

Abstract
Glycine receptors are Cys loop ligand-gated ion channels that mediate fast inhibitory synaptic transmission in the mammalian central nervous system. The functionally distinct splice variants α3L and α3K of the human glycine receptor differ by a 15-amino acid insert within the long intracellular TM3–4 loop, a region of high intersubunit diversity. In a mutational study, effects of the insert on ion channel function and secondary structure of the TM3–4 loop were investigated. Whole cell current responses and protein surface expression data indicated that the major effect of mutations within the insert was on channel gating. Changes in channel gating correlated with the distribution of charged residues about the splice region. Analysis of complex molecular weight indicated that recombinant TM3–4 loops of α3L and α3K associated into oligomers of different stoichiometry. Secondary structure analysis suggested that the insert stabilized the overall fold of the large cytoplasmic domain of α3L subunits. The absence of the insert resulted in a channel that was still functional, but the TM3–4 cytoplasmic domain appeared not stably folded. Thus, our data identified the spliced insert within the large TM 3–4 loop of α3 Gly receptors as a novel regulatory motif that serves a 2-fold role: (i) the presence of the insert stabilizes the overall spatial structure of the domain, and (ii) the insert presents a control unit that regulates gating of the receptor ion channel.
Glycine receptors (GlyRs),3 together with γ-aminobutyric acid receptors, are the principal carriers of fast synaptic inhibition in the mammalian central nervous system. They share structural and functional homology with other members of the ligand gated ion channel family (1–3). To date, four ligand binding subunits (α1–4) capable of forming homomeric functional ion channels and one β subunit have been identified (1–4). α1 subunits, prevalent in spinal cord and brain stem, are associated with the human hypertonic motor disorder, hyperekplexia (STHE, stiff baby syndrome, OMIM (Online Mendelian Inheritance in Man) 138491) (2, 5–7). In contrast, the α3 subunit was found widely distributed over the human central nervous system (7). Two human splice variants, α3K and α3L, have been identified, distinguished by an insert of 15 amino acids following a segment of eight positively charged residues within the long cytoplasmic TM3–4 loop of the receptor subunit (8). The splice variants exhibit similar glycine affinities and single channel conductances but differ in the extent and time course of desensitization (9). Three hydroxyl groups are located within the alternatively spliced insert, each within a minimum phosphorylation consensus sequence (8, 9). Replacement of all three hydroxylated residues by their hydrocarbon analogs resulted in modified desensitization kinetics but had no effect on single channel conductances or macroscopic parameters such as EC50 and maximum current amplitude (9). These observations indicate that hydroxyl functions are important, but not exclusive, determinants of receptor desensitization.
Here, we studied the contribution of the alternatively spliced region to domain folding and to the individual steps of α3 GlyR function. Replacement of hydroxylated residues and modification of insert length by deletion or duplication of six residues resulted in dramatic changes in current responses that could be accounted for by altered channel gating behavior. Changes in receptor gating correlated with the distribution of charges on the protein surface of this variable loop. Isolated TM3–4 loops of α3L and α3K formed defined oligomers, each of different stoichiometry. CD spectroscopy indicated a well defined secondary structure for the long splice variant α3L only, whereas the fold of α3K was not stable. Thus, the spliced insert was identified as a novel regulatory motif of the inhibitory α3 glycine receptor, carrying a dual function: (i) stabilization of the secondary structure of the large cytoplasmic TM3–4 loop of α3L and (ii) regulation of ion channel gating.
EXPERIMENTAL PROCEDURES
Generation of GlyR α3 Mutants
Single nucleotide exchanges, as well as deletions and duplications within the alternatively spliced insert of GlyR α3L, were introduced by PCR-mediated site-directed mutagenesis using an overlap extension PCR approach (10). Mutagenesis primers (Amersham Biosciences) contained nucleotides specific for the amino acid exchange together with a silent restriction site for a rapid check of mutated clones. PCRs were set up as follows: 1 ng of template DNA, 50 μm each dATP, dCTP, dGTP, and dTTp, 100 pmol of each primer, and 2 units of high fidelity Taq polymerase in the supplied buffer (Roche Applied Science). PCR conditions were 5 min at 95 °C for denaturation, 5 min at 50 °C for annealing, 5 min at 72 °C for elongation in the first cycle, followed by 28 cycles of 1 min at 95 °C, 2 min 50 °C, and 2.5 min at 72 °C. The last cycle ended with a 10-min 72 °C amplification step. The final fragments were cut with restriction enzymes as close as possible to the mutated site to minimize the PCR-generated sequence and reinserted into GlyRα3. All mutated clones were sequenced across the PCR-generated sequence to verify successful mutagenesis using the ABI sequencer system (ABI Systems). Note that deletion or duplication of a six-residue stretch within the insert left the hydroxylated residues and neighboring positive charges intact (see Fig. 1).
FIGURE 1.

GlyR α subunit sequences and constructs. A, alignment of various receptors in the TM3–4 loop region. The alternatively spliced region is indicated. Note the large sequence divergence in the region of the insert. Lower panel, Jpred (34) secondary structure predictions for α3L (middle row) and α3K (bottom row). Use of several other prediction routines from the EXPASY server gave similar results. The α-helical content of α3L according to the prediction is ∼38% (35 of 92 residues), in good agreement with secondary structure analysis by CD spectroscopy. B, glycine receptor a3 variants used in the study; the one-letter codes for amino acids are used. The alternatively spliced 15-residue segment (positions 358–372) is indicated by bold letters, and flanking sequences are shown in gray. Deleted residues within the spliced segment are shown as bold dashes, and flanking sequences are aligned on either side of the insert. Mutated residues are highlighted.
Cell Culture and Transfection
HEK 293 cells were grown in 80-cm2, 225-cm3 Nunc flasks at 37 °C, 5% CO2, in a water-saturated atmosphere. Minimum essential medium (Invitrogen), supplemented with penicillin (100 IU), streptomycin (100 mg/liter), l-glutamine, and 10% heat-inactivated fetal calf serum (Invitrogen). 5 × 106 cells/culture flask in 35 ml of medium were seeded for culture and passaged twice weekly. For experiments, 1 × 106 cells were seeded in 10-cm Petri dishes containing 10 ml of culture medium. Transfection was carried out using a calcium phosphate precipitation protocol (11). Briefly, 5 μg of plasmid DNA plus 5 μg of green fluorescent protein-DNA were diluted with sterile deionized water (215 μl). 25 μl of 2.5 m CaCl2 was added, followed by 250 μl of 2× BBS buffer (50 mm BES (Sigma), 280 mm NaCl, 1.5 mm Na2HPO4, pH 6.96, with NaOH). After 15 min the solution was added to cultured HEK-293 cells at 1 ml/10 ml of medium, and transfection was allowed to proceed overnight (12–18 h) at 37 °C, 3% CO2, in a water-saturated atmosphere. The cells were then washed with minimum essential medium once, fresh culture medium was added, and the cells were stored at 5% CO2. These cells could be kept up to 5 days, although they routinely were used for experiments 1–3 days after transfection.
