

To the Editor: Catatonia is a syndrome associated with psychiatric or general medical conditions.1 An auto-activation deficit is a syndrome related to lesions of the basal ganglia.2 To our knowledge, catatonia has not yet been described in association with an auto-activation deficit. Although means of successfully treating catatonia are now well known,1,3 we found no pharmacologic recommendations for treating an auto-activation deficit.
We report here a case of catatonia associated with an auto-activation deficit, which occurred in the context of a methadone overdose and improved owing to a combination of lorazepam and low-dose amisulpride.
Case report. Mr A, a 38-year-old man, was found unconscious at his home (Glasgow4 score = 3) in 2008 following a methadone overdose. He was treated by in situ resuscitation with external cardiac massage, two injections of adrenaline (2 mg, then 5 mg), intubation, and mechanical ventilation. He was hospitalized in the intensive care unit, where the examination revealed metabolic acidosis with hyperkalaemia, acute renal failure, and rhabdomyolysis, complicating a compartment syndrome. Urine tests showed the presence of methadone, and blood alcohol level was 0.10 g/L. Within 24 hours, the patient's consciousness level improved. He was transferred to the nephrology department because of acute tubular necrosis that resulted from rhabdomyolysis.
At his first psychiatric examination (72 hours after waking up), he told us that he had smoked heroin after consuming alcohol and an unknown amount of methadone. He had not previously been treated in the addictology department. We noted no comorbid psychiatric or psycho-behavioral disorders. We started liaison addictology management, with a view to sending him to our outpatients clinic once he was discharged from the hospital. Since he had no withdrawal symptoms, no substitution treatment was offered to him.
Fifteen days after our examination, as he was recovering from sepsis (superinfected skin necrosis), he presented with a confusional syndrome associated with focal neurologic deficits (right-sided blindness), and increased tendon reflexes in all 4 limbs. A cerebral computed tomography scan revealed no abnormalities. An electroencephalogram suggested diffuse cerebral distress, considered to be of metabolic origin. A clinical ophthalmologic examination was unhelpful. Cerebral blood flow measurements showed reduced flow in the bilateral frontal cortical and subcortical and the left anterior temporal regions, as well as occipital hyperfixation. Magnetic resonance imaging performed at the same time showed subtentorial leukoencephalopathy of toxic or anoxic origin and focal bipallidal lesions (Figures 1A and 1B). The transient ophthalmologic signs regressed spontaneously.
Figure 1.
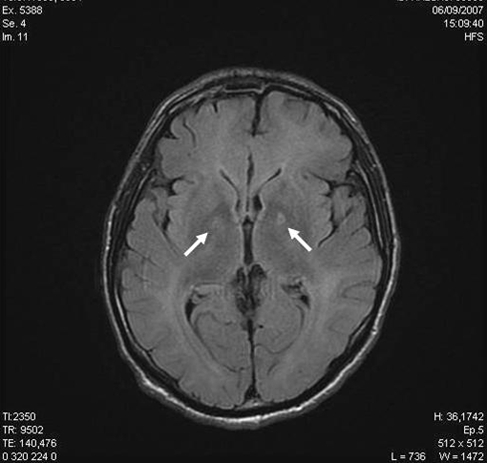
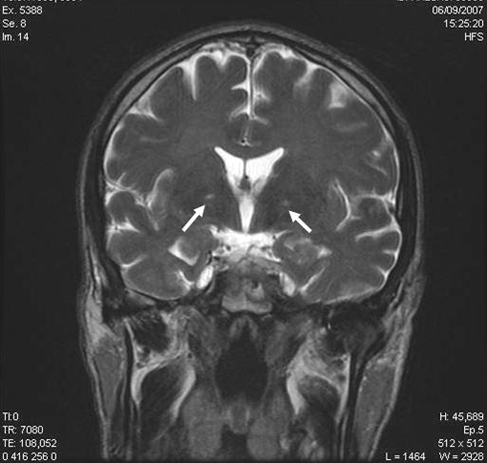
(A) T2 Flair Axial and (B) T2 Coronale Magnetic Resonance Imaging Scans at Day 15a
aArrows correspond to the focal bipallidal lesions.
From a psychobehavioral point of view, we noticed a lack of psychomotor coordination, stereotypic lesions, the maintenance of postures imposed on him, behavioral disinhibition, an absence of spontaneous speech, affective indifference, and gegenhalten (ie, resistance to passive movement that is in proportion to the strength of the stimulus and that appears automatic rather than willful) suggestive of a catatonic syndrome.5 Treatment with lorazepam 10 mg/day1,3 improved the catatonic symptoms within 72 hours, in particular the motor disorders, the behavioral disinhibition, and the gegenhalten.
Nevertheless, residual clinical signs not typical of catatonia persisted. These included a persistence of stereotypical postures, the absence of motor initiation, a total absence of affective expression, and mental emptiness. It was possible to stimulate him, but he made no attempt to initiate conversation. Only by external stimulation was it possible to get any response from him. Although he had lost all initiative, we noted that he responded appropriately to reality and had normal orientation in time and space. With this clinical picture (stereotypic activities, inertia, the positive effect of external stimulation, mental emptiness, and blunted affect), together with the cerebral lesions observed on neurologic imaging (frontal and bipallidal lesions), we put forward the diagnosis of an auto-activation deficit.2
In view of the dopaminergic agonist action of low-dose amisulpride,6 we decided to offer the patient treatment with amisulpride (50 mg/day for 3 days, then 100 mg/day). After 7 days, we observed a significant improvement in his behavior, with fluent spontaneous speech and good emotional expression, but persistence of the frontal syndrome signs (disinhibition and perseveration). After a month of treatment, we did not observe any psychobehavioral disorders, and the renal function had returned to normal. He was transferred to the functional rehabilitation department because of residual external popliteal sciatic nerve paralysis from the compartment syndrome. Executive function tests performed after 2 months of treatment showed a residual prefrontal lesion with an attention disorder (selective and divided attention) and executive function disorders (changes in cognitive flexibility, rule discovery, and maintaining or changing rules). This prefrontal lesion later improved without any changes to the treatment.
