

Abstract
Background
Array genomic hybridization is being used clinically to detect pathogenic copy number variants in children with intellectual disability and other birth defects. However, there is no agreement regarding the kind of array, the distribution of probes across the genome, or the resolution that is most appropriate for clinical use.
Results
We performed 500 K Affymetrix GeneChip® array genomic hybridization in 100 idiopathic intellectual disability trios, each comprised of a child with intellectual disability of unknown cause and both unaffected parents. We found pathogenic genomic imbalance in 16 of these 100 individuals with idiopathic intellectual disability. In comparison, we had found pathogenic genomic imbalance in 11 of 100 children with idiopathic intellectual disability in a previous cohort who had been studied by 100 K GeneChip® array genomic hybridization. Among 54 intellectual disability trios selected from the previous cohort who were re-tested with 500 K GeneChip® array genomic hybridization, we identified all 10 previously-detected pathogenic genomic alterations and at least one additional pathogenic copy number variant that had not been detected with 100 K GeneChip® array genomic hybridization. Many benign copy number variants, including one that was de novo, were also detected with 500 K array genomic hybridization, but it was possible to distinguish the benign and pathogenic copy number variants with confidence in all but 3 (1.9%) of the 154 intellectual disability trios studied.
Conclusion
Affymetrix GeneChip® 500 K array genomic hybridization detected pathogenic genomic imbalance in 10 of 10 patients with idiopathic developmental disability in whom 100 K GeneChip® array genomic hybridization had found genomic imbalance, 1 of 44 patients in whom 100 K GeneChip® array genomic hybridization had found no abnormality, and 16 of 100 patients who had not previously been tested. Effective clinical interpretation of these studies requires considerable skill and experience.
Background
Chromosomal imbalance has been recognized as the most frequent cause of intellectual disability (ID) for 50 years [1-3]. Until recently, most of this genomic imbalance was diagnosed by cytogenetic analysis, but studies over the past few years have found that ID is caused by constitutional gains or losses of submicroscopic genomic segments even more often than by microscopically-apparent chromosomal aberrations [4-6]. Our ability to recognize these submicroscopic genomic changes, which are usually called copy number variants (CNVs), as the most frequent cause of ID depends on the use of Array Genomic Hybridization (AGH) (also known as array-comparative genomic hybridization, chromosomal microarray analysis, or copy number analysis). AGH can survey the entire genome for imbalance that is 1/100th the size of that detectable by conventional cytogenetic analysis.
Although AGH is now being used routinely as a clinical test for the identification of chromosomal imbalance in people with ID and other birth defects, controversy still exists regarding the most appropriate platform to use for this purpose. Initial clinical studies were done with arrays having a few thousand BACs distributed at 1-3 Mb intervals across the genome or with BACs targeted to regions where pathogenic submicroscopic deletions or duplications were known to occur. More recent studies have shown that arrays with higher resolution and genome-wide coverage provide better detection rates for pathogenic CNVs in children with ID and normal cytogenetic analysis [7-12]. Other methods have identified pathogenic CNVs that are too small to have been detected by the array platforms used in most AGH studies [13-15], so analysis at even higher resolution may be necessary to detect all pathogenic CNVs in children with ID.
Unfortunately, use of higher resolution AGH for detection of genomic imbalance is confounded because most CNVs are not pathogenic. Estimates of the mean number of apparently benign CNVs per person range from 6-824 in various studies, depending on the technology used to identify the variants and the size range used to define a CNV [16-25]. Sequencing of the complete diploid genomes of normal individuals has shown that the number of CNVs per person is actually even greater [26-29]. Distinguishing these benign CNVs from those that cause ID and other birth defects is the most serious challenge to routine clinical use of AGH.
We previously reported our findings using 100 K Affymetrix GeneChip® AGH [10,16] to perform a genome-wide survey of benign and pathogenic CNVs in 100 idiopathic ID trios, each comprised of an affected child and both unaffected parents. Here we describe the results of a study of 100 new idiopathic ID trios, as well as 54 of the trios tested previously, using 500 K Affymetrix GeneChip® AGH. We found that higher resolution AGH detected a larger number of apparently pathogenic CNVs in both groups. Many benign CNVs, including at least one that was de novo, were also detected with the 500 K AGH, but it was possible to distinguish benign and pathogenic CNVs with confidence in almost all cases.
Results
We performed AGH with 500 K Affymetrix GeneChip® arrays on 154 children with idiopathic ID and on both parents of each affected child. Fifty-four of these trios (called "the 100 K cohort") had previously been studied with lower-resolution 100 K GeneChip® AGH [10,16]; the other 100 trios (called "the new cohort") were studied by AGH for the first time with the 500 K GeneChip® arrays. Data were analyzed to determine copy number along the length of all chromosomes except the Y (for which there are no probes on either the 100 K or 500 K GeneChip® arrays), and the findings for each child were compared with those for his or her parents. Autosomal CNVs seen in the child and in at least one parent were considered likely to be benign polymorphisms. Autosomal CNVs found in the child but not in either parent were evaluated by an independent method to confirm the presence of the CNV and its de novo occurrence. CNV calls on the X-chromosome in a female child were validated by an independent method if they appeared to have occurred de novo; CNV calls on the X-chromosome in a male child were validated by an independent method whether they appeared to be de novo or to have been inherited from the mother.
We found a total of 4577 hits (putative CNVs including at least 10 contiguous SNPs called by the SMD software with a p-value below 1 × 10-8) in the 462 samples (154 trios) analyzed by 500 K GeneChip® AGH. This is an average of about 10 hits per sample, which is an underestimate of the total number of benign CNVs present because of the stringent cutoff used to obtain a false discovery rate of less than 5% (see Methods).
Within the 54 trios who were studied with both platforms, we found four times as many hits with 500 K AGH as we did with 100 K AGH (Table 1). The ratio of the number of putative CNVs called with the 500 K platform to the number called with the 100 K platform was 11.0 for CNVs between 100 kb and 200 kb in size but was lower for both smaller and larger CNVs (4.1 for those < 100 kb and 3.4 for those between 200 kb and 500 kb).
Table 1.
Putative CNVs called on 100 K and 500 K AGH in 54 trios tested with both technologies.
| Comparison | Child to Father | Child to Mother | |||
|---|---|---|---|---|---|
| Array | 100 K | 500 K | 100 K | 500 K | |
| All CNV Calls | Number | 125 | 497 | 110 | 501 |
| Number per Child | 2.31 | 9.20 | 2.04 | 9.28 | |
| Statistical Significance | t = 7.18, p < 0.0001 | t = 6.67, p < 0.0001 | |||
| CNV Calls < 100 kb | Number | 59 | 237 | 53 | 221 |
| Number per Child | 1.06 | 4.39 | 0.98 | 4.09 | |
| Statistical Significance | t = 7.82, p < 0.0001 | t = 5.19, p < 0.0001 | |||
| CNV Calls ≥ 100 kb and < 200 kb | Number | 13 | 130 | 12 | 146 |
| Number per Child | 0.24 | 2.41 | 0.22 | 2.70 | |
| Statistical Significance | t = 5.39, p < 0.0001 | t = 6.27, p < 0.0001 | |||
| CNV Calls ≥ 200 kb and < 500 kb | Number | 33 | 101 | 25 | 98 |
| Number per Child | 0.61 | 1.87 | 0.46 | 1.81 | |
| Statistical Significance | t = 4.66, p < 0.0001 | t = 5.25, p < 0.0001 | |||
Two-tailed statistical significance was calculated with Student's t-test.
The 4577 putative CNVs called in all 154 trios were subjected to further bioinformatic analysis (see Methods) to produce a final annotated list of 58 apparently de novo CNVs and two cases of mosaic trisomy that were called in 50 patients. These apparent genomic imbalances were subjected to validation by independent methods. Thus, an apparent de novo CNV call that required independent validation was made in about 1 child in 3. Thirty-three of these CNVs in 30 patients and both cases of mosaic trisomy were confirmed to be de novo by an independent method and are described in detail below. The other putative CNVs were found to be present in both of the parents as well as in the child (false negative AGH calls in the parents) in two instances or could not be confirmed to be present in the child (false positive AGH calls) in 22 instances. Altogether, false positive CNV calls were made in 21 (13.6%) of the 154 trios studied and false negative CNV calls were made in 2 (1.3%) of the 154 trios studied. In one other trio (Family 5202), an apparent de novo deletion of chromosome 14 called on AGH in the child was found by FISH to be a duplication of the region in both parents instead.
Nineteen of 100 children with ID in the new cohort were found by Affymetrix 500 K GeneChip® AGH to have de novo genomic imbalance that was confirmed by FISH, MLPA, AGH on an Agilent® 244 K platform or cytogenetic re-analysis (Table 2). One of these children (Patient 8056) had mosaic trisomy 9, and two were found to have de novo unbalanced reciprocal translocations - a der(10)t(2;10)(q37;q26.13) in Patient 873 and a der(4)t(4;8)(p16.1;p23.1) in Patient 5814 - each producing both a terminal duplication and a terminal deletion identified by AGH. We found de novo submicroscopic deletions in 13 other patients and de novo submicroscopic duplications in three other patients. The deletions ranged in size from 89 kb to 11.0 Mb; six were less than 1 Mb. The duplications ranged in size from 362 kb to 11.1 Mb; one was less than 1 Mb.
Table 2.
Genomic imbalance and uniparental disomy detected by 500K GeneChip® AGH in the new cohort.