Protein Characterization and Radioligand Binding
For crude membrane preparations, the cells were harvested using ice-cold PBS (13.7 mm NaCl, 0.27 mm KCl, 0.43 mm Na2HPO4, 0.14 mm KH2PO4, pH 7.4) and collected by centrifugation (10 min, 1000 rpm). All of the subsequent steps were carried out on ice. The cell pellet was taken up in a 20-fold volume of 10 mm potassium phosphate buffer, pH 7.4, supplemented with protease inhibitors (C@mplete EDTA-free (Roche Applied Science), 5 mm EDTA, 5 mm EGTA), homogenized using a glass potter and an ultraturrax, and centrifuged (20 min, 35,000 × g). This step was repeated, and the membrane pellet was finally resuspended in a 5-fold volume of storage buffer (25 mm potassium phosphate, pH 7.4, 200 mm KCl, plus protease inhibitors) and stored in 400-μl aliquots. Total protein content was determined using the Lowry method. Western blot analysis from membrane preparations was carried out using the GlyR-pan-α monoclonal antibody mAb-4a, a cy5-coupled goat anti-mouse secondary antibody gαMIgG-Cy5 (Dianova) and a Storm 860 Fluoroimager (Molecular Dynamics, Krefeld, Germany) for visualization.
Surface Protein Biotinylation
COS7 cells were transfected (10 μg of plasmid/10-cm dish) using DEAE-Dextran (10 mg/ml) 48 h before labeling the cells. Following three washing steps with ice-cold PBS, the cells were incubated 15 min with 1 mg/ml EZ-link TM sulfo-N-hydroxy-sulfosuccinimide-S-S-biotin (Pierce) in cold PBS (pH 8.0) with gentle agitation at 4 °C. The cells were again washed and incubated with quenching puffer (192 mm glycine, 25 mm Tris in PBS) for 10 min at 4 °C. The cells were collected using cold PBS and centrifuged for 10 min at 1000 × g, and cell pellets were homogenized in lysis puffer (1% Triton X-100 in Tris-buffered saline containing 0.1 mm phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 10 μg/ml pepstatin A, 2 mm EDTA, pH 8.0) and centrifuged. The supernatants were incubated with 50 μl of streptavidin-Sepharose beads (Sigma) for at least 2 h at 4 °C while rotating. The beads were pelleted by brief centrifugation, and aliquots of the supernatants were taken to represent the unbound, intracellular pool. Eluted proteins were boiled in 1× sample buffer for 5 min and loaded on a SDS gel.
Radioligand Binding
Specific radioligand binding to membrane fractions was determined by a filtration assay (80 μg of protein/assay) (12). Briefly, 40 μl of cold strychnine solution were added to the membrane suspension (1 μg/μl of protein); after 45 min of incubation on ice, 30 μl of a 47.8 nm [3H]strychnine solution (PerkinElmer Life Sciences; specific activity, 47.8 mCi/mol) were added, and incubation continued for another 45 min. The final concentrations were 80 μg of membrane protein, 9.6 nm [3H]strychnine, 0–1000 nm cold strychnine in a final volume of 150 μl. All of the determinations were carried out in triplicate. Using Microcal Origin (Additive, Friedrichsdorf, Germany), the data were fitted to the following equation,
 |
where dpmspec represents specific binding, expressed as scintillator counts (decays/min), dpmobs is the observed radioactivity, dpmunspec is nonspecific binding; [Str] and [3H-Str] are the concentrations of cold strychnine (varied) and [3H]strychnine, respectively; and KD is the equilibrium dissociation constant for strychnine.
Dot Blot Expression Assay
Expression of various GlyR α3 constructs in HEK 293 was quantified using dot blot receptor assay (11). To this end, membrane suspensions (5–10 μg of total protein) were taken up in a 10-fold volume of uptake buffer (150 mm NaCl, 50 mm Tris/HCl, pH 7.4, 2% sodium desoxycholate, 40% methanol, 5 mm EDTA, 5 mm EGTA), diluted to 200 μl with dilute buffer (150 mm NaCl, 50 mm Tris/HCl, pH 7.4, 0.5% sodium desoxycholate, 20% methanol, 5 mm EDTA, 5 mm EGTA), and directly spotted onto a nitrocellulose membrane. GlyR content was determined using the same antibodies and procedure as for Western blot analysis. Duplicate determinations were carried out for each measurement, and three independent assays were performed to give n = 6 data points/construct.
Electrophysiological Recordings and Data Analysis
Whole cell currents were recorded using a HEKA EPC9 amplifier (HEKA Electronics, Lambrecht, Germany) controlled by Pulse software (HEKA Electronics) on a personal computer. Recording pipettes were pulled from borosilicate glass (World Precision Instruments, Berlin, Germany) using a Sutter P-97 horizontal puller. Ligand application using a U-tube gave a time resolution of 10–30 ms. The external buffer consisted of 137 mm NaCl, 5.4 mm KCl, 1.8 mm CaCl2, 1.0 mm MgCl2, 5.0 mm Hepes, pH adjusted to 7.2 with NaOH; the internal buffer was 120 mm CsCl, 20 mm N(Et)4Cl, 1.0 mm CaCl2, 2.0 mm MgCl2, 11 mm EGTA, 10 mm Hepes, pH adjusted to 7.2 with CsOH. Current responses were measured at room temperature of 21–23 °C, and the holding potential was −60 mV. Responses to the saturating concentration of 2 mm glycine were used for normalization of dose-response data. Maximum current responses from each individual cell were also recorded and averaged for each subunit (see Table 1).
TABLE 1.