We note that when amisulpride was prematurely stopped for a few days, the patient once again presented with symptoms of an auto-activation deficit, which disappeared again in a few days after reintroducing amisulpride (100 mg/day). He continues to be followed up in the addictology outpatients department, and no longer shows any signs of executive dysfunction. His treatment remains unchanged.
Bipallidal cerebral lesions have for a long time been linked to carbon monoxide poisoning, a symptom-free initial period, and characteristic radiologic signs being considered as a pathognomonic. Studies have shown heroin consumption to have been responsible for a reduction in neuronal density in the globus pallidus.7 It can also cause bilateral symmetrical necrosis of the globus pallidus in 5% to 10% of cases.8 In our case, the most likely physiopathologic hypothesis seems to be an ischemic origin, although direct neurotoxicity cannot be completely ruled out.9
In the most severe cases, these lesions may cause an auto-activation deficit,2 characterized by cognitive-behavioral inertia (disappearing after hetero-stimulation), mental emptiness, stereotypic activities, and a blunted affect, suggesting that the cortico-subcortical pathways have been damaged as a result of damage to the prefrontal cortex and/or the basal ganglia.10 The sensorimotor, emotional, and motivational symptomatology are suggestive of disruption of the motor, limbic, and associative loops, where they converge in the globus pallidus.11
We found no associated catatonic symptomatology with auto-activation deficit in the literature. Catatonia seems to be due to damage to the GABAergic transmission pathways, as shown by a rapid complete response to lorazepam,1,3 and to psychological and organic stress (sepsis).12
We decided to treat the auto-activation deficit with low-dose amisulpride, in view of its clinical and cerebral blood flow similarities with deficit forms of schizophrenia.5 At a dose of 100 mg/day, amisulpride preferentially blocks the high-affinity presynaptic dopaminergic receptors. The pharmacologic effect is probably due to an increase in dopaminergic transmission.5 The clinical efficacy of low-dose amisulpride (a dopamine agonist) seems to indicate changes in the dopaminergic pathways in an auto-activation deficit, and also of the existence of a functional loop, in the case of a partial lesion.13
REFERENCES
- 1.Fink M, Taylor MA. Catatonia: A Clinician's Guide to Diagnosis and Treatment. Cambridge, UK: Cambridge University Press; 2003. [Google Scholar]
- 2.Laplane D, Dubois B. Auto-activation deficit: a basal ganglia related syndrome. Mov Disord. 2001 Sep;16(5):810–814. doi: 10.1002/mds.1185. [DOI] [PubMed] [Google Scholar]
- 3.Caroff S, Mann S, Francis A, et al. Catatonia: From Psychopathology to Neurobiology. Washington DC: American Psychiatric Publishing, Inc; 2004. [Google Scholar]
- 4.Teasdale G, Jennett B. Assessment of coma and impaired consciousness: a practical scale. Lancet. 1974;2:81–83. doi: 10.1016/s0140-6736(74)91639-0. [DOI] [PubMed] [Google Scholar]
- 5.Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition. Washington, DC: American Psychiatric Association; 1994. American Psychiatric Association. [Google Scholar]
- 6.Vaiva G, Thomas P, Llorca PM, et al. SPECT imaging, clinical features, and cognition before and after low doses of amisulpride in schizophrenic patients with the deficit syndrome. Psychiatry Res. 2002 Aug;115(1–2):37–48. doi: 10.1016/s0925-4927(02)00031-8. [DOI] [PubMed] [Google Scholar]
- 7.Pearson J, Baden MB, Richter RW. Neuronal depletion in the globus pallidus of heroin addicts. Drug Alcohol Depend. 1976;1(5):349–356. doi: 10.1016/0376-8716(76)90037-5. [DOI] [PubMed] [Google Scholar]
- 8.Andersen SN, Skullerud K. Hypoxic/ischaemic brain damage, especially pallidal lesions, in heroin addicts. Forensic Sci Int. 1999 May;102(1):51–59. doi: 10.1016/s0379-0738(99)00040-7. [DOI] [PubMed] [Google Scholar]
- 9.Buttner A, Mall G, Penning R, et al. The neuropathology of heroin abuse. Forensic Sci Int. 2000 Sep;113(1–3):435–442. doi: 10.1016/s0379-0738(00)00204-8. [DOI] [PubMed] [Google Scholar]
- 10.Levy R, Dubois B. Apathy and the functional anatomy of the prefrontal cortex-basal ganglia circuits. Cereb Cortex. 2006 Jul;16(7):916–928. doi: 10.1093/cercor/bhj043. [DOI] [PubMed] [Google Scholar]
- 11.Yelnik J. Structural anatomy and basal ganglia function. Encephale. 2006 Apr;32:S3–S9. doi: 10.1016/s0013-7006(06)78684-5. [DOI] [PubMed] [Google Scholar]
- 12.Cottencin O, Warembourg F, de Chouly de Lenclave MB, et al. Catatonia and consultation-liaison psychiatry study of 12 cases. Prog Neuropsychopharmacol Biol Psychiatry. 2007 Aug;31(6):1170–1176. doi: 10.1016/j.pnpbp.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 13.Starkstein SE, Berthier ML, Leiguarda R. Psychic akinesia following bilateral pallidal lesions. Int J Psychiatry Med. 1989;19(2):155–164. doi: 10.2190/ae1q-qy7b-lu2f-gjhf. [DOI] [PubMed] [Google Scholar]