| Patient | Change | Location | SNP Count | Start → End |
Size (bp) | Validation | Phenotype | RefSeq Genes Involved* | Comments | Interpretation |
|---|---|---|---|---|---|---|---|---|---|---|
| 9133 | Deletion | 1p36.32-p36.33 | 198 | 769,185 → 3,581,308 |
2,812,123 | FISH, MLPA | 9 year-old female with obesity, moderate cognitive impairment, myoclonus, polyphagia, hypotonia, narrow frontal area, deep-set eyes, prominent orbital rims, short nose with low nasal bridge and upturned nasal tip, midface retrusion, short philtrum, tented upper lip, thoracic kyphosis, small distal phalanges of the toes, strabismus, and 11 ribs | ~70 genes including AGRN, GNB1, PEX10, PRKCZ, SKI, and TP73 | This CNV is included in the 1p36 deletion syndrome critical region, and the patient's clinical features are compatible with that syndrome [73]. | Pathogenic |
| 873 | Duplication† | 2q37 | 1801 | 231,577,285 → 242,663,303 |
11,086,018 | FISH, MLPA | 15 year-old male with severe cognitive impairment, birth weight < 1st centile, birth length < 1st centile, head circumference at birth < < 10th centile, hypotonia, microcephaly, contractures of hips and knees, hypoplastic scrotum, undescended testes, prominent, cup-shaped ears, narrow bifrontal diameter, broad nasal root, prominent epicanthal folds, bilateral clinodactyly V, ataxic gait, progressive joint contractures and muscle wasting of lower extremities, mixed hearing loss and hypoplastic inferior cerebellar vermis, partial dysgenesis of corpus callosum, and narrow pons and brain stem on MRI | ~100 genes including AGXT, ATG16L1, CAPN10, CHRND, CHRNG, COL6A3, D2HGDH, GBX2, HDAC4, MLPH, PDCD1, PER2, SAG and UGT1A1 | Pathogenic | |
| Deletion† | 10q26.13 | 1920 | 126,415,527 → 134,032,911 |
7,617,384 | ~75 genes including ADAM12, CTBP2, DOCK1, DPYSL4, FGFR2, OAT, and UROS | |||||
| 6473 | Deletion | 4p16.3 | 337 | 190,631 → 3,277,436 |
3,086,805 | Cytogenetic re-analysis, FISH | 3 year-old male with fetal growth retardation, length 2 standard deviations below the mean for age, weight 3-4 standard deviations below the mean, head circumference 3-4 standard deviations below the mean, global developmental delay, seizures, triangular face, small jaw, posteriorly rotated ears, 2° hypospadias, and ataxia | ~45 genes including ADD1, FGFR3, HTT, IDUA, LETM1, PDE6B, SH3BP2, and WHSC1 | The deletion in this patient includes the Wolf- Hirschhorn syndrome critical region, and the clinical features are compatible with that syndrome [74]. | Pathogenic |
| 5814 | Deletion† | 4p16.1-p16.3 | 1520 | 13,255 → 8,472,657 |
8,459,402 | Cytogenetic re-analysis, MLPA | 13 month-old female with length below the 3rd centile, microcephaly, developmental delay, bilateral preauricular pits, and submucus cleft palate | ~90 genes including ADD1, ADRA2C, CRMP1, EVC | The deletion includes the Wolf- Hirschorn syndrome critical region. | Pathogenic |
| Duplication† | 8p23.1-p23.3 | 2579 | 180,568 → 6,898,076 |
6,717,508 | 20 genes including ARHGEF10, CLN8, DLGAP2, and MCPH1 | |||||
| 216 | Deletion | 6p21.33 | 42 | 29,937,087 → 30,026,517 |
89,430 | MLPA | 18 year-old male with postnatal onset growth retardation, unilateral sensorineural deafness, narrow face, bulbous nasal tip and mild intellectual disability. | HCG9, MICD and 5 pseudogenes | Deletion is homozygous in child, heterozygous in both parents (not de novo mutation) | Not pathogenic for ID |
| 3160 | Deletion | 6 q21-q22.31 | 1596 | 111,979,175 → 121,506,916 |
9,527,741 | FISH | 8 year-old female with moderate developmental delay, hypotonia, microcephaly, brachycephaly, epicanthic folds, small ears with hypoplastic lobes, and micrognathia | ~40 genes including ASF1A, COL10A1, FRK, FYN, GOPC, HDAC2, LAMA4, MCM9, PLN, TSPYL1, and WISP3 | Pathogenic | |
| 2200 | Deletion | 7p15.3 | 2320 | 14,141,506 → 24,950,414 |
10,808,908 | FISH | 11 1/2 year-old female with head circumference at the 2nd centile, mild cognitive impairment, sensorineural hearing loss, cleft palate, craniosynostosis, unilateral ptosis and esotropia, orbital rim hypoplasia, malar and midface hypoplasia, low-set ears with incomplete out-folding of superior helix, brachydactyly and syndactyly of digits, broad thumbs, decreased range of motion in elbows, and leg length discrepancy | ~40 genes including DFNA5, DGKB, DNAH11, FAM126A, HDAC9, IL6, and RAPGEF5 | Pathogenic | |
| 9938 | Deletion | 7q22.1 | 170 | 98,211,585 → 100,553,755 |
2,342,170 | FISH | 14 year-old female with height < 5th centile, weight < 5th centile, head circumference < 5th centile, severe cognitive impairment, left sensorineural hearing loss, close-set eyes, broad nasal root, marked retrognathia, high-arched palate, small and narrow feet, short 2-5th toes with hypoplastic nails, atrio-vetricular septal defect, and polyarticular arthritis | ~70 genes including ACHE, ACTL6B, CYP3A5, EPO, MUC3A, SERPINE1, SMURF1, and TFR2 | Pathogenic | |
| 1594 | Duplication | 8 q12 | 1220 | 58,388,614 → 65,306,097 |
6,917,483 | FISH | 1 1/2 year-old female with height > 97th centile, head circumference at 2nd-5th centile, developmental delay, Duane anomaly, broad glabella, epicanthic folds with telecanthus, upslanting palpebral fissures, pre-auricular pits, large ears, atrial and ventricular septal defects, and renal reflux | 15 genes including ASPH, CHD7, RAB2A, RLBP1L1, TOX, and TTPA | Pathogenic | |
| 663 | Deletion | 9 p13.3 | 800 | 34,144,847 → 38,736,451 |
4,591,604 | FISH | 5 1/2 year-old female with height at the 5th centile, developmental delay, tremor, ocular hypertelorism, epicathal folds, double hair whorl, bilateral ptosis, short upturned nose, flattened philtrum, underdeveloped genitalia, and pigmentary retinal changes | ~75 genes including CNTFR, DNAI1, DNAJB5, FANCG, GALT, GBA2, GNE, GRHPR, NPR2, PAX5, RECK, SHB, TPM2, UNC13B, and VCP | The deleted region in this patient is completely included in the region deleted in patient 9346. | Pathogenic |
| 9346 | Deletion | 9p11.2-p13.3 | 880 | 33,702,471 → 44,744,675 |
11,042,204 | FISH | 9 1/2 year-old female with moderate cognitive impairment, seizures, tremor, cataract, broad frontal area with bossing, arched eyebrows, low nasal bridge, and short, upturned nose | ~85 genes including CNTFR, DNAI1, DNAJB5, FANCG, GALT, GBA2, GNE, GRHPR, NPR2, PAX5, RECK, SHB, TPM2, UNC13B, and VCP | The deleted region in this patient includes the entire segment deleted in Patient 663. | Pathogenic |
| 523 | Deletion | 9q34.3 | 36 | 139,516,033 → 139,814,485 |
298,452 | FISH | 4 year-old female with moderate developmental delay, hypotonia, microcephaly, flat face with upslanting palpebral fissures, ocular hypotelorism, synophrys, and anteverted nares | 7 genes including EHMT1 | This deletion is within the critical region for the 9q subtelomeric deletion syndrome[75], and the child's clinical features are compatible with that syndrome. | Pathogenic |
| 8056 | Mosaic Trisomy | 9 | 27,641 | whole chromosome | FISH | 2 1/2 year-old male with weight < 5th centile, developmental delay, preauricular skin tags, hypospadias, and cryptorchidism | Numerous | Clinical features compatible with mosaic trisomy 9 syndrome[76,77] | Pathogenic | |
| 6904 | Uniparental Disomy | 11p11.2-pter | See Table 3 | 196,767 → 44,589,530 |
44,392,763 | Micro-satellite markers | 11 year-old female with height < 5th centile, gross and fine motor delay, hypotonia, and moderate mental handicap | Numerous | Mosaic paternal isodisomy; phenotype not compatible with Beckwith-Wiedemann syndrome | Uncertain |
| 9897 | Deletion | 13q33.3-q34 | 530 | 107,271,189 → 109,368,996 |
2,097,807 | FISH | 10 year-old female with fetal growth retardation, moderate cognitive impairment, upslanting palpebral fissures, and retrognathia | 6 genes including IRS2, LIG4, and MYO16 | Pathogenic | |
| 818 | Deletion | 14q11.2 | 24 | 21,929,710 → >22,036,502 |
106,792 | Fosmid FISH (variable)§ | 6 1/2 year-old male with weight < 5th centile, height at 5th centile, mild cognitive impairment with particular delay in language, mild mid-face hypoplasia with narrow high-arched palate, mild micrognathia, pre-auricular pit, joint laxity, bilateral clinodactyly of hands, and bilateral 2-3 syndactyly of toes | Multiple T-cell receptor alpha-chain V, J, and region genes | Highly polymorphic region | Not pathogenic for ID |
| 1658 | Uniparental Disomy | 16 | See Table 3 | Whole chromosome | Whole chromosome | Micro-satellite markers | 5 year-old female with normal growth, severe mental handicap, seizures, self-abusive behaviour, deep and dark creases under the eyes, mild mid-face hypoplasia, and large mouth | Numerous | Uncertain | |
| 2106 | Deletion | 17q21.31 | 149 | 41,049,321 → 41,564,451 |
515,130 | FISH | 15 year-old male with fetal growth retardation, mild cognitive impairment, attention deficit disorder, sagittal craniosynostosis, long face with malar hypoplasia and mild rectrognathia, short and upslanting palpebral fissures, low-set ears, unilateral cryptorchidism, partial agenesis of the corpus callosum, and dilatation of the aortic root | 5 genes including MAPT | The deleted segment includes the critical region for the 17q21.31 deletion syndrome, [78] and this patient's clinical features are compatible with that syndrome. | Pathogenic |
| 8619 | Deletion | 21q22.11 | 13 | 33,902,218 → 34,087,893 |
185,675 | Agilent 244 K AGH | 24 year-old male with prenatal and postnatal growth retardation, moderate to severe intellectual disability, severe hypotonia, microcephaly, metopic craniosynostosis, cleft palate, down-slanting palpebral fissures, low-set ears, wide nasal base, retrognathia, tetralogy of Fallot, cryptorchidism, and joint hyperextensibility | 5 genes including SYNJ1 | No polymorphisms of region reported | Uncertain |
| Deletion | 22q11.2 | 110 | 19,062,809 → 19,785,125 |
722,316 | FISH | 14 genes including BCR, DGCR8, HIRA, MAPK1, PRODH, SNAP29, SEPT5, SERPIND1, and TBX1 | The deleted segment is included in the 22q11 deletion syndrome critical region, and the phenotype is compatible with other reported cases of distal 22q11.2 microdeletion [79-81] | Pathogenic | ||
| 9979 | Duplication | 22q11.21 | 57 | 19,429,297 → 19,791,607 |
362,310 | FISH | 20 year-old female with short stature, mild mental deficiency, cleft palate, and micrognathia | 9 genes including PI4KA SERPIND1, LZTR1, SNAP29 | Polymorphic region | Uncertain |
| 1128 | Duplication | Xq12-q21.1 | 475 | 67,088,023 → 76,204,344 |
9,116,321 | FISH | 11 year-old male with normal growth, severe developmental delay, hypotonia, brachycephaly, bilateral epicanthic folds, and posteriorly rotated ears with hypoplastic helices | ~60 genes including ABCB7, DLG3, EDA, EFNB1, GJB1, IGBP1, IL2RG, OPHN1 NAP1L2, NLGN3, PHKA1, SLC16A2, TAF1, and ZDHHC15 |
Pathogenic | |
The table includes all de novo CNVs, mosaic trisomy and UPD detected by 500 K AGH and confirmed by an independent method in 100 children with idiopathic ID. Breakpoints are shown on Human Genome Assembly Build 36.1.
* The approximate number of RefSeq genes for each CNV is given, but only the most likely genes for the phenotype are named.
† Unbalanced reciprocal translocation.
§ Interphase FISH in patient 818 showed some cells with no signals, some with 1 signal and some with two signals for a probe in the region of the deletion detected by AGH. This was interpreted as evidence of somatic mosaicism for this deletion.
Using 500 K Affymetrix GeneChip® AGH, we also confirmed the genomic imbalance that had previously been identified in 10 of the 54 ID trios from the 100 K cohort (Table 3 and Additional File 1: Supplemental Table S1). In addition, we identified and confirmed by FISH two de novo CNVs that were not called on the 100 K assay - a 1.6 Mb duplication of 8q23.2-23.3 in Patient 3890 and a 1.5 Mb deletion of 4p16.3 in Patient 4840 (Table 3 and Figure 1).
Table 3.