Dose-response properties of GlyR α3 mutants
The data are given as the means ± S.D.
| Subunit | EC50a | nHilla | Isata | Calculated EC50b | KDb | Φb | Imaxb | Number of cellsc | KD ([3H]Stry)d |
|---|---|---|---|---|---|---|---|---|---|
| μm | pA | μm | μm | pA | nm | ||||
| α3L | 35 ± 4 | 1.8 ± 0.3 | 2410 ± 410 | 40 | 56 ± 14 | 0.23 ± 0.11 | 3251 ± 317 | 8 | 12.2 ± 1.7 |
| α3K | 24 ± 2 | 2.8 ± 0.4 | 2088 ± 240 | 30 | 43 ± 9 | 0.23 ± 0.09 | 2838 ± 237 | 7 | 5.3 ± 0.4 |
| α3LΔ6 | 3.9 ± 0.5 | 1.8 ± 0.4 | 3870 ± 400 | 3.6 | 39 ± 16 | 0.007 ± 0.005 | 3938 ± 452 | 5 | 30.1 ± 2.2 |
| α3L+6 | 26 ± 1 | 4.7 ± 0.7 | 1850 ± 150 | 23 | 19 ± 3 | 0.30 ± 0.06 | 2540 ± 148 | 6 | |
| α3LT358A | 86 ± 10 | 1.3 ± 0.3 | 570 ± 50 | 91 | 33 ± 7 | 3.24 ± 0.34 | 1161 ± 132 | 6 | |
| α3LY367F | 8.9 ± 0.4 | 1.6 ± 0.4 | 1790 ± 220 | 8.1 | 7 ± 2 | 0.67 ± 0.04 | 2896 ± 205 | 5 | |
| α3LS370A | 28 ± 4 | 1.2 ± 0.2 | 1350 ± 220 | 17 | 16 ± 3 | 1.04 ± 0.50 | 2738 ± 142 | 5 | |
| α3LT358A/Y367F | 157 ± 23 | 1.1 ± 0.2 | 680 ± 120 | 148 | 51 ± 12 | 5.38 ± 1.93 | 1589 ± 145 | 6 | |
| α3LT358A/S370A | 89 ± 7 | 1.8 ± 0.3 | 360 ± 30 | 93 | 53 ± 16 | 3.11 ± 0.30 | 1215 ± 72 | 5 | 27.8 ± 5.9 |
| α3LY358F/S370A | 16 ± 2 | 1.6 ± 0.3 | 1370 ± 140 | 23 | 11 ± 2 | 1.40 ± 0.90 | 3340 ± 145 | 8 | |
| α3LT358A/Y358F/S370A | 51 ± 5 | 1.6 ± 0.3 | 2020 ± 110 | 54 | 52 ± 12 | 0.55 ± 0.10 | 2451 ± 275 | 5 |
a Data from dose-response curves.
b Data from two-ligand model.
c Data for whole cell recording.
d Data from the radioligand binding assay.
Dose-response curves were constructed from the peak current amplitudes obtained with at least seven appropriately spaced glycine concentrations in the range 5–10,000 μm glycine. Using a nonlinear algorithm (Microcal Origin), dose-response data were first analyzed using the following Hill equation,
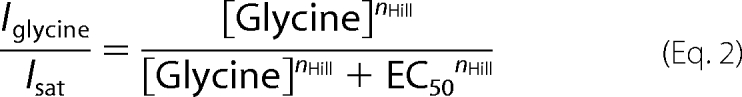 |
where Iglycine is the current amplitude at a given glycine concentration, Isat is the current amplitude at saturating concentrations of glycine, EC50 is the glycine concentration producing half-maximal current responses, and nHill is the Hill coefficient. Currents from each individual cell were normalized to the maximum response at saturating glycine concentrations. For each construct, the mean maximum current at saturating glycine concentration (2 mm) was calculated from all cells that were used for analysis. Dose-response curves were then scaled to this mean current amplitude. EC50 values are listed in Table 1. Differences of EC50 and Imax between wild type (α3L and α3K) and high and low affinity receptor variants were tested using one-way analysis of variance, values for p ≤ 0.05 were considered significant.
A kinetic model that requires binding of two ligands to independent sites prior to channel opening and considers channel gating (13–15) was used for subsequent analysis of dose-response data. The average maximum current at saturating glycine concentrations (2 mm) was used to scale current response data for each construct; dose-response data were then fit to Equation 3.
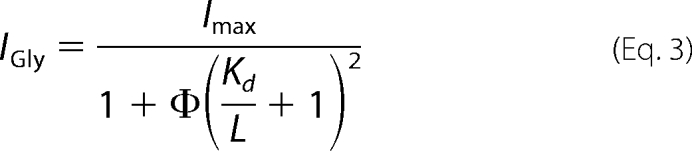 |
Here, Imax is the theoretical maximum current that would be observed if all channels were open simultaneously, IGly is the current obtained at a given glycine concentration, Kd is the dissociation constant of glycine from the receptor, L is the concentration of ligand (i.e. glycine), and Φ−1 = kop/kcl is the gating constant. The solid lines through the dose-response data (see Fig. 3, B and C) were computed using this model.
FIGURE 3.

Electrophysiological characterization of GlyR α3 subunit variants. A and B, dose-response analysis of GlyR α3 subunit variants. Current responses to fast glycine application were scaled using the average current response obtained with each subunit (Table 1). The curves through the data points were constructed using a two-ligand model of receptor activation. See Table 1 for constants. A, α3LΔ6 (solid squares, dashed and dotted line), α3L (open diamonds, dashed line), α3K (solid diamonds, dotted line), α3L+6 (crosses, solid line), α3LT358A/Y367F (solid circles, short-dotted line), α3LT358A/S370A (open circles, dashed and double-dotted line). B, α3LY367F (solid squares, solid line), α3LS370A (crosses, dashed line), α3LY367F/S370A (open circles, dotted line), α3LT358A/Y367F/S370A (solid triangles, short-dashed line), and α3LT358A (open triangles, dashed and dotted line). C, summary of receptor expression and functional data of GlyR α3 subunit variants. The subunit and the number of cells used is indicated on top of the bars. The subunits are: 1, α3LY367F; 2, α3LS370A; 3, α3LY367F/S370A; 4, α3LT358A; 5, α3LT358A/Y367F/S370A; 6, α3LΔ6; 7, α3LT358A/Y367F; 8, α3LT358A/S370A; 9, α3L+6; 10, α3L; 11, α3K. Upper panel, EC50 values (means ± S.D.). Differences between wild type (α3L and α3K) and various mutant receptors were tested for significance using one-way analysis of variance; a value of p ≤ 0.05 was considered significant. *, p ≤ 0.05; ns, not significant. Lower panel, Isat values (Table 1); maximum responses of HEK 293 cells transfected with the indicated subunit (means ± S.E.). D, correlation plot of Imax versus EC50 data of the GlyR α3 subunit variants. See Table 1 for constants. The correlation coefficient from a linear fit of the data points was r = −0.79.
