1093/bioinformatics/18.suppl_2.s110. [DOI] [PubMed] [Google Scholar]
- 35.Huang D., Chow T.W. Identifying the biologically relevant gene categories based on gene expression and biological data: an example on prostate cancer. Bioinformatics. 2007;23:1503–10. doi: 10.1093/bioinformatics/btm141. [DOI] [PubMed] [Google Scholar]
- 36.Kohler S., Bauer S., Horn D., Robinson P.N. Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Genet. 2008;82:949–58. doi: 10.1016/j.ajhg.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perez-Iratxeta C., Bork P., Andrade M.A. Association of genes to genetically inherited diseases using data mining. Nat. Genet. 2002;31:316–9. doi: 10.1038/ng895. [DOI] [PubMed] [Google Scholar]
- 38.Segal E., Wang H., Koller D. Discovering molecular pathways from protein interaction and gene expression data. Bioinformatics. 2003;19(Suppl 1):i264–71. doi: 10.1093/bioinformatics/btg1037. [DOI] [PubMed] [Google Scholar]
- 39.Wang K., Li M., Bucan M. Pathway-based approaches for analysis of genomewide association studies. Am. J. Hum. Genet. 2007;81:1278–1283. doi: 10.1086/522374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang H.H., Ahn J., Lin X., Park C. Gene selection using support vector machines with non-convex penalty. Bioinformatics. 2006;22:88–95. doi: 10.1093/bioinformatics/bti736. [DOI] [PubMed] [Google Scholar]
- 41.Lewis D.P., Jebara T., Noble W.S. Support vector machine learning from heterogeneous data: an empirical analysis using protein sequence and structure. Bioinformatics. 2006;22:2753–60. doi: 10.1093/bioinformatics/btl475. [DOI] [PubMed] [Google Scholar]
- 42.Radivojac P., Peng K., Clark W.T., et al. An integrated approach to inferring gene-disease associations in humans. Proteins. 2008;72:1030–7. doi: 10.1002/prot.21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boztug K., Appaswamy G., Ashikov A., et al. A syndrome with congenital neutropenia and mutations in G6PC3. N. Engl. J. Med. 2009;360:32–43. doi: 10.1056/NEJMoa0805051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van der Burg M., Ijspeert H., Verkaik N.S., et al. A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J. Clin. Invest. 2009;119:91–8. doi: 10.1172/JCI37141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.von Bernuth H., Picard C., Jin Z., et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science. 2008;321:691–6. doi: 10.1126/science.1158298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aksentijevich I., Masters S.L., Ferguson P.J., et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N. Engl. J. Med. 2009;360:2426–37. doi: 10.1056/NEJMoa0807865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huck K., Feyen O., Niehues T., et al. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J. Clin. Invest. 2009;119:1350–8. doi: 10.1172/JCI37901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reddy S., Jia S., Geoffrey R., et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N. Engl. J. Med. 2009;360:2438–44. doi: 10.1056/NEJMoa0809568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shiow L.R., Roadcap D.W., Paris K., et al. The actin regulator coronin 1A is mutant in a thymic egress-deficient mouse strain and in a patient with severe combined immunodeficiency. Nat. Immunol. 2008;9:1307–15. doi: 10.1038/ni.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kenny E.F., O'Neill L.A. Signalling adaptors used by toll-like receptors: an update. Cytokine. 2008;43:342–9. doi: 10.1016/j.cyto.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 51.Chen J., Aronow B.J., Jegga A.G. Disease candidate gene identification and prioritization using protein interaction networks. BMC Bioinformatics. 2009;10:73. doi: 10.1186/1471-2105-10-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ortutay C., Vihinen M. Identification of candidate disease genes by integrating Gene Ontologies and protein-interaction networks: case study of primary immunodeficiencies. Nucleic Acids Res. 2009;37:622–8. doi: 10.1093/nar/gkn982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bader G.D., Betel D., Hogue C.W. BIND: the biomolecular interaction network database. Nucleic Acids Res. 2003;31:248–50. doi: 10.1093/nar/gkg056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Breitkreutz B.J., Stark C., Reguly T., et al. The BioGRID interaction database: 2008 update. Nucleic Acids Res. 2008;36:D637–40. doi: 10.1093/nar/gkm1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harley J.B., Alarcon-Riquelme M.E., Criswell L.A., et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat. Genet. 2008;40:204–10. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nath S.K., Han S., Kim-Howard X., et al. A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat. Genet. 2008;40:152–4. doi: 10.1038/ng.71. [DOI] [PubMed] [Google Scholar]
- 57.Kozyrev S.V., Abelson A.K., Wojcik J., et al. Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat. Genet. 2008;40:211–6. doi: 10.1038/ng.79. [DOI] [PubMed] [Google Scholar]
- 58.Goyette P., Lefebvre C., Ng A., et al. Gene-centric association mapping of chromosome 3p implicates MST1 in IBD pathogenesis. Mucosal Immunol. 2008;1:131–8. doi: 10.1038/mi.2007.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johnson A.D., O'Donnell C.J. An open access database of genome-wide association results. BMC Med. Genet. 2009;10:6. doi: 10.1186/1471-2350-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Todd J.A., Walker N.M., Cooper J.D., et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet. 2007;39:857–64. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Plenge R.M., Cotsapas C., Davies L., et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat. Genet. 2007;39:1477–82. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Remmers E.F., Plenge R.M., Lee A.T., et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N. Engl. J. Med. 2007;357:977–86. doi: 10.1056/NEJMoa073003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Graham D.S., Graham R.R., Manku H., et al. Polymorphism at the TNF superfamily gene TNFSF4 confers susceptibility to systemic lupus erythematosus. Nat. Genet. 2008;40:83–9. doi: 10.1038/ng.2007.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ueda H., Howson J.M., Esposito L., et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–11. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- 66.Ikegami H., Awata T., Kawasaki E., et al. The association of CTLA4 polymorphism with type 1 diabetes is concentrated in patients complicated with autoimmune thyroid disease: a multicenter collaborative study in Japan. J. Clin. Endocrinol. Metab. 2006;91:1087–92. doi: 10.1210/jc.2005-1407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.