Genomic imbalance detected by 500K AGH in 54 idiopathic ID trios from the 100K cohort.
| Affymetrix 100K AGH | Affymetrix 500K AGH | |||||||
|---|---|---|---|---|---|---|---|---|
| Family | Change | Location | Start → End | Size (bp) | Change | Location | Start → End | Size (bp) |
| 1895 | Deletion | 13q12.11-q12.13 | 18,867,056 → 24,517,730 |
5,650,674 | Deletion | 13q12.11-q12.12 | 18,876,037 → 24,330,232 |
5,454,195 |
| 3476 | Deletion | 4q21.21-q22.1 | 82,008,594 → 93,076,278 |
11,067,684 | Deletion | 4q21.21-q22.1 | 82,429,950 → 91,434,337 |
9,004,387 |
| 3890 | Normal | Duplication | 8q23.2-q23.3 | 111,442,951 → 113,003,770 |
1,560,819 | |||
| 4794 | Duplication | 16p13.3 | 925,718 → 3,864,938 |
2,939,220 | Duplication | 16p13.3 | 2,681,813 → 3,927,524 |
1,245,711 |
| 4818 | Deletion | 12q14.2-q15 | 63,342,649 → 66,780,095 |
3,437,446 | Deletion | 12q14.2-q15 | 63,362,084 → 66,737,699 |
3,375,615 |
| 4840 | Normal | Deletion | 4p16.3 | 1,346,924 → 2,846,261 |
1,499,337 | |||
| 5003 | Deletion | 2p16.3 | 50,799,281 → >51,120,644 |
321,363 | Deletion | 2p16.3 | 50,829,675 → 51,120,302 |
290,627 |
| 5566 | Deletion | 14q11.2 | 20,741,117 → 20,918,741 |
177,624 | Deletion | 14q11.2 | 20,787,740 → 20,988,716 |
200,976 |
| 5994 | Mosaic Trisomy 9 | Whole Chromosome | Mosaic Trisomy 9 | Whole Chromosome | ||||
| 6545 | Deletion | 7p22.2-p22.1 | 3,498,135 → 7,134,218 |
3,636,083 | Deletion | 7p22.2-p22.1 | 3,657,805 → 6,165,597 |
2,507,792 |
| 7807 | Deletion | 22q12.1 | 26,144,210 → 27,557,971 |
1,413,761 | Deletion | 22q12.1 | 26,293,416 → 27,462,458 |
1,169,042 |
| 8326 | Deletion | 14q11.2 | 19,584,863 → 21,207,935 |
1,623,072 | Deletion | 14q11.2 | 19,592,409 → 21,256,822 |
1,664,413 |
The table includes all de novo CNVs and mosaic trisomy detected by 500 K AGH and confirmed by an independent method in a selected group of 54 trios who had previously been tested by 100 K GeneChip® AGH [10]. Breakpoints are shown on Human Genome Assembly Build 36.1.
Figure 1.
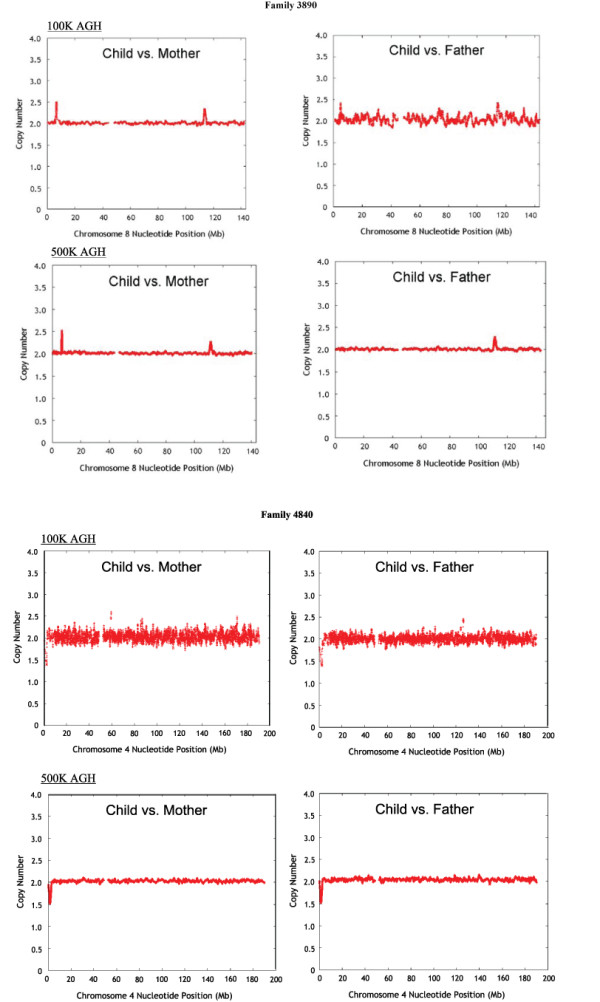
De novo CNVs detected with 500 K but not 100 K AGH in children with idiopathic ID. The plots show in silico comparison of estimated copy number in child versus mother (left) and child versus father (right) at each position along the chromosome. Upper panel: Smoothed copy number plots for chromosome 8 in Family 3890. Affymetrix 100 K AGH is shown with a 59 SNP window, and Affymetrix 500 K AGH is shown with a 170 SNP window. Note duplication at 111,442,951 to 113,003,770 bp that is apparent on 500 K AGH but was not called on our original analysis of the 100 K AGH data. The CNV is represented by 59 SNPs on the 100 K array. Lower panel: Smoothed copy number plot for chromosome 4 in Family 4840. Affymetrix 100 K AGH is shown with a 13 SNP window, and Affymetrix 500 K AGH is shown with a 145 SNP window. Note deletion at 1,346,924 to 2,846,261 bp that is apparent on 500 K AGH but was not called as de novo by 100 K AGH on our initial analysis. This CNV is represented by 17 SNPs on the 100 K array.
We found two instances of uniparental disomy (UPD), diagnosed by the occurrence of mendelian inconsistency in a region with a normal copy number of 2 [30], among the 100 ID trios in the new cohort studied by 500 K Affymetrix GeneChip® AGH (Table 4 and Figures 2A and 2B). Patient 6904 has mosaic paternal isodisomy of most of the short arm of chromosome 11. Patient 1658 has maternal UPD 16, being heterodisomic for the central portion of the chromosome and isodisomic for both ends. Both cases were confirmed to be disomic with microsatellite markers (Figures 2C and 2D).
Table 4.
UPD detected in 100 children with idiopathic ID from the new cohort.
| Patient | Chromosome | Affected Region | SNPs in Region of UPD | Interpretation | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Start | End | Size (bp) | Total | Paternal UPD | Maternal UPD | BPI* | MI* | |||
| 6904 | 11 | 196,767 | 44,589,530 | 44,392,763 | 9,857 | h = 192* i = 456* |
h = 89 i = 1 |
59 | 8 | Paternal Isodisomy |
| 1658 | 16 | 14,139 | 11,559,620 | 11,545,481 | 2,837 | h = 0 i = 1 |
h = 193 i = 188 |
5 | 19 | Maternal Isodisomy |
| 11,559,620 | 84,641,380 | 73,081,760 | 11,698 | h = 0 i = 0 |
h = 638 i = 3 |
12 | 95 | Maternal Heterodisomy | ||
| 84,641,383 | 88,668,856 | 4,027,473 | 772 | h = 0 i = 0 |
h = 25 i = 56 |
1 | 7 | Maternal Isodisomy | ||
The table includes all UPD detected by 500 K GeneChip® AGH and confirmed by an independent method in 100 children with idiopathic ID. All mendelian inconsistencies observed were single. SNPs that are not listed as paternal UPD, maternal UPD, BPI* or MI* were uninformative with respect to UPD. Breakpoints are shown on Human Genome Assembly Build 36.1.
* Abbreviations used: h = heterodisomy, i = isodisomy, BPI = biparental inheritance, MI = mendelian inconsistency.
Figure 2.

Uniparental disomy detected with Affymetrix 500 K AGH in two patients with idiopathic ID. A) The child in Family 6904 was found to have mosaic paternal uniparental disomy, probably isodisomy, of chromosome 11 p15.5-p11.2. SNP genotypes obtained by Affymetrix 500 K AGH and interpreted for the trio as described in the Methods are shown along the length of chromosome 11. B) The child in Family 1658 was found to have maternal uniparental disomy for all of chromosome 16. The ends of both chromosome arms (proximal to 11,559,620 bp and distal to 84,641,383 bp) appear to be isodisomic; the central portion of the chromosome is heterodisomic. SNP genotypes obtained by Affymetrix 500 K AGH and interpreted for the trio as described in the Methods are shown along the length of chromosome 16. C) Allelic imbalance, compatible with paternal isodisomy and mosaicism, for two informative microsatellite markers in the involved region of chromosome 11 in the child in Family 6904. The location of each marker is shown in brackets. D) Maternal heterodisomy for two informative microsatellite markers in the involved region of chromosome 16 in the child in Family 1658. The location of each marker is shown in brackets.
We judged the mosaic trisomy 9, both unbalanced reciprocal translocations, 11 of the other de novo deletions, and two of the other de novo duplications found in the new cohort to be pathogenic (Table 2). A mosaic 107 kb de novo deletion of chromosome 14 q11.2 (Patient 818) and a homozygous 89 kb deletion of the HLA-G region that resulted from transmission by parents who both carried heterozygous deletions of the same region (Patient 216) were judged to be benign variants. A 186 kb deletion of chromosome 21q22.11 (Patient 8619), a 362 kb duplication of chromosome 22 q11.21 (Patient 9979) and both cases of UPD (Patients 1658 and 6904) were of uncertain clinical significance.
Of the two de novo CNVs identified by 500 K Affymetrix GeneChip® AGH in the 44 children with ID whose 100 K GeneChip® AGH studies had been interpreted as normal (Table 3), one is likely to be pathogenic and the other is of uncertain clinical significance. A 1.5 Mb deletion of 4p16.3 in Patient 4840 is pathogenic because of its size, the inclusion of two genes that have been implicated in the Wolf-Hirschhorn syndrome (WHSC1 and WHSC2), overlap with known pathogenic CNVs, and a compatible clinical phenotype. The 1.6 Mb duplication of 8q23.2-23.3 in Patient 3890 is of unknown clinical significance. It includes no RefSeq genes but involves a large (1.6 Mb) genomic region, most of which has never been reported to be polymorphic in normal individuals.
Discussion
Because its detection rate for pathogenic genomic imbalance is much higher than that of conventional cytogenetic analysis, a consensus has developed that AGH should be used clinically for the evaluation of patients with ID and other birth defects [31-38]. It is clear that AGH using "targeted" arrays that only include probes for genomic regions known to be involved in microdeletion or microduplication syndromes has substantially lower detection rates for CNVs that cause ID than AGH using arrays that provide genome-wide coverage [37,39-41]. Beyond this, however, there is no agreement regarding the kind of array, the distribution of probes across the genome, or the resolution that is most appropriate for clinical use. Although BAC arrays were initially used, most clinical laboratories now prefer oligonucleotide arrays because high-quality platforms that produce consistent results are reliably available from commercial sources. In addition, the use of larger numbers of smaller probes on oligonucleotide arrays permits more precise delineation of the breakpoints of CNVs that are detected, which facilitates genotype-phenotype correlation and clinical interpretation. AGH with a SNP array provides the additional advantage of generating genotypes that can be used to verify family relationships and find uniparental disomy as well as a second method (in addition to hybridization intensity) for identifying genomic imbalance [10,42-44].