EC50 was related to the Kd value obtained from the two-ligand model as follows: a glycine concentration of EC50 gives a half-maximum response; thus, with [Glycine] = EC50 and
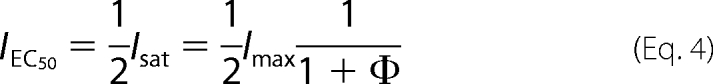 |
one obtains
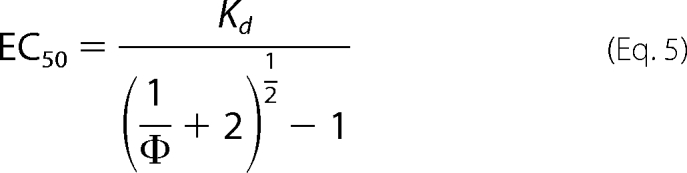 |
Protein Expression and CD Spectroscopy of Native Recombinant TM 3–4 Loops
DNA constructs encoding the large cytoplasmic TM3–4 loop of the human GlyR subunits α3L (amino acids 309–400) and α3K (amino acids 309–385) were cloned into the pET30a vector (Novagen, Darmstadt, Germany). After transformation and expression, Escherichia coli BL21 cells (Novagen) were harvested by centrifugation, resuspended in lysis buffer (50 mm NaH2PO4 pH 8.0, 300 mm NaCl, 10 mm imidazole), treated with lysozyme (0.1 mg/ml, 30 min, 0 °C), and sonicated (eight 10-s pulses) on ice. Sonification was repeated, and the supernatant was collected. Native protein was purified by chromatography on a nickel-nitrilotriacetic acid-agarose column and elution with increasing concentrations of imidazole. If needed, the protein was further purified by chromatography on a UnoQ anion exchange column. Protein was then dialyzed against 10 mm sodium phosphate buffer, pH 7.4, concentrated to 4–10 mg/ml.
Protein size and oligomerization was tested by size exclusion chromatography on a Sephacryl S-200 column (Bio-Rad). Marker proteins (Amersham Biosciences) were ribonuclease A (14 kDa), chymotrypsinogen (25 kDa), ovalbumin (43 kDa), albumin (67 kDa), and γ-globulin (150 kDa).
Measurements of CD were performed on a JASCO-J810 spectrometer (JASCO, Groβ-Umstadt, Germany) equipped with a Peltier thermostat in a 0.1-cm-path length quartz cell. Samples were degassed before measurements, and all of the spectra were base line-corrected by subtracting buffer runs. Four to eight individual scans at 20 °C from 260 to 190 nm with a scan speed of 10 nm/min, a 0.2-nm step size, and a response time of 1 s were taken and averaged. The protein concentrations were determined by measuring the absorbance at 280 nm using the equation c = A280/(ϵ × l), where A280 is absorbance at 280 nm, ϵ is the extinction coefficient, and l is the path length of the measurement cell. Extinction coefficients ϵ were determined from the amino acid sequences (16) to be ϵ = 2560 lmol−1 cm−1 for α3L, and ϵ = 1280 lmol−1 cm−1 for α3K.
Peptide Synthesis and CD Spectroscopy of Synthetic α3L Polypeptide
The peptide was synthesized by a solid phase synthesis on a 9050 Plus Peptide Synthesizer (Millipore, Freiburg, Germany) using Fmoc-PALPEG-PS-resin. The lyophilized crude peptide was analyzed for integrity and purity by reversed phase HPLC analysis and matrix-assisted laser desorption ionization-time of flight mass spectrometry on a Bruker Autoflex Spectrometer (Bruker Daltonics, Bremen, Germany). Purification of the final polypeptide was performed by reversed phase chromatography using a Perseptive BioCAD HPLC apparatus and a PD10 column (PerSeptive Biosystems, Freiburg, Germany). The purified polypeptide was lyophilized and taken up in 10 mm Na+ phosphate buffer, pH 8.0. Amino acid analysis was performed on a Merck-Hitachi L-6200 system, equipped with a F-1050 fluorometer and a D-2500 integrator. The concentration of synthetic α3L polypeptide was determined after acid hydrolysis in 6 m HCl at 155 °C for 1 h and precolumn derivatization with o-phthaldialdehyde/3-mercaptopropionic acid (17). CD spectra were normalized to the protein concentration and normalized spectra deconvoluted using the CDSSTR algorithm and the Reference set 7 at Dichroweb (18, 19). The values for helix 1 and helix 2 as well as sheet 1 and sheet 2 were added to obtain total helix and total sheet content (20). The concentration of the synthetic α3L polypeptide was 0.0452 mg/ml. Thermal denaturation was carried out by a heating the sample from 30 to 80 °C and cooling back to 30 °C at a rate of 2 K/min.
RESULTS
Design and Expression of GlyR α3 Constructs
The large cytoplasmic TM3–4 loop is a region of high sequence diversity between different glycine receptor subunits (Fig. 1A). For the long splice variant of the human α3 GlyR subunit, α3L, secondary structure prediction algorithms suggest an extension of a cytosolic α-helix, covering the alternatively spliced exon. In the homologous cytosolic region, the propensity to attain an α-helical fold appears to be less pronounced in α3K. To identify the influence of alterations of the spliced 15-residue insert within the TM3–4 loop, glycine receptor α3 constructs were generated using site-directed mutagenesis. The three hydroxyl-bearing residues Thr358, Tyr367, and Ser370 were replaced by their closest hydrocarbon analogs, covering all possible permutations (Fig. 1B). Because the length of the spliced protein region itself may be a determinant of receptor function, the 15-amino acid insert was further shortened or elongated by removal (α3Δ6) or duplication (α3+6) of the 6-amino acid segment 359EAFALE364 (Fig. 1B). This segment was chosen to leave the minimum phosphorylation consensus motif (+/X/-OH) intact in both constructs. A cluster of six positive charges, which precedes the alternatively spliced region (7), has been shown to be critical for endoplasmic reticulum trafficking and membrane insertion of recombinant α1 GlyR (21). This cluster was not modified in our study.
Western blot analysis revealed expression of all constructs in HEK 293 cells (Fig. 2A). Quantification of receptor antigen content in HEK 293 membranes using a dot blot receptor assay (12) indicated similar expression efficiency for all subunits (Fig. 2B). Relative amounts of GlyR antigen (arbitrary units) for the different constructs varied between 2.7 ± 0.5 (α3LY367F) and 6.1 ± 1.9 (α3L), i.e. by a factor of 2.3 (Fig. 2B). Because crude membrane preparations may also contain endoplasmic reticulum membranes, surface protein was determined taking advantage of the high affinity between biotin and streptavidin.
FIGURE 2.

