

Abstract
Elevated levels of plasma low density lipoprotein (LDL)-cholesterol, leading to familial hypercholesterolemia, are enhanced by mutations in at least three major genes, the LDL receptor (LDLR), its ligand apolipoprotein B, and the proprotein convertase PCSK9. Single point mutations in PCSK9 are associated with either hyper- or hypocholesterolemia. Accordingly, PCSK9 is an attractive target for treatment of dyslipidemia. PCSK9 binds the epidermal growth factor domain A (EGF-A) of the LDLR and directs it to endosomes/lysosomes for destruction. Although the mechanism by which PCSK9 regulates LDLR degradation is not fully resolved, it seems to involve both intracellular and extracellular pathways. Here, we show that clathrin light chain small interfering RNAs that block intracellular trafficking from the trans-Golgi network to lysosomes rapidly increased LDLR levels within HepG2 cells in a PCSK9-dependent fashion without affecting the ability of exogenous PCSK9 to enhance LDLR degradation. In contrast, blocking the extracellular LDLR endocytosis/degradation pathway by a 4-, 6-, or 24-h incubation of cells with Dynasore or an EGF-AB peptide or by knockdown of endogenous autosomal recessive hypercholesterolemia did not significantly affect LDLR levels. The present data from HepG2 cells and mouse primary hepatocytes favor a model whereby depending on the dose and/or incubation period, endogenous PCSK9 enhances the degradation of the LDLR both extra- and intracellularly. Therefore, targeting either pathway, or both, would be an effective method to reduce PCSK9 activity in the treatment of hypercholesterolemia and coronary heart disease.
High levels of circulating low-density lipoprotein (LDL)3-cholesterol represent a major risk factor that leads to coronary heart disease, the main cause of death and morbidity worldwide (1). LDL particles are cleared mainly from the bloodstream by the hepatic cell surface LDL receptor (LDLR) (2). Genetics studies demonstrated that loss-of-function mutations in either LDLR or apolipoprotein B, the protein component of LDL that binds LDLR, result in familial hypercholesterolemia and premature coronary heart disease (3). More recently, the proprotein convertases subtilisin kexin 9 (PCSK9) gene (4), which is highly expressed in liver and small intestine (5), was identified as the third locus associated with familial hypercholesterolemia (6). It is now clear that PCSK9 binds the LDLR and triggers its intracellular degradation in acidic compartments, resulting in increased circulating plasma cholesterol (7–10).
After its autocatalytic cleavage, PCSK9 is secreted as a stable noncovalent complex with its prosegment (pro·PCSK9) (5, 7). This cleavage results in a conformational change (11) that favors the binding of PCSK9 to the epidermal growth factor A domain (EGF-A) of the LDLR (12), with increased affinity at acidic pH values (11). Although the C-terminal Cys-His-rich domain of PCSK9 is a spatially separate domain (11) that does not participate directly in the PCSK9-EGF-A interaction (12), it is a critical determinant for the PCSK9-enhanced cellular degradation of the LDLR (13). In agreement, we recently demonstrated that annexin A2, which binds the Cys-His-rich domain of PCSK9, blocks its effect on LDLR degradation (14).
Overexpression studies in liver suggested that both intra- and extracellular PCSK9 target the LDLR (9, 15, 16) toward degradation in late endosomes/lysosomes (LE/L) (7–10). It was shown that the adaptor protein ARH, which interacts with the cytosolic tail of the LDLR, is essential for the endocytosis and degradation of the cell surface PCSK9·LDLR complex in vivo (16). However, hepatic LDLR protein levels were also reduced upon overexpression of PCSK9 in Arh −/− mice (9), suggesting the presence of an ARH-independent intracellular pathway. Intriguingly, at endogenous levels of PCSK9, the absence of ARH did not affect hepatic LDLR subcellular localization in LE/L or protein levels (17). This is not the expected result if PCSK9 mostly targets LDLR by the extracellular pathway (18), as one would have expected that in Arh−/− mice total LDLR levels should have been more elevated.