We previously reported that 100 K Affymetrix GeneChip® AGH is a robust platform for the detection of pathogenic CNVs in patients with ID [10]. Here we show that the detection rate of CNVs among such patients is higher with 500 K GeneChip® AGH than with 100 K GeneChip® AGH. We made about four times as many CNV calls overall with the 500 K platform as with the 100 K platform when using the same method of bioinformatic analysis in 54 trios studied with both technologies (Table 1). We also found 18 instances of pathogenic genomic imbalance in 16 of 100 children with ID and normal cytogenetic analysis studied by 500 K GeneChip® AGH (Table 2), compared to 11 instances of pathogenic genomic imbalance in 11 of 100 similarly-ascertained children tested by 100 K GeneChip® AGH in our previous study. Although the higher detection rate we observed with the 500 K platform may have occurred by chance because we just happened to include a few more patients with such genomic changes in the new cohort than in the 100 K cohort, we also found two additional de novo CNVs by 500 K GeneChip® AGH among the 44 children whose 100 K AGH was interpreted as normal in our earlier studies (Figure 1 and Table 3).
We detected three apparently de novo CNVs smaller than 200 kb among the 100 trios tested with 500 K GeneChip® AGH in the new cohort (a 107 kb deletion of chromosome 14 in Patient 818, a 186 kb deletion of chromosome 21 in Patient 8619, and an 89 kb deletion of chromosome 6 in Patient 216 that was actually a homozygous loss inherited from two heterozygous parents), but none of these CNVs was clearly pathogenic. The overall size distribution of pathogenic CNVs detected by 500 K GeneChip® AGH among 154 children with idiopathic ID in the present study is similar to that observed by 100 K GeneChip® AGH among 100 children with idiopathic ID whom we studied previously [10] (see Additional File 2: Supplemental Figure S1). The higher detection rate on the 500 K array therefore appears to be related more to better probe coverage in relevant genomic regions and an improved ability to distinguish CNVs from background noise, rather than to a capacity to identify much smaller pathogenic CNVs. This is illustrated in Families 3890 and 4840 (Figure 1), in which a 1.6 Mb duplication of 8q23.2q23.3 and a 1.5 Mb deletion of 4p16.3, respectively, are obvious on the 500 K AGH but were not called on the 100 K analysis. In retrospect, the 4p16.3 deletion in Patient 4840 can be seen on the 100 K AGH copy number plot despite the noisy data, but it was not called by either the automated analysis or visual inspection of these plots when the initial study was done. Our failure to detect the de novo duplication of 8q23.2q23.3 in Patient 3890 was probably caused by the noisy data in the father's study.
Distinguishing benign CNVs from those that cause ID and other birth defects is a critical issue in routine clinical use of AGH. Benign CNVs occur in all people and are a major source of genetic variation in the normal population [21,27,29]. Most apparently benign CNVs over 2 kb in size occur as polymorphisms with minor allele frequencies of at least 5% [21] and are inherited from a parent [21,23,45,46].
Benign and pathogenic CNVs can usually be distinguished in patients with ID and other birth defects by inheritance and genotype-phenotype correlation [5,33,47]. In this study, we identified a mean of about 10 CNVs per subject in the 154 ID trios tested by 500 K AGH. The vast majority of these CNVs were characterized as benign because they were inherited from a normal parent. Genomic imbalance that occurs de novo in a patient with ID whose parents are normal is more likely to be pathogenic than genomic imbalance that was inherited unchanged from a normal parent. We performed AGH on both parents of every child with ID to determine the inheritance of the CNVs found in the child, but this is sometimes not possible in clinical practice. In such instances it is necessary to infer likely de novo occurrence by information obtained from populations that have previously been studied [19,22-24,48,49]. Great care must be taken to avoid misinterpretation when this is done, especially if the available data were obtained with lower resolution AGH, the phenotypic characteristics of the comparison population are uncertain, reported polymorphic CNVs do not have exactly the same breakpoints as the CNV of interest, or the population frequency of a previously-reported CNV is unknown.
Compelling evidence that a CNV in a person with ID is pathogenic exists if the genomic imbalance is known to cause the patient's phenotype in other individuals, e.g., if a child with del 9q34.3 has features of the 9q subtelomeric deletion syndrome (Patient 523), a child with del 1p36.32p36.33 has features of the 1p36 deletion syndrome (Patient 9133), or a child with del 17q21.31 has features of the syndrome associated with this deletion (Patient 2106). Pathogenicity is also supported when a CNV includes a gene that is known to cause the patient's phenotype when inactivated (if the CNV is a deletion) or over-expressed (if the CNV is a duplication). On the other hand, a CNV is unlikely to be pathogenic if it involves a highly polymorphic region in which genomic loss (or gain, whichever is present in the patient) of the entire involved segment is known to occur in normal people.
If a direct genotype-phenotype correlation of this kind cannot be made in a particular case, certain genetic features of the CNV may provide clues to its pathogenicity. CNVs that are larger and those that involve gene-rich regions are more likely to be pathogenic than CNVs that are smaller and involve only gene-poor regions [5,47]. In addition, clinical experience suggests that deletions are more likely to be pathogenic than duplications [47]. The genetic content of a CNV may also make pathogenicity more or less likely. For example, involvement of a gene that lies within a pathway that is known to contain other dosage-sensitive genes associated with a similar phenotype strengthens the possibility of pathogenicity, while a CNV that does not contain any genes that are expressed in relevant tissues during embryogenesis is unlikely to be pathogenic for ID.
There are, of course, exceptions to each of these "rules". Some benign CNVs arise de novo [21,23,45,46,50], as appears to have occurred in the de novo deletion of chromosome 14q11.2 we found in Patient 818. The 107 kb region involved is highly polymorphic and contains several T-cell receptor variable region genes. On the other hand, some CNVs that are inherited from a normal parent are pathogenic for ID. Examples include maternal transmission of a UBE3A deletion to a child with Angelman syndrome [51], maternal transmission of a MECP2 duplication to a son [52], and CNVs such as dup 22q11.2 [53,54] or del 1q21.2 [55] that can cause ID but exhibit incomplete penetrance.
Although large (> 250 kb) CNVs are often pathogenic, they may be benign [19,29]. Most benign CNVs are small (< 250 kb) [19,27,29], and it seems probable that the smaller a CNV, the more likely it is to be benign. Nevertheless, no clear size distinction exists between benign and pathogenic CNVs. We found pathogenic CNVs as small as the 298 kb deletion of 9q34.3 in Patient 523 in this study, and others have reported even smaller pathogenic CNVs [13-15,56-58]. Expression patterns, functional annotation and animal models can provide important clues to pathogenicity in some cases, but without knowledge of the phenotypic effects of a copy number alteration in humans, one can rarely, if ever, be certain whether a novel gain or loss of a particular genomic region can produce ID or other birth defects.
In our 500 K AGH study of 154 ID trios, 58 de novo CNVs were called by bioinformatic analysis, and 33 of these CNVs were confirmed and shown to be de novo by an independent method. Because we could assess the phenotypes of our patients in detail and correlate the findings with those obtained by AGH, we were able to determine with confidence whether the genomic imbalance we observed was pathogenic or not in every case studied except three (Tables 2 and 3). Such genotype-phenotype correlation is critical to determining the effects of novel CNVs detected by AGH in patients with ID.
A CNV of uncertain clinical significance was encountered in three (1.9%) of the 154 trios analyzed in this series - a 362 kb duplication of 22q11.21 in Patient 9979 (Table 2), a 186 kb deletion of 21q22.11 in Patient 8619 (Table 2) and a 1.6 Mb duplication of chromosome 8q23.2q23.3 in Patient 3890 (Table 3). This rate of CNVs of uncertain clinical significance is similar to that reported in large series of patients with ID and other birth defects studied by AGH with "targeted" chips [31,35,59].
We were uncertain of the clinical significance of either case of UPD that we detected. Although only a few liveborn children with UPD 16 have been recognized, the reported experience does not suggest that UPD 16 can cause the abnormalities observed in Patient 1658 [60,61]. Paternal UPD 11p15 can produce Beckwith-Wiedemann syndrome [62], but this phenotype is very different from that observed in the affected child in Family 6904. However, as both of these cases involved isodisomy of a portion of the chromosome, we cannot rule out the possibility that the abnormal phenotype was produced by homozygosity for a recessive mutant allele [63,64]. Although the detection of UPD in addition to alterations of copy number is a theoretical benefit of using an array that includes probes for SNPs, the clinical utility of genome-wide screening for UPD in patients with idiopathic ID and other birth defects is uncertain.
We detected more pathogenic CNVs with 500 K AGH than with 100 K AGH, but some CNVs that were present among our patients were not detected using the 500 K assay. For example, our 500 K GeneChip® analysis failed to identify a pathogenic 83 kb deletion of chromosome 16p13.3 (3,862,993 bp to 3,945,522 bp) involving the CREBBP gene (Patient 5121). This de novo deletion was found by AGH on the Agilent® 244 K platform and was confirmed by MLPA. The patient is an 8 year-old boy whose clinical features are characteristic of the Rubinstein-Taybi syndrome, which has been associated with deletions and other mutations of CREBBP in other patients [65,66]. The 83 kb genomic region deleted in our patient is poorly represented on the 500 K GeneChip® arrays, with a total of only 15 SNPs. SNP arrays have uneven genomic coverage, and the addition of non-polymorphic oligonucleotide probes to the design of arrays like the one used in this study has been shown to provide substantially better detection of CNVs [21].
Conclusion
Affymetrix GeneChip® 500 K array genomic hybridization performed in individuals with idiopathic intellectual disability detected pathogenic genomic imbalance in 10 of 10 patients in whom 100 K GeneChip® array genomic hybridization found genomic imbalance, 1 of 44 patients in whom 100 K GeneChip® array genomic hybridization had found no abnormality, and 16 of 100 patients who had not previously been tested. Further improvements in array design, ongoing improvements in AGH software, and continuing enhancement of resources like DECIPHER https://decipher.sanger.ac.uk/ and the Toronto Database of Genomic Variants http://projects.tcag.ca/variation/ are helping to establish AGH as the primary clinical tool for recognition of genomic imbalance that causes ID and other birth defects [33,34,37,41,67-70]. It seems likely, however, that no perfect AGH platform for detection of pathogenic CNVs may ever exist and that effective clinical interpretation of these studies will continue to require considerable skill and experience [33,50,71].
Methods
Patients and Families
We studied 100 children with idiopathic ID who had not been studied by AGH before ("the new cohort") and 54 of the idiopathic ID patients whom we had previously tested with 100 K Affymetrix GeneChip® AGH ("the 100 K cohort"). Ten of the 54 patients in the 100 K cohort had previously been found to have pathogenic genomic imbalance; the other 44 patients had previously been reported to have normal 100 K GeneChip® AGH (see Additional File 1: Supplemental Table S1) [10,16]. We also performed 500 K AGH on both unaffected parents of each child.
All of the children were assessed by a clinical geneticist who was unable to determine the cause of the ID despite thorough clinical evaluation and testing that included routine karyotyping with at least 450-band resolution. Subjects were selected for AGH testing because they had ID or developmental delay and at least one of the following additional clinical features: one major malformation, microcephaly, abnormal growth, or multiple minor anomalies. Informed consent was obtained from each family, and assent was also obtained from the child, if possible. The study was approved by the University of British Columbia Clinical Research Ethics Board.
DNA Preparation
DNA was extracted from whole blood with a Gentra Puregene DNA Purification Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The DNA was precipitated in 70% alcohol, resuspended in hydration solution, and stored at 4°C.