Surface expression and radioligand binding of GlyR α3 subunit variants. A, HEK 293 cells were transfected with the indicated subunits, and crude membrane preparations were analyzed using the pan-α antibody mAb-4a and detected with a cy5-coupled goat anti-mouse antibody. The subunits are: 1, α3LY367F; 2, α3LS370A; 3, α3LY367F/S370A; 4, α3LT358A; 5, α3LT358A/Y367F/S370A; 6, α3LΔ6; 7, α1(wt); 8, α3LT358A/Y367F; 9, α3LT358A/S370A; 10, α3L; 11, α3K; 12, water transfected cells; and 13, α3L+6. The arrows indicate the 48-kDa band, and the arrowheads indicate 45- and 36-kDa bands. B, quantitation of receptor expression via dot blot receptor assay (means ± S.D.). The entire assay was performed three times using duplicate determinations for each data point to give n = 6 measurements/subunit. C, comparison of surface expression (upper panel) and intracellular protein (lower panel) of GlyRα3 variants (α3K, α3L, α3LΔ6, α3L+6, and α3LT358A/S370A) differing within the splice cassette of α3L. Untransfected COS7 cells (lane UT) as well as a mock transfection using a green fluorescent protein (lane GFP) plasmid were used as negative controls. GlyRα1 wild type served as positive control. The arrows point to the specific GlyR protein band at 48 kDa. Double bands in the surface fraction are most likely due to varying degrees of glycosylation. D, radioligand binding assays. Displacement of [3H]strychnine (KD = ∼9.6 nm) by cold strychnine was measured. For each tested subunit, specific binding was normalized to the maximum binding (at zero concentration of cold strychnine) and plotted. Unspecific binding was determined at the maximum concentration of cold strychnine. All of the determinations were carried out in triplicate, see Table 1 for constants. The data are shown for α3L (■, solid line), α3K (○, dashed line), α3LT358A/S370A (◇, dotted line), and α3LΔ6 (▲, dashed and dotted line). The specific counts measured for each subunit were: α3L (dpmmax = 11118 ± 890, dpmnonspec = 613 ± 45), α3K (dpmmax = 20711 ± 910, dpmnonspec = 1524 ± 230), α3LT358A/S370A (dpmmax = 18626 ± 674, dpmnonspec = 1224 ± 68), and α3LΔ6 (dpmmax = 22714 ± 1427, dpmnonspec = 1546 ± 105).
To determine the fraction of recombinant α3 subunit variants expressed at the plasma membrane in COS7 cells, surface proteins were labeled by biotinylation using sulfo-N-hydroxy-sulfosuccinimide-S-S-coupled biotin and subjected to subsequent precipitation. Plasma membrane integration and intracellular protein accumulation levels were analyzed by immunoblotting of biotinylated precipitates and supernatants using the pan-α antibody, mAb-4a (Fig. 2C). The supernatants represent the unbiotinylated intracellular pool (Fig. 2C, lower panel). No differences were detectable between extra- and intracellular fractions from cells transfected with α3L, α3K, α3LΔ6, α3L+6, and α3LT358A/S370A mutants as compared with cells carrying the wild type α1 subunit protein (Fig. 2C, upper panel). The glycine receptor-specific antibody revealed double bands in the surface fraction, most likely because of different degrees of glycosylation; the ratio of these double bands, however, was similar for all of the constructs tested. These observations indicated that alterations in the spliced insert within the TM3–4 loop did not affect receptor biosynthesis or surface expression in the recombinant system.
Radioligand Binding
To assess whether TM3–4 mutations had an effect on equilibrium antagonist binding, displacement of [3H]strychnine binding by cold strychnine was examined (Fig. 2D). The tested constructs showed KD values for strychnine ranging from 5.3 ± 0.4 nm (α3K) to 30.1 ± 2.2 nm (α3LΔ6). It should be noted that although these constants varied ∼6-fold, they did not show any trend, in contrast to current dose-response data. In fact, the two subunits that differed most in their current response characteristics (α3LΔ6 and α3LT358A/S370A) had quite similar KD values in the ligand displacement test (Table 1). Thus, radioligand studies supported the hypothesis that ligand binding properties of GlyR α3 constructs were not notably affected by the modifications of the TM3–4 loop under study (Fig. 2D and Table 1). Although equilibrium radioligand binding gives only limited information about ligand affinity of the resting receptor, these results nevertheless suggested that mutations in the TM3–4 loop had no significant effect on the ligand-binding pocket of α3 glycine receptors.
Whole Cell Current Recordings
In whole cell current recordings from HEK 293 cells transfected with GlyR α3 constructs (Fig. 3, A and B), a clear correlation between maximum current amplitudes and EC50 values was observed (Fig. 3D), where high sensitivity (low EC50) was associated with large current amplitudes (Fig. 3C and Table 1). The responses could be classified into three groups (Fig. 3, A and B): (i) high affinity, high Imax (α3LΔ6); (ii) intermediate affinity and Imax (α3L, α3K, α3L+6, α3LT358A/Y367F/S370A, α3LY367F, α3LS370A, and α3LY367F/S370A); and (iii) low affinity and Imax (α3LT358A, α3LT358A/Y367F, and α3LT358A/S370A). The extremes for EC50 and Imax were 3.9 ± 0.5 μm (α3LΔ6) and 157 ± 23 μm (α3LT358A/Y367F) and 360 ± 30 pA (α3LT358A/S370A) and 3870 ± 400 pA (α3LΔ6), respectively. Thus, the EC50 and Imax values observed differed by factors of 40 and 11, respectively, compared with <2.3-fold variability of protein expression levels. Furthermore, those constructs that differed most in their electrophysiological properties showed similar protein expression in HEK 293 cells (Fig. 2B). Current responses of both the high activity mutant (α3LΔ6), as well as the low activity mutants (α3LT358A, α3LT358A/Y367F, and α3LT358A/S370A), differed markedly from the wild types (α3L and α3K).
Dose-response data (Fig. 3) were fit to a two-ligand model of receptor activation to estimate which of the constants describing receptor channel activation, Imax, KD, and the gating constant Φ, were most affected. Fitted KD values ranged from 7 to 56 μm, i.e. 8-fold; Imax values varied 3.4-fold (1161–3938 pA), whereas the value for the gating constant Φ = kcl/kop was found to vary between 0.007 and 5.38, i.e. over 750-fold (Table 1). The EC50 values calculated using constants obtained from the two-ligand mechanism agreed well with those found in a direct fit of the dose-response data (Table 1). Thus, the dominant effect of TM3–4 mutations appeared to be on channel gating. It should be noted that dose-response data from whole cell recordings alone do not allow determination of the gating constant Φ. Nevertheless, the analysis performed here showed that when all parameters were allowed to float during the fitting procedure, the largest variation was in the value of Φ. Indeed, dose-response curves could well be fitted using the same KD and Imax values for all subunit variants and only varying the gating constant Φ. Consistently, recordings from outside/out patches indicated that high and low activity subunits had similar single-channel conductances (data not shown).