In this study, we focused on the relative contribution of the intra- versus extracellular pathways of endogenous PCSK9-induced LDLR degradation. This information should guide the choice of therapeutic approaches that will best target the site of PCSK9-LDLR interaction to control hypercholesterolemia and coronary heart disease.
EXPERIMENTAL PROCEDURES
Plasmids and Reagents
Human PCSK9 and its mutant cDNAs were cloned into pIRES2-EGFP (Clontech, Mountain View CA) as described (5). HepG2 and HEK293 cells (American Type Culture Collection) were routinely cultivated in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS; Wisent). LDL coupled with 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate (diI-LDL) and lipoprotein-deficient serum (LPDS) were from Biomedical Technologies. 3-Hydroxy-naphthalene-2-carboxylic acid (3,4-dihydroxy-benzylidene)-hydrazide monohydrate (Dynasore) was purchased from Sigma. Purified recombinant human hPCSK9 was provided by Bristol-Myers Squibb.
siRNA- and shRNA-mediated Knockdowns
Using the siPORT NeoFx reagent (Ambion), 3 × 105 HepG2 cells were plated in a 35-mm dish and simultaneously transfected either with 50 nm Alexa Fluor 555 nonsilencing siRNA (siCtl; Qiagen), both specific siRNAs (siCLCa, 5′-GGAAAGUAAUGGUCCAACA and siCLCb, 5′-GGAACCAGCGCCAGAGUGA; Dharmacon) or 7.5 nm Silencer Select (siARH, 5′-GGCUGUUACCCUCACCGUA; Ambion) and maintained in complete media for 72 h. Unless stated otherwise, at 48 h after transfection cells were washed twice, incubated overnight in 1 ml of DMEM. HEK293 cells were transfected with specified 60 nm siRNAs using Lipofectamine (Invitrogen), as described by the manufacturer. After 6 h, complete media were replaced, and cells were collected 72 h after transfection.
Mission® shRNA constructs in pLKO.1-puro vector (Sigma-Aldrich) targeting hPCSK9 (shPCSK9-1, 5′-CCGGGAATGCAAAGTCAAGGAGCATCTCGAGATGCTCCTTGACTTTGCATTCTTTTTG; and shPCSK9-2, 5′-CCGGGCCAGCAAGTGTGACAGTCATCTCGAGATGACTGTCACACTTGCTGGCTTTTTG) and controls (nontarget shNT, and empty vector pLKO) were used to generate lentiviral particles in the packaging cell line HEK293 FT. This was done by co-transfecting the recombinant lentiviral vectors with packaging plasmids (Invitrogen) using Lipofectamine 2000. Viral supernatants were filtered through a 0.45-μm filter 3 days after transfections. A total of 1.5 × 105 of HepG2 cells were plated in a 35-mm dish 1 day before infection. Transductions with lentivirus were made at various multiplicities of infection in the presence of Polybrene (4 μg/μl). Infections were made in duplicate to increase accuracy. Stable selection with puromycin (2 μg/μl) was done to obtain a pool of transduced cells for each shRNA sequence and control. Total RNA was isolated and analyzed with real time PCR using SYBR Green.
Western Blot Analysis
Cells were washed three times in phosphate-buffered saline (PBS) and lysed in complete radioimmune precipitation assay buffer (50 mm Tris-HCl, pH 8.0, 1% (v/v) Nonidet P-40, 0.5% sodium deoxycholate, 150 mm NaCl, and 0.1% (v/v) SDS) supplemented with 1× complete protease inhibitor mixture (Roche Applied Science). Proteins were separated by 10% SDS-PAGE and blotted on HyBond nitrocellulose membranes (GE Healthcare), which were blocked for 1 h in TBS-T (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.1% Tween 20) containing 5% nonfat dry milk. Membranes were then incubated 3 h in 1% nonfat milk with the respective antibodies: m,hLDLR (1:1000, R&D Systems), hTfR (1:1000, BD Biosciences), hPCSK9 (1:2500) (10), hCLCa and b (1:10,000) (19), hARH (1:1000, Abcam), β-actin (1:5000, Sigma). Appropriate horseradish peroxidase-conjugated secondary antibodies (1:10,000, Sigma) were used for detection with enhanced chemiluminescence using the ECL plus kit (GE Healthcare).