Hybridization to GeneChip® Mapping 500 K Arrays
DNA degradation, labeling, hybridization, and scanning were performed according to the manufacturer's protocols http://www.affymetrix.com using an Affymetrix Fluidics Station 450, Affymetrix Hybridization Oven 640 and Affymetrix GeneChip Scanner 3000 (Affymetrix, Inc., Santa Clara, CA, USA).
Copy Number Analysis
Chip-to-chip normalization, standardization to a reference, genotype detection, and copy number estimation on a single SNP basis were performed using the Affymetrix Power Tools (version 1.6.0) software suite http://www.affymetrix.com, as previously described [30]. Estimation of CNV boundary positions was done using Significance of Mean Difference (SMD), a method that we developed. Briefly, the mean of SNP copy number estimates (or log2 ratios) within a CNV was compared to the mean of those on the rest of the chromosome, and the probability that the null hypothesis of Student's t-test was true, i.e., that the means were from the same distribution, was calculated. A search using this statistic was conducted over different CNV lengths to find the position and length (in number of SNPs) that yielded the lowest probability, defining the boundaries of a putative CNV. For each sample, a random data set was produced by shuffling the genomic positions of the data, and an identical search was conducted. The results of this search represent false discoveries due to the random variation of the individual SNP data.
The p-value distribution of apparent CNVs detected by SMD in all 462 samples (both normal parents and the affected child from 154 ID trios) is shown in Figure 3. We observed that a CNV call with a p-value of 1 × 10-8 usually had a false discovery rate of less than 5%, while a call with a p-value of 1 × 10-7 often had a false discovery rate of 30%, with some variation from sample to sample.
Figure 3.

P-value distribution of apparent CNVs detected by SMD in 462 samples from 154 ID trios. Data are from the analysis performed by 500 K GeneChip® AGH. Some of these apparent CNVs were merged into larger CNVs at a later stage of the analysis, but most represent individual aberrations. Apparent CNVs with p-values less than 1 × 10-8 were analysed further to determine if they were inherited or had occurred de novo. 4577 apparent CNVs were found with p-values below this threshold (shown in blue in the figure). The bin plotted at 10-17 actually contains all CNVs with p < 10-16, which is the lower limit of the tables used to calculate p-values.
Since SMD compares a putative CNV to the rest of the chromosome, aberrations on the X chromosome are detected equivalently well for males and females. The pseudo-autosomal regions of the X-chromosomes are exceptions, but CNVs there can be detected as a function of the reference set used in the analysis. GeneChip® 500 K arrays do not contain Y-chromosome probes.
The SMD search was performed on each child with each parent as a reference. Since our goal was to find duplications and deletions that were de novo, a criterion for selection of CNVs for further analysis was that the child have the same aberration with each parent as reference. Correct parental relationships were confirmed in all trios by use of the SNP genotyping calls.
Every putative de novo CNV call was evaluated by another analysis conducted using a large reference set of individuals. Early in the study, the 48 sample reference set available from Affymetrix was used, but later a set of 50 mothers from our own data was used. The use of a local reference set significantly reduced noise. The use of the large reference set was required to detect the occurrence of aberrations in both parents that were not inherited by the child. Putative de novo CNVs were further evaluated by visual examination of the copy number plots in comparison to both parents as well as of the plots for the child and both parents in comparison to the large reference set.
Validation of De Novo CNVs
Putative de novo CNVs identified by 500 K AGH were validated by fluorescence in situ hybridization (FISH) or multiplex ligation-dependant probe amplification (MLPA), Agilent® 244 K AGH, or repeat cytogenetic analysis. FISH was performed with BAC or fosmid probes selected using the University of California at Santa Cruz Genome Browser [72] and the May 2006 assembly of the human genome sequence. BAC or fosmid DNA was isolated by small-scale (miniprep) preparation and was labeled with Spectrum Red or Green (Vysis, Abbott Molecular, Abbott Park, IL, USA) by use of a Vysis nick translation reagent kit. The labeled product was mixed with 3 mg of human Cot-1 DNA (Invitrogen, Life Technologies Corporation, Carlsbad, CA, USA) and was isolated by means of a standard DNA precipitation method. Cytogenetic pellets were prepared according to standard clinical procedures, and chromosomes and nuclei were visualized by counterstaining with 4',6-diamidino-2-phenylindole. For deletions, at least 10 metaphase cells were analyzed, and interphase nuclei were examined but not counted. For duplications, at least 10 metaphase cells and at least 50 interphase nuclei were analyzed. All FISH probes were tested on metaphase spreads from unaffected individuals to assure proper hybridization.
MLPA was performed using the P070 subtelomeric kit (MRC Holland, Amsterdam, The Netherlands); all positive results were confirmed with another kit (P036B). The procedure was conducted according to the manufacturer's recommendations. Briefly, the patient's DNA was diluted in PCR-grade water and quality was assessed via spectrophotometry (Nanodrop®, Thermo Scientific, Wilmington, DE, USA). The hybridization solution (SALSA probe-mix and MLPA buffer) was added to a final DNA concentration of 60 ng/μl. DNA was denatured at 90°C, then hybridized for 16-20 hours at 60°C. Ligation was performed at 54°C for 15 minutes, and the ligated product was denatured at 98°C for 5 minutes and then amplified by PCR (SALSA PCR buffer, PCR-primers and polymerase). The PCR product was run for fragment analysis on an ABI 3130 sequencer (Applied Biosystems, Life Technologies, Carlsbad, CA, USA). Normalization of the data and analysis of the MLPA results were conducted using Coffalyser v3.5 software provided by MRC Holland (Amsterdam, The Netherlands).
AGH was performed with Agilent® 244 K oligonucleotide arrays (Agilent Technologies Inc., Santa Clara, CA, USA) according to the manufacturer's instructions. Two arrays were used for each trio, one in which the child's DNA was hybridized against the father's DNA, and another in which the child's DNA was hybridized against the mother's DNA. Captured images were analysed with Feature Extraction v 9.1 and CGH Analytics 3.5.14 (Agilent Technologies Inc., Santa Clara, CA, USA).
Cytogenetic analysis was performed on peripheral blood cultures after preparation and G-banding of metaphase chromosomes using standard clinical methods.
Uniparental Disomy (UPD)
Given the genotypes for the child, mother, and father, errors in mendelian transmission were identified and their frequency compared to normal or technical error rates, which were very low. Essentially, the occurrences of an AA parent with a BB child, or vice versa, were counted. Graphical tools were developed to distinguish heterodisomy and isodisomy by visualization and to determine the parent of origin [30].
Confirmation of uniparental disomy was obtained by genotyping microsatellite markers chosen for high heterozygosity values. The following markers were genotyped and were informative for chromosome 11: D11S1363 (AFMA134WH5), D11S4046 (AFMB042YF5), D11S1318 (AFM218XE1), D11S4088 (AFMA155TE9) and D11S4146 (AFMB072WE5). The following markers were genotyped and were informative for chromosome 16: D16S3093 (AFMB308ZH9), D16S409 (AFM161XA1), D16S515 (AFM340YE5) and D16S402 (AFM031XA5). 25 ng of DNA diluted in 10:1 TE buffer were amplified for each PCR reaction. Primers fluorescently labeled with either 6-fam or HEX (Applied Biosystems, Life Technologies, Carlsbad, CA, USA) were used in conjunction with AmpliTaq Gold® PCR kit reagents (Applied Biosystems, Life Technologies, Carlsbad, CA, USA). PCR was performed using the following steps: 95°C for 10 minutes; 30 cycles of 95°C for 30 seconds, 55°C for 30 seconds, and 72°C for 90 seconds; and a final step at 72°C for 7 minutes. The resulting PCR product in a volume of 1 μl was combined with 9 μl formamide (Applied Biosystems, Life Technologies, Carlsbad, CA, USA) and 0.3 ul GeneScan ROX 500 (Applied Biosystems, Life Technologies, Carlsbad, CA, USA), denatured at 95°C for 5 minutes, quickly chilled in ice and loaded onto a Genetic Analyzer 3130XL (Applied Biosystems, Life Technologies, Carlsbad, CA, USA). Data were visualized using GeneMapper v4.0 (Applied Biosystems, Life Technologies, Carlsbad, CA, USA).
List of Abbreviations Used
AGH: array genomic hybridization; BAC: bacterial artificial chromosome; bp: base pairs; CNV: copy number variant; del: deletion; FISH: fluorescence in situ hybridization; ID: intellectual disability; K: thousand; Mb: megabase; MLPA: multiplex ligation-dependant probe amplification; PCR: polymerase chain reaction; SMD: significance of mean difference; SNP: single nucleotide polymorphism; UPD: uniparental disomy.
Authors' contributions
JMF and MAM conceived, designed and coordinated this study. PB developed and implemented the consent process, and JMF, SA, LA, LA, PB, CB, WTG, SL, EL, PM, JLM, BCM, MSP, GAR, MIVA and S-LY recruited patients. Clinical evaluations were performed by LA, LA, CB, JMF, WTG, SL, EL, PM, BCM, MSP, MIVA and S-LY. AB and SC performed the array genomic hybridizations, and AB, ADD, PE, SF, SL, HIL, HQ, MAM and JMF interpreted the AGH data. Confirmatory FISH and MLPA studies were done by PE, DC, EL and JM. LA, LA, CV, PE, WTG, SL, EL, PM, JLM, BCM, MSP, MIVA, S-LY, FRZ and JMF performed the genotype-phenotype correlations. JMF drafted the manuscript, which was critically reviewed and edited by AB, ADD, PE, SL, JLM, MSP, FRZ and MAM. All authors read and approved the final manuscript.
Supplementary Material
Supplemental Table S1. 54 ID trios from the 100 K cohort on whom 500 K GeneChip® AGH was performed. The findings in families in whom de novo CNVs were found are summarized in Table 3.
Supplemental Figure S1. Size distribution of pathogenic CNVs detected by 100 K and 500 K AGH in idiopathic ID trios. The number of deletions (red bars) or duplications (blue bars) in each size class is shown for 8 submicroscopic deletions and 2 submicroscopic duplications found in 100 idiopathic ID trios studied with 100 K AGH (empty and stippled bars) and for 22 submicroscopic deletions and 5 submicroscopic duplications found in 154 idiopathic ID trios studied with 500 K AGH (filled bars and stippled bars). 46 trios in the 100 K cohort were studied only with 100 K GeneChip® AGH (empty bars), 54 other trios from the 100 K cohort were studied with both 100 K and 500 K GeneChip® AGH (stippled bars), and 100 additional trios constituted a new cohort who were studied only with 500 K GeneChip® AGH (solid bars). Data for the 100 K trios are from [10]. The 500 K data include two unbalanced reciprocal translocations, for each of which both a deletion and a duplication are shown in the figure.
{kind=link}
Contributor Information
JM Friedman, Email: frid@interchange.ubc.ca.
Shelin Adam, Email: shelinadam@gmail.com.
Laura Arbour, Email: Laura.Arbour@viha.ca.
Linlea Armstrong, Email: llarmstrong@cw.bc.ca.
Agnes Baross, Email: abaross@genomebc.ca.
Patricia Birch, Email: birch@interchange.ubc.ca.
Cornelius Boerkoel, Email: boerkoel@interchange.ubc.ca.
Susanna Chan, Email: schan@bcgsc.ca.
David Chai, Email: dchai33@hotmail.com.
Allen D Delaney, Email: adelaney@bcgsc.ca.
Stephane Flibotte, Email: sflibotte@bcgsc.ca.
William T Gibson, Email: wtgibson@cmmt.ubc.ca.
Sylvie Langlois, Email: SLANGLOIS@cw.bc.ca.