Association and Secondary Structure Analysis of Isolated TM 3–4 Loops
GlyR α3 TM3–4 loops of the long (α3L) and short (α3K) variant were overexpressed in E. coli. The native soluble protein was purified by chromatography on nickel-nitrilotriacetic acid-agarose and ion exchange columns (22). Size exclusion chromatography on a Sephacryl S-200 column indicated association of α3L into 5- and 3-mers, whereas α3K gave oligomers of ten and five subunits as well as monomers (Fig. 4, B and C). Because the same protocol for expression and purification had been followed for both subunits, the differences in domain association were significant. CD spectroscopy of the recombinant cytoplasmic loops indicated a well ordered structure for the α3L loop construct (Fig. 4D). Thus, defined secondary structures would be expected for the α3L TM3–4 loop, whereas the markedly different CD spectrum of the α3K loop construct (Fig. 4D) was characteristic of an unfolded protein.
FIGURE 4.

Secondary structure and association of recombinant and synthesized TM3–4 loops. A, SDS-polyacrylamide gel electrophoresis and Coomassie stain to verify integrity and purity of expressed TM3–4 loops. The subunits and size markers are indicated. B, size exclusion chromatography of recombinant TM3–4 loops on a Sephacryl S-200 column. Peaks indicating different oligomerization states are labeled. C, summary of size exclusion chromatography data. D, CD spectra of TM3–4 loops of recombinant α3L and α3K from expression in E. coli. E, SDS-polyacrylamide gel electrophoresis and Coomassie stain to verify integrity and purity of the synthesized α3L TM3–4 loop. The size markers are indicated. F, CD spectra of the synthetic α3L TM3–4 loop before and after thermal denaturation. Inset, thermal unfolding and cooling curves of synthetic α3L TM3–4 loop. Ellipticity at 200 nm is plotted versus temperature. Note melting point of highly ordered structure at ∼58 °C. Dashed line, heating, solid line, cooling. G, summary of secondary structure analysis data of synthetic α3L TM3–4 loop. MW, molecular weight.
The TM3–4 loop of α3L was synthesized chemically, yielding a stable polypeptide of correct mass (Fig. 4E). The CD spectrum (Fig. 4F) indicated a secondary structure similar to the native TM3–4 loop obtained from recombinant expression. Secondary structure analysis indicated ∼41% of α-helix and 25% of β-sheet (Fig. 4G), in good agreement with the values found for the native protein. Thermal unfolding (Fig. 4F, inset) gave a melting temperature for the fold of 58 °C, with a predominant loss of helical structures. Data from the synthetic protein confirmed the presence of stable secondary structures in the TM3–4 loop of the α3L loop polypeptide.
DISCUSSION
Ligand-gated ion channels of the Cys loop receptor type share both pentameric architecture of the protein complex as well as transmembrane topology of subunits (2, 3, 23, 24). Amino acid sequence homology between mammalian GlyR α subunit orthologs ranges from 80 to 98%, whereas intersubunit homology between α and β sequences is on the order of 60% (2). The cytoplasmic TM3–4 loop is the region of highest sequence diversity between GlyR α subunits, as well as other members of the Cys loop receptor superfamily (25). This domain carries motifs for phosphorylation (26), ubiquitination (27), as well as putative Ca2+-dependent intracellular factors (28), and a basic cluster thought to ensure correct transmembrane topology (21). Two natural splice variants of the human GlyR α3 subunit differ in desensitization kinetics (8); hydroxyl functions within this insert are relevant, but not exclusive, determinants of receptor kinetics (9). Here, the impact of the 15-residue insert on channel function was probed by removal of its hydroxyl groups and varying its length. Comparison of ion channel properties using patch-clamp recording techniques and secondary structure analysis of isolated TM3–4 loops identified a novel regulatory motif within this cytoplasmic domain of GlyR α3 subunits.
GlyR α3L Mutations Affect Ion Channel Function
Although cell surface protein expression levels were largely unaffected, whole cell current responses of recombinant α3 GlyRs revealed a striking sensitivity of ion channel function to alterations within the 15-residue insert in α3L. Maximum current amplitudes (Imax) and EC50 were found to differ 11- and 40-fold, respectively, despite almost identical protein expression. Based on current responses, GlyR α3 mutants could be classified into three groups: (i) one high sensitivity mutant (α3LΔ6), characterized by large Imax and small EC50; (ii) constructs of intermediate Imax and EC50 values (α3L, α3K, α3L+Δ6, α3LT358A/Y367F/S370A, α3LY367F,α3LS370A, and α3LY367F/S370A); and (iii) low sensitivity mutants (α3LT358A, α3LT358A/Y367F, and α3LT358A/S370A), displaying low Imax and large EC50 values. Subunit expression was not different between the various α3 constructs, as determined by dot-blot assay as well as surface protein expression by biotinylation. Thus, because receptor density was excluded as a cause for the variations in Imax, differences in whole cell current responses could be attributed to alterations in (i) ligand affinity, (ii) single channel conductance, or (iii) ion channel gating. The value of EC50 is composed of two equilibria, namely ligand binding and gating (13), whereas Imax depends on the number of receptors on the cell surface, as well as channel conductance and gating. We tested, therefore, to which extent other steps of channel function were affected by α3L mutations. Radioligand displacement of [3H]strychnine by its cold analog showed no difference in antagonist affinity for the mutants. Because binding domains for strychnine and glycine on the receptor are thought to overlap (3), one would have expected at least some effect on strychnine binding, if ligand binding were responsible for the observed 40-fold shift in EC50.
The concomitant changes of EC50 and Imax were consistent with modification of ion channel gating (14) as the main effect of the mutations. Indeed, when dose-response data were analyzed using a two-ligand model of receptor activation (29), the Imax and KD values were found to differ 3.4- and 8-fold, respectively. In contrast, the value for the gating constant Φ varied ∼770-fold (Table 1), consistent with variations channel gating.
The hyperekplexia mutants GlyR α1(K276E) (30) and GlyR α1(Q266H) (31), located within the TM2–3 loop and TM2, respectively, impaired channel gating, leading to a reduced duration of channel openings. Alterations in channel gating were also demonstrated for recombinant glycine receptors from zebrafish, where a soluble Ca2+-binding factor was proposed to mediate these effects (28). The marked difference in open channel dwell times between cell-attached and outside-out recordings would be consistent with an intracellular mode of ion channel regulation. In the glycine receptor α1 subunit, mutations within the TM1–2 loop resulted in altered receptor desensitization (9, 15, 32) and ion permeation (33). Similar effects have been observed with γ-aminobutyric acid, type A receptor mutants (34), also belonging to the Cys loop family of amino acid gated anion channels.
Ligand binding appeared least sensitive to alterations within the large intracellular loop, yet desensitization and channel gating were both affected by changes within the TM3–4 loop. Removal of all OH groups of the spliced insert resulted in altered receptor desensitization (9), whereas the results described here identified changes in channel gating caused by structure modifications within the insert.