Fluorescence-activated Cell Sorting (FACS)
Using FuGENE HD reagent (Roche Applied Science), 3 h after passage 1 × 106 HepG2 cells were transiently transfected with a bicistronic vector (pIRES-PCSK9, Clontech), which encodes both different human PCSK9 constructs (H226A, wild type (WT), D374Y) and the enhanced green fluorescent protein (EGFP). Twenty-four hours after transfection, cells were washed three times with calcium/magnesium-free PBS containing 0.5% bovine serum albumin (solution A; Sigma). Cells were then incubated 5 min at 37 °C with 500 μl of 1× Versene solution (Invitrogen) and diluted in 15 ml of solution A. Cells were then centrifuged for 3 min at 1000 rpm and resuspended in 1 ml of solution A together with 1:200 of LDLR antibody (R&D Systems) for 45 min. Cells were washed once with 5 ml of solution A, centrifuged, and resuspended for 15 min in 1 ml of PBS containing 1:300 of Alexa Fluor 647 donkey anti-goat antibody (Molecular Probes). Cells were washed and resuspended in 300 μl of PBS and 0.3% propidium iodide. Viable cells (propidium iodide-negative) were then analyzed by FACS for both EGFP and Alexa Fluor 647 using the FACS BD LSR (BD Biosciences). HEK293 cells were transfected with pIRES-PCSK9 using Lipofectamine reagent. Twenty-four hours after transfection, the cells were then co-cultured in an increasing amount together with untransfected HEK293 cells. After overnight incubation, cells were analyzed by FACS as described above.
Immunocytochemistry
Cells were washed three times with PBS, fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.1% Triton X-100/PBS for 10 min, and incubated with 150 mm glycine to stabilize the aldehydes. The cells were then incubated for 30 min with 1% bovine serum albumin (fraction V, Sigma) containing 0.1% Triton X-100, followed by overnight incubation at 4 °C with selected antibodies (1:100 goat polyclonal anti-LDLR; R&D Systems, 1:1000 rabbit polyclonal CLCs (19); 1:500 mouse monoclonal cation-independent mannose 6-phosphate receptor (CI-MPR; Abcam). The cells were then incubated for 60 min with corresponding Alexa Fluor-conjugated secondary antibodies (Molecular Probes) and mounted in 90% glycerol containing 1% 1,4-diazabicyclo[2.2.2]octane (DABCO; Sigma). Immunofluorescence analyses were performed with a Zeiss LSM-510 confocal microscope coupled with a Nikon Eclipse TE2000-U laser-scanning microscope with 408-, 488-, and 543-nm laser lines. Images were processed with Adobe Photoshop CS2, version 9.0 (Adobe Systems).
Preparation and Incubation of HepG2 Cells with EGF-AB
Glutathione S-transferase-EGF-AB was captured and washed on a glutathione-Sepharose 4B resin and cleaved on the column using human α-thrombin. Cleaved EGF-AB was then applied to a size exclusion column, preequilibrated, and eluted in refolding buffer. After incubation in open tubes at 20 °C for 2 days, properly refolded EGF-AB was isolated as a trifluoroacetic acid salt using preparative reversed phase chromatography followed by lyophilization. Refolded EGF-AB conformer, containing six disulfide bridges, was recovered from preparative reversed phase chromatography at 94.5% purity, as determined by analytical reversed phase chromatography and mass spectrometry. Lyophilized EGF-AB peptide was resuspended in PBS at 1 mm and frozen at −80 °C until added into DMEM with or without purified human PCSK9 and incubated with HepG2 cells, as described.