Emmanuelle Lemyre, Email: emmanuelle.lemyre@recherche-ste-justine.qc.ca.
H Irene Li, Email: ili@bcgsc.ca.
Patrick MacLeod, Email: patrick.macleod@viha.ca.
Joan Mathers, Email: JMathers@cw.bc.ca.
Jacques L Michaud, Email: jacques.michaud@recherche-ste-justine.qc.ca.
Barbara C McGillivray, Email: bmcgillivray@cw.bc.ca.
Millan S Patel, Email: mPatel@cw.bc.ca.
Hong Qian, Email: hqian@bcgsc.ca.
Guy A Rouleau, Email: guy.rouleau@umontreal.ca.
Margot I Van Allen, Email: mvallen@cw.bc.ca.
Siu-Li Yong, Email: slyong@cw.bc.ca.
Farah R Zahir, Email: farahz@interchange.ubc.ca.
Patrice Eydoux, Email: PEydoux@cw.bc.ca.
Marco A Marra, Email: mmarra@bcgsc.bc.ca.
Acknowledgements
This work was supported by grants from Genome Canada (JMF, MAM),Genome British Columbia (JMF, MAM), the Canada Foundation for Innovation (JMF), British Columbia Knowledge Development Fund (JMF), Réseau de Médecine Génétique Appliquée of the Fonds de la Recherche en Santé du Québec (JLM, EL) and by the Fonds d'encouragement à la recherche clinique du CHU Sainte-Justine (JLM, EL). The authors thank Jacquie Schein for providing the BACs used for FISH confirmation and Tracy Tucker for her helpful comments regarding the manuscript.
References
- Ford C, Miller O, Polani P, de Almeida J, Briggs J. A sex-chromosome anomaly in a case of gonadal dysgenesis (Turner's syndrome) Lancet. 1959;1:711–3. doi: 10.1016/S0140-6736(59)91893-8. [DOI] [PubMed] [Google Scholar]
- Jacobs P, Strong J. A case of human intersexuality having a possible XXY sex-determining mechanism. Nature. 1959;183:302–3. doi: 10.1038/183302a0. [DOI] [PubMed] [Google Scholar]
- Lejeune J, Gautier M, Turpin M. Étude des chromosomes somatiques de neuf enfants mongoliens. CR Acad Sci (Paris) 1959;248:1721–2. [PubMed] [Google Scholar]
- de Ravel TJ, Devriendt K, Fryns JP, Vermeesch JR. What's new in karyotyping? The move towards array comparative genomic hybridisation (CGH) Eur J Pediatr. 2007;166:637–43. doi: 10.1007/s00431-007-0463-6. [DOI] [PubMed] [Google Scholar]
- Vermeesch JR, Fiegler H, de Leeuw N, Szuhai K, Schoumans J, Ciccone R, Speleman F, Rauch A, Clayton-Smith J, Van Ravenswaaij C, Sanlaville D, Patsalis PC, Firth H, Devriendt K, Zuffardi O. Guidelines for molecular karyotyping in constitutional genetic diagnosis. Eur J Hum Genet. 2007;15:1105–1114. doi: 10.1038/sj.ejhg.5201896. [DOI] [PubMed] [Google Scholar]
- Zahir F, Friedman J. The impact of array genomic hybridization on mental retardation research: a review of current technologies and their clinical utility. Clin Genet. 2007;72:271–87. doi: 10.1111/j.1399-0004.2007.00847.x. [DOI] [PubMed] [Google Scholar]
- Bruno D, Ganesamoorthy D, Schoumans J, Bankier A, Coman D, Delatycki M, Gardner M, Hunter M, James P, Kannu P, McGillivray G, Pachter N, Peters H, Rieubland C, Savarirayan R, Scheffer I, Sheffield L, Tan T, White S, Yeung A, Bowman Z, Ngo C, Choy K, Cacheux V, Wong L, Amor D, Slater H. Detection of cryptic pathogenic copy number variations and constitutional loss of heterozygosity using high resolution SNP microarray analysis in 117 patients referred for cytogenetic analysis and impact on clinical practice. J Med Genet. 2009;46:123–31. doi: 10.1136/jmg.2008.062604. [DOI] [PubMed] [Google Scholar]
- de Vries BB, Pfundt R, Leisink M, Koolen DA, Vissers LE, Janssen IM, Reijmersdal S, Nillesen WM, Huys EH, Leeuw N, Smeets D, Sistermans EA, Feuth T, van Ravenswaaij-Arts CM, van Kessel AG, Schoenmakers EF, Brunner HG, Veltman JA. Diagnostic genome profiling in mental retardation. Am J Hum Genet. 2005;77:606–16. doi: 10.1086/491719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan YS, Jayakar P, Zhu H, Barbouth D, Sacharow S, Morales A, Carver V, Benke P, Mundy P, Elsas LJ. Detection of pathogenic gene copy number variations in patients with mental retardation by genomewide oligonucleotide array comparative genomic hybridization. Hum Mutat. 2007;28:1124–32. doi: 10.1002/humu.20581. [DOI] [PubMed] [Google Scholar]
- Friedman JM, Baross A, Delaney AD, Ally A, Arbour L, Armstrong L, Asano J, Bailey DK, Barber S, Birch P, Brown-John M, Cao M, Chan S, Charest DL, Farnoud N, Fernandes N, Flibotte S, Go A, Gibson WT, Holt RA, Jones SJ, Kennedy GC, Krzywinski M, Langlois S, Li HI, McGillivray BC, Nayar T, Pugh TJ, Rajcan-Separovic E, Schein JE, Schnerch A, Siddiqui A, Van Allen MI, Wilson G, Yong SL, Zahir F, Eydoux P, Marra MA. Oligonucleotide microarray analysis of genomic imbalance in children with mental retardation. Am J Hum Genet. 2006;79:500–13. doi: 10.1086/507471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer J, Dreweke A, Becker C, Göhring I, Thiel C, Peippo M, Rauch R, Hofbeck M, Trautmann U, Zweier C, Zenker M, Hüffmeier U, Kraus C, Ekici A, Rüschendorf F, Nürnberg P, Reis A, A R. Molecular karyotyping in patients with mental retardation using 100 K single-nucleotide polymorphism arrays. J Med Genet. 2007;44:629–36. doi: 10.1136/jmg.2007.050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menten B, Maas N, Thienpont B, Buysse K, Vandesompele J, Melotte C, de Ravel T, Van Vooren S, Balikova I, Backx L, Janssens S, De Paepe A, De Moor B, Moreau Y, Marynen P, Fryns JP, Mortier G, Devriendt K, Speleman F, Vermeesch JR. Emerging patterns of cryptic chromosomal imbalance in patients with idiopathic mental retardation and multiple congenital anomalies: a new series of 140 patients and review of published reports. J Med Genet. 2006;43:625–33. doi: 10.1136/jmg.2005.039453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinning E, Tufarelli C, Winship W, Aldred M, Trembath R. Genomic duplication in Dyggve Melchior Clausen syndrome, a novel mutation mechanism in an autosomal recessive disorder. J Med Genet. 2005;42:e70. doi: 10.1136/jmg.2005.033829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman C, Hosomi N, Varela M, Souza A, Fukai K, Koiffmann C. Angelman syndrome associated with oculocutaneous albinism due to an intragenic deletion of the P gene. Am J Med Genet A. 2003;119A:180–183. doi: 10.1002/ajmg.a.20105. [DOI] [PubMed] [Google Scholar]
- Brooks E, Branda R, Nicklas J, O'Neill J. Molecular description of three macro-deletions and an Alu-Alu recombination-mediated duplication in the HPRT gene in four patients with Lesch-Nyhan disease. Mutat Res. 2001;476:43–54. doi: 10.1016/s0027-5107(01)00065-3. [DOI] [PubMed] [Google Scholar]
- Baross A, Delaney AD, Li HI, Nayar T, Flibotte S, Qian H, Chan SY, Asano J, Ally A, Cao M, Birch P, Brown-John M, Fernandes N, Go A, Kennedy G, Langlois S, Eydoux P, Friedman JM, Marra MA. Assessment of algorithms for high throughput detection of genomic copy number variation in oligonucleotide microarray data. BMC Bioinformatics. 2007;8:368. doi: 10.1186/1471-2105-8-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Smith AJ, Tsalenko A, Sampas N, Scheffer A, Yamada NA, Tsang P, Ben-Dor A, Yakhini Z, Ellis RJ, Bruhn L, Laderman S, Froguel P, Blakemore AI. Array CGH analysis of copy number variation identifies 1284 new genes variant in healthy white males: implications for association studies of complex diseases. Hum Mol Genet. 2007;16:2783–94. doi: 10.1093/hmg/ddm208. [DOI] [PubMed] [Google Scholar]
- de Stahl TD, Sandgren J, Piotrowski A, Nord H, Andersson R, Menzel U, Bogdan A, Thuresson AC, Poplawski A, von Tell D, Hansson CM, Elshafie AI, Elghazali G, Imreh S, Nordenskjold M, Upadhyaya M, Komorowski J, Bruder CE, Dumanski JP. Profiling of copy number variations (CNVs) in healthy individuals from three ethnic groups using a human genome 32 K BAC-clone-based array. Hum Mutat. 2008;29:398–408. doi: 10.1002/humu.20659. [DOI] [PubMed] [Google Scholar]
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, Haugen E, Zerr T, Yamada NA, Tsang P, Newman TL, Tuzun E, Cheng Z, Ebling HM, Tusneem N, David R, Gillett W, Phelps KA, Weaver M, Saranga D, Brand A, Tao W, Gustafson E, McKernan K, Chen L, Malig M, Smith JD, Korn JM, McCarroll SA, Altshuler DA, Peiffer DA, Dorschner M, Stamatoyannopoulos J, Schwartz D, Nickerson DA, Mullikin JC, Wilson RK, Bruhn L, Olson MV, Kaul R, Smith DR, Eichler EE. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, Taillon BE, Chen Z, Tanzer A, Saunders AC, Chi J, Yang F, Carter NP, Hurles ME, Weissman SM, Harkins TT, Gerstein MB, Egholm M, Snyder M. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–6. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, de Bakker PI, Maller JB, Kirby A, Elliott AL, Parkin M, Hubbell E, Webster T, Mei R, Veitch J, Collins PJ, Handsaker R, Lincoln S, Nizzari M, Blume J, Jones KW, Rava R, Daly MJ, Gabriel SB, Altshuler D. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;40:1166–74. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- Perry GH, Ben-Dor A, Tsalenko A, Sampas N, Rodriguez-Revenga L, Tran CW, Scheffer A, Steinfeld I, Tsang P, Yamada NA, Park HS, Kim JI, Seo JS, Yakhini Z, Laderman S, Bruhn L, Lee C. The fine-scale and complex architecture of human copy-number variation. Am J Hum Genet. 2008;82:685–95. doi: 10.1016/j.ajhg.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, Gonzalez JR, Gratacos M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Zhang J, Armengol L, Conrad DF, Estivill X, Tyler-Smith C, Carter NP, Aburatani H, Lee C, Jones KW, Scherer SW, Hurles ME. Global variation in copy number in the human genome. Nature. 2006;444:444–54. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KK, deLeeuw RJ, Dosanjh NS, Kimm LR, Cheng Z, Horsman DE, MacAulay C, Ng RT, Brown CJ, Eichler EE, Lam WL. A comprehensive analysis of common copy-number variations in the human genome. Am J Hum Genet. 