Mutations in the GlyR α3L Splice Insert Affect Protein Surface Polarity
How can the gating differences observed for GlyR α3 mutants be attributed to changes in protein structure? An alignment of the amino acid sequences around the alternatively spliced region allowed a subdivision of this region into four blocks, each five to six residues long. These blocks were characterized by distinct distributions of charged and hydrophobic side chains (Fig. 5). Common to all constructs is an eight-residue block containing six positive charges immediately preceding the splice site, which is also found in the homologous position of the GlyR α1 subunit (21). This is followed by two six-amino acid segments and one stretch of five residues. The distribution of hydrophobic residues over the entire region is similar for all constructs, but the net charge of each segment is different between mutants. The high efficacy mutation, α3LΔ6, is characterized by a (+)-(+)-(−)-(+) sequence of charged blocks, whereas intermediate and low efficacy mutants display the sequence (+)-(−)-(+)-(−). This sequence of net charges is also retained in the short splice variant, α3K (Fig. 5). Furthermore, Thr358 is absent in the low efficacy mutants with the exception of the triple mutant α3LT258A/Y367F/S370A. Taken together, an extended stretch of positive charges after the alternative splice site and the presence of a hydroxyl group were associated with efficient channel gating, whereas insertion of a short segment carrying a net negative charge appeared to dramatically reduce the efficacy of channel opening. The absence of the hydroxyl function at or near position GlyR α3(358) further impaired gating.
FIGURE 5.

Sequence alignment and chemical properties of the splice site in GlyR α3 variants. The starting position is 349, before the conserved cluster of positive charges. A, aligned sequences and representation of residue properties. Symbols to describe side chains are: +, positive charge; −, negative charge; N, polar, neutral; ○, large hydrophobic; ⊗, -OH; ●, small. The sequence can be subdivided into distinct blocks of five or six residues. B, scheme indicating electrostatic properties of the alternatively spliced region in various α3 constructs. The gray color indicates the positive charge. Note that the overall hydrophobicity of each block remains constant, but the sequence of charged blocks is reversed between high and low activity mutants.
Spliced Insert Determines Stability of TM 3–4 Loop Structure and Controls Channel Function
CD spectroscopy of native and synthesized TM3–4 loops of α3L and α3K indicated a defined and stable fold only for α3L, whereas α3K appeared less ordered. A sequence comparison shows that the insert is located in a region of pronounced diversity among glycine receptor subunits (Fig. 1A). However, secondary structure predictions (35) using various algorithms suggested only minor differences between the TM 3–4 loops of α3L and α3K. Generally, a lower amount of α-helix was predicted for the latter, in qualitative agreement with CD spectroscopy (Fig. 4D). Also, no major differences in secondary structure were predicted for the TM 3–4 loops of the mutant constructs. Thus, the pronounced changes in ion channel function and the differences in secondary structure analysis from CD spectroscopy exceeded bioinformatic predictions. This suggested that the spliced insert stabilizes secondary structures of the TM 3–4 loop, whereas removal of the insert, as in α3K, renders the domain more flexible. In nicotinic acetylcholine receptors, an unfolded (or unstably folded) structure appears compatible with receptor function (36) and has been observed for the cytoplasmic loop of nicotinic acetylcholine receptors (37). Indeed, the sensitivity of channel gating to mutations within the variable insert indicates that this site is an effective regulatory motif for channel function.
The relevance of the intracellular TM3–4 loop of Cys loop receptors to ion channel properties has only recently become a topic of recognition. An arginine residue in 5-HT3A serotonin receptors had been shown to affect receptor desensitization (38), and charged residues within the TM3–4 loop have been implicated in modulation of ion channel conductance (39–41) and ion selectivity (42). In fact, the previous study of the spliced insert on α3 glycine receptor currents had shown that hydroxyl groups were contributing but not exclusive determinants of receptor desensitization (9). These findings are in agreement with observations that charged (glutamate) residues, introduced into the TM3–4 loop of α1 glycine receptors, affected single channel conductances, but the major contribution to conductance still was mediated through the channel-lining TM2 domain (43). Overall, it appears that both charges and structural alterations within the cytoplasmic TM3–4 domain are indeed contributing but not exclusive determinants of ion channel function.
Secondary structure analysis and structure prediction using bioinformatics tools consistently indicated that the spliced insert in α3 GlyRs determines the folding of large parts of the intracellular TM3–4 domain. Alterations of critical positions within the insert as well as insert size confirmed a correlation between the structure of the insert and the function of the corresponding ion channel.
Thus, the role of the spliced insert in human α3 GlyRs appeared 2-fold: (i) the insert also stabilizes the fold of the TM3–4 domain, and (ii) the insert is a determinant of ion channel desensitization (8) and gating. Further studies are needed to identify the molecular partners and mechanisms of ion channel regulation. Yet, GlyR function is remote-controlled by intracellular protein domains that, although physically distant from the receptor ion pore, appear to contribute to ion permeation and channel gating.
Acknowledgments
Helpful discussions with Drs. H. Sticht, P. Bregestowski, K. Becker, and C. Kluck and cell culture maintenance by Rosa Weber are gratefully acknowledged. We thank Dr. E. Hannappel for help with amino acid analysis of the synthesized proteins, Dr. Finn Bauer for help with CD spectroscopy, and P. Wenzeler for performing protein synthesis.
This work was supported by Deutsche Forschungsgemeinschaft Grants SFB 359-A13 (to H.-G. B. and C.-M. B.), SPG 1026, and BR 1507/4 and by the Fonds der Chemischen Industrie.
- GlyR
- glycine receptor
- BES
- 2-[bis(2-hydroxyethyl)amino]ethanesulfonic acid
- PBS
- phosphate-buffered saline
- HPLC
- high pressure liquid chromatography
- CD
- circular dichroism.