LDLR Activity Assay
HepG2 cells were plated in a 96-well poly-d-lysine-coated plate (Becton Dickinson) at a density of 15,000 cells/well in RPMI 1640 medium (Invitrogen), containing 5% lipoprotein-deficient serum (Intracel). After 18 h, purified PCSK9 protein with or without EGF-AB was added to the cells in 30 μl of medium (RPMI 1640 medium containing 5% LPDS). After 2 h or 24 h of incubation at 37 °C, 5 μg/ml diI-LDL was added to the cell media, and cells were returned to tissue culture incubator for another 2 h. Cells were then fixed in the presence of a 2% paraformaldehyde solution containing 4 μg/ml Hoechst (Molecular Probes). After three washes of the cell layer, plates were scanned on Cellomics Array-scan (Thermo). DiI-LDL uptake measured as the mean total intensity of fluorescence per cell was obtained from quadruplicate wells and an average read of 500–700 cells/well for each treatment condition. Data were analyzed using GraphPad Prism 4.
PCSK9 ELISA
LumiNunc Maxisorp white assay plates (Nunc, Denmark) were coated with 0.5 μg/well PCSK9-Ab by incubation at 37 °C for 3 h in PBS (NaPO4 10 mm, NaCl 0.15 m, pH 7.4) containing sodium azide (1 g/liter) and then stored at 4 °C. The plates were washed six times before use with PBS containing Tween 20 (0.5 ml/liter) (PBST) and then incubated for 1 h at room temperature with blocking buffer (PBS, 0.1% casein, 0.01% merthiolate). Calibrators were prepared using serial dilutions of recombinant PCSK9 in dilution buffer (PBS, 1.8 m urea, 0.25% bovine serum albumin, 0.5 ml/liter Tween 20, and 0.01% merthiolate). Samples were diluted 1:20 in dilution buffer without bovine serum albumin. Calibrators and samples were incubated for 30 min in a water bath at 46 °C before application (100 μl) in duplicate. The plates were incubated overnight at 37 °C with shaking. After washing, 100 μl of hPCSK9-antibody-horseradish peroxidase-diluted 1:750 was added for 3 h at 37 °C with shaking. Finally, after washing, 100 μl of substrate (SuperSignalTM ELISA Femto Substrate; Pierce) was applied to each well. Chemiluminescence was quantified on a Pherastar luminometer (BMG Labtech). A standard curve was established using a conditioned medium containing recombinant human PCSK9. This standard medium was calibrated by comparison with a full-length secreted and purified PCSK9. The peptide purity and concentration were determined by quantitative amino acid analysis after a 18–24-h hydrolysis in the presence of 5.7 n HCl in vacuo at 110 °C on a Beckman autoanalyzer (model 6300) with a postcolumn ninhydrin detection system coupled to a Varian DS604 data station (performed by Dr. C. Lazure, Clinical Research Institute of Montreal). Plasma PCSK9 concentration was calculated by comparing sample luminescence with the standard luminescence curve.
Primary Hepatocyte Isolation
Hepatocytes were prepared from livers of WT C57BL/6 or pure genetic background Pcsk9−/− (20) mice maintained on a normal chow diet in a 12-h light/12-h dark schedule and used between the ages of 8 and 12 weeks. All experiments performed were in accordance with protocols approved by the bioethics committee of the Clinical Research Institute of Montreal. According to established protocols (21), livers were perfused using a peristaltic pump with 50 ml of pH 7.4 HEPES buffer I (0.83% (w/v) NaCl, 0.05% (w/v) KCl, 0.24% (w/v) HEPES, 0.019% (w/v) EGTA) at a flow rate of 8 ml/min and then switched to 50 ml of pH 7.6 HEPES buffer II (0.39% (w/v) NaCl, 0.05% (w/v) KCl, 2.4% (w/v) HEPES, 0.07% (w/v) CaCl2, 236 units/ml collagenase type IV; Sigma) at a flow rate of 6 ml/min. Digested livers were filtered using 70–100 μm cell strainers (Falcon). Isolated hepatocytes were then sequentially resuspended into 20 ml of Williams' medium E (Invitrogen) containing 10% FBS and 1× antibiotic-antimycotic mixture (Invitrogen) and centrifuged at 1000× rpm for 5 min. This procedure was repeated twice. Highly viable hepatocytes preparations were plated on fibronectin (Sigma) precoated Primaria 6-well plates (Falcon) at an initial density of 1 × 106 cells/well in complete Williams' medium. Fresh serum-free hepatoZYME medium (Invitrogen) was added to hepatocytes 4 h after plating.