2007;80:91–104. doi: 10.1086/510560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zogopoulos G, Ha KC, Naqib F, Moore S, Kim H, Montpetit A, Robidoux F, Laflamme P, Cotterchio M, Greenwood C, Scherer SW, Zanke B, Hudson TJ, Bader GD, Gallinger S. Germ-line DNA copy number variation frequencies in a large North American population. Hum Genet. 2007;122:345–53. doi: 10.1007/s00439-007-0404-5. [DOI] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR, Rasolonjatovo IM, Reed MT, Rigatti R, Rodighiero C, Ross MT, Sabot A, Sankar SV, Scally A, Schroth GP, Smith ME, Smith VP, Spiridou A, Torrance PE, Tzonev SS, Vermaas EH, Walter K, Wu X, Zhang L, Alam MD, Anastasi C, Aniebo IC, Bailey DM, Bancarz IR, Banerjee S, Barbour SG, Baybayan PA, Benoit VA, Benson KF, Bevis C, Black PJ, Boodhun A, Brennan JS, Bridgham JA, Brown RC, Brown AA, Buermann DH, Bundu AA, Burrows JC, Carter NP, Castillo N, Chiara ECM, Chang S, Neil Cooley R, Crake NR, Dada OO, Diakoumakos KD, Dominguez-Fernandez B, Earnshaw DJ, Egbujor UC, Elmore DW, Etchin SS, Ewan MR, Fedurco M, Fraser LJ, Fuentes Fajardo KV, Scott Furey W, George D, Gietzen KJ, Goddard CP, Golda GS, Granieri PA, Green DE, Gustafson DL, Hansen NF, Harnish K, Haudenschild CD, Heyer NI, Hims MM, Ho JT, Horgan AM, Hoschler K, Hurwitz S, Ivanov DV, Johnson MQ, James T, Huw Jones TA, Kang GD, Kerelska TH, Kersey AD, Khrebtukova I, Kindwall AP, Kingsbury Z, Kokko-Gonzales PI, Kumar A, Laurent MA, Lawley CT, Lee SE, Lee X, Liao AK, Loch JA, Lok M, Luo S, Mammen RM, Martin JW, McCauley PG, McNitt P, Mehta P, Moon KW, Mullens JW, Newington T, Ning Z, Ling Ng B, Novo SM, O'Neill MJ, Osborne MA, Osnowski A, Ostadan O, Paraschos LL, Pickering L, Pike AC, Pike AC, Chris Pinkard D, Pliskin DP, Podhasky J, Quijano VJ, Raczy C, Rae VH, Rawlings SR, Chiva Rodriguez A, Roe PM, Rogers J, Rogert Bacigalupo MC, Romanov N, Romieu A, Roth RK, Rourke NJ, Ruediger ST, Rusman E, Sanches-Kuiper RM, Schenker MR, Seoane JM, Shaw RJ, Shiver MK, Short SW, Sizto NL, Sluis JP, Smith MA, Ernest Sohna Sohna J, Spence EJ, Stevens K, Sutton N, Szajkowski L, Tregidgo CL, Turcatti G, Vandevondele S, Verhovsky Y, Virk SM, Wakelin S, Walcott GC, Wang J, Worsley GJ, Yan J, Yau L, Zuerlein M, Rogers J, Mullikin JC, Hurles ME, McCooke NJ, West JS, Oaks FL, Lundberg PL, Klenerman D, Durbin R, Smith AJ. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–9. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, Lin Y, MacDonald JR, Pang AW, Shago M, Stockwell TB, Tsiamouri A, Bafna V, Bansal V, Kravitz SA, Busam DA, Beeson KY, McIntosh TC, Remington KA, Abril JF, Gill J, Borman J, Rogers YH, Frazier ME, Scherer SW, Strausberg RL, Venter JC. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, Fan W, Zhang J, Li J, Zhang J, Guo Y, Feng B, Li H, Lu Y, Fang X, Liang H, Du Z, Li D, Zhao Y, Hu Y, Yang Z, Zheng H, Hellmann I, Inouye M, Pool J, Yi X, Zhao J, Duan J, Zhou Y, Qin J, Ma L, Li G, Yang Z, Zhang G, Yang B, Yu C, Liang F, Li W, Li S, Li D, Ni P, Ruan J, Li Q, Zhu H, Liu D, Lu Z, Li N, Guo G, Zhang J, Ye J, Fang L, Hao Q, Chen Q, Liang Y, Su Y, San A, Ping C, Yang S, Chen F, Li L, Zhou K, Zheng H, Ren Y, Yang L, Gao Y, Yang G, Li Z, Feng X, Kristiansen K, Wong GK, Nielsen R, Durbin R, Bolund L, Zhang X, Li S, Yang H, Wang J. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–5. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, Gomes X, Tartaro K, Niazi F, Turcotte CL, Irzyk GP, Lupski JR, Chinault C, Song XZ, Liu Y, Yuan Y, Nazareth L, Qin X, Muzny DM, Margulies M, Weinstock GM, Gibbs RA, Rothberg JM. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–6. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- Delaney A, Qian H, Friedman J, Marra M. Use of Affymetrix mapping arrays in the diagnosis of gene copy number variation. Curr Protoc Hum Genet. 2008;Chapter 8(Unit 8.13) doi: 10.1002/0471142905.hg0813s59. [DOI] [PubMed] [Google Scholar]
- Baldwin EL, Lee JY, Blake DM, Bunke BP, Alexander CR, Kogan AL, Ledbetter DH, Martin CL. Enhanced detection of clinically relevant genomic imbalances using a targeted plus whole genome oligonucleotide microarray. Genet Med. 2008;10:415–29. doi: 10.1097/GIM.0b013e318177015c. [DOI] [PubMed] [Google Scholar]
- Lu XY, Phung MT, Shaw CA, Pham K, Neil SE, Patel A, Sahoo T, Bacino CA, Stankiewicz P, Kang SH, Lalani S, Chinault AC, Lupski JR, Cheung SW, Beaudet AL. Genomic imbalances in neonates with birth defects: high detection rates by using chromosomal microarray analysis. Pediatrics. 2008;122:1310–8. doi: 10.1542/peds.2008-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koolen D, Pfundt R, de Leeuw N, Hehir-Kwa J, Nillesen W, Neefs I, Scheltinga I, Sistermans E, Smeets D, Brunner H, van Kessel A, Veltman J, de Vries B. Genomic microarrays in mental retardation: a practical workflow for diagnostic applications. Hum Mutat. 2008;30:283–92. doi: 10.1002/humu.20883. [DOI] [PubMed] [Google Scholar]
- Manning M, Hudgins L. Use of array-based technology in the practice of medical genetics. Genet Med. 2007;9:650–3. doi: 10.1097/gim.0b013e31814cec3a. [DOI] [PubMed] [Google Scholar]
- Shaffer L, Bejjani B, Torchia B, Kirkpatrick S, Coppinger J, Ballif B. The identification of microdeletion syndromes and other chromosome abnormalities: Cytogenetic methods of the past, new technologies for the future. Am J Med Genet C Semin Med Genet. 2007;145C:335–345. doi: 10.1002/ajmg.c.30152. [DOI] [PubMed] [Google Scholar]
- Shevell MI, Bejjani BA, Srour M, Rorem EA, Hall N, Shaffer LG. Array comparative genomic hybridization in global developmental delay. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1101–8. doi: 10.1002/ajmg.b.30730. [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Beaudet AL. Use of array CGH in the evaluation of dysmorphology, malformations, developmental delay, and idiopathic mental retardation. Curr Opin Genet Dev. 2007;17:182–92. doi: 10.1016/j.gde.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Wordsworth S, Buchanan J, Regan R, Davison V, Smith K, Dyer S, Campbell C, Blair E, Maher E, Taylor J, Knight SJ. Diagnosing idiopathic learning disability: a cost-effectiveness analysis of microarray technology in the National Health Service of the United Kingdom. Genomic Med. 2007;1:35–45. doi: 10.1007/s11568-007-9005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aston E, Whitby H, Maxwell T, Glaus N, Cowley B, Lowry D, Zhu XL, Issa B, South ST, Brothman AR. Comparison of targeted and whole genome analysis of postnatal specimens using a commercially available array based comparative genomic hybridisation (aCGH) microarray platform. J Med Genet. 2008;45:268–74. doi: 10.1136/jmg.2007.055319. [DOI] [PubMed] [Google Scholar]
- Friedman JM. High-resolution array genomic hybridization in prenatal diagnosis. Prenat Diagn. 2009;29:20–8. doi: 10.1002/pd.2129. [DOI] [PubMed] [Google Scholar]
- Veltman JA, de Vries BB. Diagnostic genome profiling: unbiased whole genome or targeted analysis? J Mol Diagn. 2006;8:534–7. doi: 10.2353/jmoldx.2006.060131. discussion 537-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater HR, Bailey DK, Ren H, Cao M, Bell K, Nasioulas S, Henke R, Choo KH, Kennedy GC. High-resolution identification of chromosomal abnormalities using oligonucleotide arrays containing 116,204 SNPs. Am J Hum Genet. 2005;77:709–26. doi: 10.1086/497343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenstaller J, Spranger S, Lorenz-Depiereux B, Kazmierczak B, Nathrath M, Wahl D, Heye B, Glaser D, Liebscher V, Meitinger T, Strom TM. Copy-number variations measured by single-nucleotide-polymorphism oligonucleotide arrays in patients with mental retardation. Am J Hum Genet. 2007;81:768–79. doi: 10.1086/521274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiffer DA, Le JM, Steemers FJ, Chang W, Jenniges T, Garcia F, Haden K, Li J, Shaw CA, Belmont J, Cheung SW, Shen RM, Barker DL, Gunderson KL. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16:1136–48. doi: 10.1101/gr.5402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski J. Genomic rearrangements and sporadic disease. Nat Genet. 2007;39:S43–7. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee YH, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimaki T, Ledbetter D, Gregersen PK, Bregman J, Sutcliffe JS, Jobanputra V, Chung W, Warburton D, King MC, Skuse D, Geschwind DH, Gilliam TC, Ye K, Wigler M. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–9. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Iafrate A, Brothman A. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat Genet. 2007;39:S48–S54. doi: 10.1038/ng2092. [DOI] [PubMed] [Google Scholar]
- Pinto D, Marshall C, Feuk L, Scherer S. Copy-number variation in control population cohorts. Hum Mol Genet. 2007;16:R168–73. doi: 10.1093/hmg/ddm241. [DOI] [PubMed] [Google Scholar]
- Zhang J, Feuk L, Duggan G, Khaja R, Scherer S. Development of bioinformatics resources for display and analysis of copy number and other structural variants in the human genome. Cytogenet Genome Res. 2006;115:205–14. doi: 10.1159/000095916. [DOI] [PubMed] [Google Scholar]
- Sharp AJ. Emerging themes and new challenges in defining the role of structural variation in human disease. Hum Mutat. 2009;30:135–44. doi: 10.1002/humu.20843. [DOI] [PubMed] [Google Scholar]
- Burger J, Horn D, Tonnies H, Neitzel H, Reis A. Familial interstitial 570 kbp deletion of the UBE3A gene region causing Angelman syndrome but not Prader-Willi syndrome. Am J Med Genet. 2002;111:233–7. doi: 10.1002/ajmg.10498. [DOI] [PubMed] [Google Scholar]
- Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gecz J, Moraine C, Marynen P, Fryns JP, Froyen G. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. 2005;77:442–53. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzel C, Fernstrom M, Ohrner Y, Anneren G, Thuresson AC. Clinical variability of the 22q11.2 duplication syndrome. Eur J Med Genet. 2008;51:501–10. doi: 10.1016/j.ejmg.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Ou Z, Berg JS, Yonath H, Enciso VB, Miller DT, Picker J, Lenzi T, Keegan CE, Sutton VR, Belmont J, Chinault AC, Lupski JR, Cheung SW, Roeder E, Patel A. Microduplications of 22q11.2 are frequently inherited and are associated with variable phenotypes. Genet Med. 2008;10:267–77. doi: 10.1097/GIM.0b013e31816b64c2. [DOI] [PubMed] [Google Scholar]
- Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, Huang S, Maloney VK, Crolla JA, Baralle D, Collins A, Mercer C, Norga K, de Ravel T, Devriendt K, Bongers EM, de Leeuw N, Reardon W, Gimelli S, Bena F, Hennekam RC, Male A, Gaunt L, Clayton-Smith J, Simonic I, Park SM, Mehta SG, Nik-Zainal S, Woods CG, Firth HV, Parkin G, Fichera M, Reitano S, Lo Giudice M, Li KE, Casuga I, Broomer A, Conrad B, Schwerzmann M, Raber L, Gallati S, Striano P, Coppola A, Tolmie JL, Tobias ES, Lilley C, Armengol L, Spysschaert Y, Verloo P, De Coene A, Goossens L, Mortier G, Speleman F, van Binsbergen E, Nelen MR, Hochstenbach R, Poot M, Gallagher L, Gill M, McClellan J, King MC, Regan R, Skinner C, Stevenson RE, Antonarakis SE, Chen C, Estivill X, Menten B, Gimelli G, Gribble S, Schwartz S, Sutcliffe JS, Walsh T, Knight SJ, Sebat J, Romano C, Schwartz CE, Veltman JA, de Vries BB, Vermeesch JR, Barber JC, Willatt L, Tassabehji M, Eichler EE. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. 2008;359:1685–99. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Saijo M, Nakatsu Y, Nakai H, Yamaizumi M, Tanaka K. Three novel mutations responsible for Cockayne syndrome group A. Genes Genet Syst. 2003;78:93–102. doi: 10.1266/ggs.78.93. [DOI] [PubMed] [Google Scholar]
- Vissers LE, Veltman JA, van Kessel AG, Brunner HG. Identification of disease genes by whole genome CGH arrays. Hum Mol Genet. 2005;14(Spec No. 2):R215–23. doi: 10.1093/hmg/ddi268. [DOI] [PubMed] [Google Scholar]
- Zanni G, Saillour Y, Nagara M, Billuart P, Castelnau L, Moraine C, Faivre L, Bertini E, Durr A, Guichet A, Rodriguez D, des Portes V, Beldjord C, Chelly J. Oligophrenin 1 mutations frequently cause X-linked mental retardation with cerebellar hypoplasia. Neurology. 2005;65:1364–9. doi: 10.1212/01.wnl.0000182813.94713.ee. [DOI] [PubMed] [Google Scholar]
- Shaffer LG, Coppinger J, Alliman S, Torchia BA, Theisen A, Ballif BC, Bejjani BA. Comparison of microarray-based detection rates for cytogenetic abnormalities in prenatal and neonatal specimens. Prenat Diagn. 2008;28:789–95. doi: 10.1002/pd.2053. [DOI] [PubMed] [Google Scholar]
- Langlois S, Yong PJ, Yong SL, Barrett I, Kalousek DK, Miny P, Exeler R, Morris K, Robinson WP. Postnatal follow-up of prenatally diagnosed trisomy 16 mosaicism. Prenat Diagn. 2006;26:548–58. doi: 10.1002/pd.1457. [DOI] [PubMed] [Google Scholar]
- Neiswanger K, Hohler PM, Hively-Thomas LB, McPherson EW, Hogge WA, Surti U. Variable outcomes in mosaic trisomy 16: five case reports and literature analysis. Prenat Diagn. 2006;26:454–61. doi: 10.1002/pd.1437. [DOI] [PubMed] [Google Scholar]
- Cytrynbaum CS, Smith AC, Rubin T, Weksberg R. Advances in overgrowth syndromes: clinical classification to molecular delineation in Sotos syndrome and Beckwith-Wiedemann syndrome. Curr Opin Pediatr. 2005;17:740–6. doi: 10.1097/01.mop.0000187191.74295.97. [DOI] [PubMed] [Google Scholar]
- Malvagia S, Papi L, Morrone A, Donati MA, Ciani F, Pasquini E, la Marca G, Scholte HR, Genuardi M, Zammarchi E. Fatal malonyl CoA decarboxylase deficiency due to maternal uniparental isodisomy of the telomeric end of chromosome 16. Ann Hum Genet. 2007;71:705–12. doi: 10.1111/j.1469-1809.2007.00373.x. [DOI] [PubMed] [Google Scholar]
- Zlotogora J. Parents of children with autosomal recessive diseases are not always carriers of the respective mutant alleles. Hum Genet. 2004;114:521–6. doi: 10.1007/s00439-004-1105-y. [DOI] [PubMed] [Google Scholar]
- Hennekam RC. Rubinstein-Taybi syndrome. Eur J Hum Genet. 2006;14:981–5. doi: 10.1038/sj.ejhg.5201594. [DOI] [PubMed] [Google Scholar]
- Roelfsema JH, Peters DJ. Rubinstein-Taybi syndrome: clinical and molecular overview. Expert Rev Mol Med. 2007;9:1–16. doi: 10.1017/S1462399407000415. [DOI] [PubMed] [Google Scholar]
- Baris HN, Tan WH, Kimonis VE, Irons MB. Diagnostic utility of array-based comparative genomic hybridization in a clinical setting. Am J Med Genet A. 2007;143A:2523–33. doi: 10.1002/ajmg.a.31988. [DOI] [PubMed] [Google Scholar]
- Bejjani BA, Shaffer LG. Application of array-based comparative genomic hybridization to clinical diagnostics. J Mol Diagn. 2006;8:528–33. doi: 10.2353/jmoldx.2006.060029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeschler J. Medical genetics diagnostic evaluation of the child with global developmental delay or intellectual disability. Curr Opin Neurol. 2008;21:117–22. doi: 10.1097/WCO.0b013e3282f82c2d. [DOI] [PubMed] [Google Scholar]
- Shaffer LG, Beaudet AL, Brothman AR, Hirsch B, Levy B, Martin CL, Mascarello JT, Rao KW. Microarray analysis for constitutional cytogenetic abnormalities. Genet Med. 2007;9:654–62. doi: 10.1097/gim.0b013e31814ce3d9. [DOI] [PubMed] [Google Scholar]
- Cook EH Jr, Scherer SW. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455:919–23. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajecka M, Mackay KL, Shaffer LG. Monosomy 1p36 deletion syndrome. Am J Med Genet C Semin Med Genet. 2007;145C:346–56. doi: 10.1002/ajmg.c.30154. [DOI] [PubMed] [Google Scholar]
- Zollino M, Murdolo M, Marangi G, Pecile V, Galasso C, Mazzanti L, Neri G. On the nosology and pathogenesis of Wolf-Hirschhorn syndrome: genotype-phenotype correlation analysis of 80 patients and literature review. Am J Med Genet C Semin Med Genet. 2008;148C:257–69. doi: 10.1002/ajmg.c.30190. [DOI] [PubMed] [Google Scholar]
- Stewart D, Huang A, Faravelli F, Anderlid B, Medne L, Ciprero K, Kaur M, Rossi E, Tenconi R, Nordenskjöld M, Gripp K, Nicholson L, Meschino W, Capua E, Quarrell O, Flint J, Irons M, Giampietro P, Schowalter D, Zaleski C, Malacarne M, Zackai E, Spinner N, Krantz I. Subtelomeric deletions of chromosome 9q: a novel microdeletion syndrome. Am J Med Genet A. 2004;128A:340–51. doi: 10.1002/ajmg.a.30136. [DOI] [PubMed] [Google Scholar]
- Cantu ES, Eicher DJ, Pai GS, Donahue CJ, Harley RA. Mosaic vs. nonmosaic trisomy 9: report of a liveborn infant evaluated by fluorescence in situ hybridization and review of the literature. Am J Med Genet. 1996;62:330–5. doi: 10.1002/(SICI)1096-8628(19960424)62:4<330::AID-AJMG1>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Wooldridge J, Zunich J. Trisomy 9 syndrome: report of a case with Crohn disease and review of the literature. Am J Med Genet. 1995;56:258–64. doi: 10.1002/ajmg.1320560304. [DOI] [PubMed] [Google Scholar]
- Koolen DA, Sharp AJ, Hurst JA, Firth HV, Knight SJ, Goldenberg A, Saugier-Veber P, Pfundt R, Vissers LE, Destree A, Grisart B, Rooms L, Aa N Van der, Field M, Hackett A, Bell K, Nowaczyk MJ, Mancini GM, Poddighe PJ, Schwartz CE, Rossi E, De Gregori M, Antonacci-Fulton LL, McLellan MD, Garrett JM, Wiechert MA, Miner TL, Crosby S, Ciccone R, Willatt L, Rauch A, Zenker M, Aradhya S, Manning MA, Strom TM, Wagenstaller J, Krepischi-Santos AC, Vianna-Morgante AM, Rosenberg C, Price SM, Stewart H, Shaw-Smith C, Brunner HG, Wilkie AO, Veltman JA, Zuffardi O, Eichler EE, de Vries BB. Clinical and molecular delineation of the 17q21.31 microdeletion syndrome. J Med Genet. 2008;45:710–20. doi: 10.1136/jmg.2008.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurahashi H, Nakayama T, Osugi Y, Tsuda E, Masuno M, Imaizumi K, Kamiya T, Sano T, Okada S, Nishisho I. Deletion mapping of 22q11 in CATCH22 syndrome: identification of a second critical region. Am J Hum Genet. 1996;58:1377–81. [PMC free article] [PubMed] [Google Scholar]
- Garcia-Miñaur S, Fantes J, Murray R, Porteous M, Strain L, Burns J, Stephen J, Warner J. A novel atypical 22q11.2 distal deletion in father and son. J Med Genet. 2002;39:E62. doi: 10.1136/jmg.39.10.e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Zink S, Zweier C, Thiel CT, Koch A, Rauch R, Lascorz J, Huffmeier U, Weyand M, Singer H, Hofbeck M. Systematic assessment of atypical deletions reveals genotype-phenotype correlation in 22q11.2. J Med Genet. 2005;42:871–6. doi: 10.1136/jmg.2004.030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1. 54 ID trios from the 100 K cohort on whom 500 K GeneChip® AGH was performed. The findings in families in whom de novo CNVs were found are summarized in Table 3.
Supplemental Figure S1. Size distribution of pathogenic CNVs detected by 100 K and 500 K AGH in idiopathic ID trios. The number of deletions (red bars) or duplications (blue bars) in each size class is shown for 8 submicroscopic deletions and 2 submicroscopic duplications found in 100 idiopathic ID trios studied with 100 K AGH (empty and stippled bars) and for 22 submicroscopic deletions and 5 submicroscopic duplications found in 154 idiopathic ID trios studied with 500 K AGH (filled bars and stippled bars). 46 trios in the 100 K cohort were studied only with 100 K GeneChip® AGH (empty bars), 54 other trios from the 100 K cohort were studied with both 100 K and 500 K GeneChip® AGH (stippled bars), and 100 additional trios constituted a new cohort who were studied only with 500 K GeneChip® AGH (solid bars). Data for the 100 K trios are from [10]. The 500 K data include two unbalanced reciprocal translocations, for each of which both a deletion and a duplication are shown in the figure.