REFERENCES
- 1.Betz H., Kuhse J., Schmieden V., Laube B., Kirsch J., Harvey R. J. (1999) Ann. N.Y. Acad. Sci. 868, 667–676 [DOI] [PubMed] [Google Scholar]
- 2.Becker K., Becker C. M., Breitinger H. G. (2000) in Channelopathies (Lehmann-Horn F. ed) pp. 199–224, Elsevier, Amsterdam, The Netherlands [Google Scholar]
- 3.Breitinger H. G., Becker C. M. (2002) ChemBioChem. 3, 1042–1052 [DOI] [PubMed] [Google Scholar]
- 4.Grudzinska J., Schemm R., Haeger S., Nicke A., Schmalzing G., Betz H., Laube B. (2005) Neuron 45, 727–739 [DOI] [PubMed] [Google Scholar]
- 5.Shiang R., Ryan S. G., Zhu Y. Z., Hahn A. F., O'Connell P., Wasmuth J. J. (1993) Nat. Genet. 5, 351–358 [DOI] [PubMed] [Google Scholar]
- 6.Becker C. M. (1995) Neuroscientist 1, 130–141 [Google Scholar]
- 7.Brune W., Weber R. G., Saul B., von Knebel Doeberitz M., Grond-Ginsbach C., Kellerman K., Meinck H. M., Becker C. M. (1996) Am. J. Hum. Genet. 58, 989–997 [PMC free article] [PubMed] [Google Scholar]
- 8.Nikolic Z., Laube B., Weber R. G., Lichter P., Kioschis P., Poustka A., Mülhardt C., Becker C. M. (1998) J. Biol. Chem. 273, 19708–19714 [DOI] [PubMed] [Google Scholar]
- 9.Breitinger H. G., Villmann C., Rennert J., Ballhausen D., Becker C. M. (2002) J. Neurochem. 83, 30–36 [DOI] [PubMed] [Google Scholar]
- 10.Higuchi R., Krummel B., Saiki R. K. (1988) Nucleic Acids Res. 16, 7351–7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen C., Okayama H. (1987) Mol. Cell Biol. 7, 2745–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Becker C. M., Hoch W., Betz H. (1989) J. Neurochem. 53, 124–131 [DOI] [PubMed] [Google Scholar]
- 13.Hess G. P. (1993) Biochemistry 32, 989–1000 [DOI] [PubMed] [Google Scholar]
- 14.Colquhoun D. (1998) Br. J. Pharmacol. 125, 924–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Breitinger H. G., Villmann C., Becker K., Becker C. M. (2001) J. Biol. Chem. 276, 29657–29663 [DOI] [PubMed] [Google Scholar]
- 16.Gill S. C., von Hippel P. H. (1989) Anal. Biochem. 182, 319–326 [DOI] [PubMed] [Google Scholar]
- 17.Hannappel E., Kalbacher H., Voelter W. (1988) Arch. Biochem. Biophys. 260, 546–551 [DOI] [PubMed] [Google Scholar]
- 18.Lobley A., Whitmore L., Wallace B. A. (2002) Bioinformatics 18, 211–212 [DOI] [PubMed] [Google Scholar]
- 19.Whitmore L., Wallace B. A. (2004) Nucleic Acids Res. 32, W668–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sreerama N., Venyaminov S. Y., Woody R. W. (1999) Protein Sci. 8, 370–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sadtler S., Laube B., Lashub A., Nicke A., Betz H., Schmalzing G. (2003) J. Biol. Chem. 278, 16782–16790 [DOI] [PubMed] [Google Scholar]
- 22.Breitinger U., Breitinger H. G., Bauer F., Fahmy K., Glockenhammer D., Becker C. M. (2004) J. Biol. Chem. 279, 1627–1636 [DOI] [PubMed] [Google Scholar]
- 23.Betz H. (1992) Q. Rev. Biophys. 25, 381–394 [DOI] [PubMed] [Google Scholar]
- 24.Lynch J. W. (2004) Physiol. Rev. 84, 1051–1095 [DOI] [PubMed] [Google Scholar]
- 25.Rajendra S., Lynch J. W., Schofield P. R. (1997) Pharmacol. Ther. 73, 121–146 [DOI] [PubMed] [Google Scholar]
- 26.Harvey R. J., Depner U. B., Wässle H., Ahmadi S., Heindl C., Reinold H., Smart T. G., Harvey K., Schütz B., Abo-Salem O. M., Zimmer A., Poisbeau P., Welzl H., Wolfer D. P., Betz H., Zeilhofer H. U., Müller U. (2004) Science 304, 884–887 [DOI] [PubMed] [Google Scholar]
- 27.Büttner C., Sadtler S., Leyendecker A., Laube B., Griffon N., Betz H., Schmalzing G. (2001) J. Biol. Chem. 276, 42978–42985 [DOI] [PubMed] [Google Scholar]
- 28.Fucile S., De Saint Jan D., de Carvalho L. P., Bregestovski P. (2000) Neuron 28, 571–583 [DOI] [PubMed] [Google Scholar]
- 29.del Castillo J., Katz B. (1957) Proc. Roy. Soc. B 146, 339–356 [DOI] [PubMed] [Google Scholar]
- 30.Lewis T. M., Sivilotti L. G., Colquhoun D., Gardiner R. M., Schoepfer R., Rees M. (1998) J. Physiol. 507, 25–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moorhouse A. J., Jacques P., Barry P. H., Schofield P. R. (1999) Mol. Pharmacol. 55, 386–395 [DOI] [PubMed] [Google Scholar]
- 32.Saul B., Kuner T., Sobetzko D., Brune W., Hanefeld F., Meinck H. M., Becker C. M. (1999) J. Neurosci. 19, 869–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee D. J., Keramidas A., Moorhouse A. J., Schofield P. R., Barry P. H. (2003) Neurosci. Lett. 351, 196–200 [DOI] [PubMed] [Google Scholar]
- 34.Wotring V. E., Miller T. S., Weiss D. S. (2003) J. Physiol. 548, 527–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cuff J. A., Barton G. J. (2000) Proteins 40, 502–511 [DOI] [PubMed] [Google Scholar]
- 36.Kukhtina V., Kottwitz D., Strauss H., Heise B., Chebotareva N., Tsetlin V., Hucho F. (2006) J. Neurochem. 97, 63–67 [DOI] [PubMed] [Google Scholar]
- 37.Unwin N. (2005) J. Mol. Biol. 346, 967–989 [DOI] [PubMed] [Google Scholar]
- 38.Hu X. Q., Sun H., Peoples R. W., Hong R., Zhang L. (2006) J. Biol. Chem. 281, 21781–21788 [DOI] [PubMed] [Google Scholar]
- 39.Deeb T. Z., Carland J. E., Cooper M. A., Livesey M. R., Lambert J. J., Peters J. A., Hales T. G. (2007) J. Biol. Chem. 282, 6172–6182 [DOI] [PubMed] [Google Scholar]
- 40.Gee V. J., Kracun S., Cooper S. T., Gibb A. J., Millar N. S. (2007) Br. J. Pharmacol. 152, 501–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jansen M., Bali M., Akabas M. H. (2008) J. Gen. Physiol. 131, 137–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Livesey M. R., Cooper M. A., Deeb T. Z., Carland J. E., Kozuska J., Hales T. G., Lambert J. J., Peters J. A. (2008) J. Biol. Chem. 283, 19301–19313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carland J. E., Cooper M. A., Sugiharto S., Jeong H. J., Lewis T. M., Barry P. H., Peters J. A., Lambert J. J., Moorhouse A. J. (2009) J. Biol. Chem. 284, 2023–2030 [DOI] [PubMed] [Google Scholar]