Immunoprecipitation of PCSK9
Secreted mouse PCSK9 derived from isolated primary hepatocytes was detected, as described (20). Briefly, 1 × 106 hepatocytes were plated on 6-well plates in serum-free hepatoZYME medium and incubated at 37 °C for 48 h. Using the rabbit IgG TrueBlot kit (eBioscience), 500 μl of conditioned medium (1:4), 500 μl of complete radioimmune precipitation assay buffer (0.5×), 50 μl of anti-rabbit IgG beads, and 3 μl of primary mouse PCSK9 antibody were incubated overnight on a rocking platform at 4 °C. Samples were then centrifuged three times at 7000 × rpm and washed with 1 ml of complete 0.5× radioimmune precipitation assay buffer. Supernatants were completely removed, and pellets were resuspended in 60 μl of Laemmli buffer, and 30 μl of immunoprecipitated PCSK9 was analyzed by SDS-PAGE. Membranes were then incubated with mouse PCSK9 antibodies (1:2500) and probed with TrueBlot horseradish peroxidase anti-rabbit IgG (1:2000; eBioscience). A total of 50 μl of plasma (pooled from three 6-month old male mice) was used as above to analyze the level of circulating PCSK9.
RESULTS
Blockade of Endocytosis in HepG2 Cells Does Not Affect Endogenous LDLR Levels
To examine the contribution of the extracellular pathway in PCSK9-induced LDLR degradation directly, we used two different approaches that aim to prevent endocytosis of the LDLR (Fig. 1). For this, we selected the human hepatic HepG2 cell line, which has been commonly used to study LDLR degradation by PCSK9 because it endogenously expresses both proteins. (i) Upon 6-h incubation of naïve HepG2 cells with Dynasore, a cell-permeable inhibitor of dynamin GTPase activity (22), which virtually eliminated the internalization of diI-LDL (Fig. 1a), no significant change in total LDLR levels was observed by Western blotting (Fig. 1b). (ii) Although siRNA-mediated knockdown of ARH (KDARH) in HepG2 cells resulted in >80% reduction of its protein levels and as expected enriched cell surface LDLR levels (Fig. 1c), it did not affect total LDLR levels (Fig. 1d), in agreement with the reported unchanged levels of LDLR in the liver of Arh−/− mice (17). We conclude that, in HepG2 cells, none of the above treatments that block LDLR endocytosis affected its total protein levels.
FIGURE 1.

Minor contribution of endogenous secreted PCSK9 to LDLR intracellular degradation. a, HepG2 cells were incubated for 4 h with 0.3% v/v of dimethyl sulfoxide (DMSO) (vehicle) or 80 μm Dynasore monohydrate, a cell-permeable inhibitor of dynamin together with 4 μg/ml diI-LDL (red) and analyzed by immunocytochemistry. b, after a 6-h incubation with dimethyl sulfoxide or 80 μm Dynasore, protein extracts were analyzed by Western blotting. c, immunocytochemistry of nonpermeabilized HepG2 cells that were transfected with either a nonsilencing siRNA (siCtl) or an ARH siRNA at 72 h after transfection is shown. Arrows emphasize the increased cell surface LDLR. (d) At 72 h after transfection, protein extracts were analyzed by Western blotting. These data are representative of three to six separate experiments. Scale bars, 20 μm.
Inhibition of the PCSK9-induced LDLR Degradation by EGF-AB
An EGF-AB peptide containing the LDLR binding site to PCSK9 was reported to inhibit the effect of exogenously added PCSK9 on LDLR degradation in either HEK293 or HepG2 cells (18, 23, 24). Although the effect of WT EGF-AB on naïve HepG2 cells was not reported, the inhibitory effect of the gain-of-function EGF-AB H306Y is best seen with exogenous PCSK9, with little effect on naïve HepG2 cells (18). Therefore, we tested the antagonistic effects of various concentrations of EGF-AB on the ability of PCSK9 to induce LDLR degradation when added extracellularly (Fig. 2). The data show that in a dose-dependent manner (0.1–10 μm) EGF-AB significantly reduced exogenous PCSK9 (67–200 nm) activity on LDLR with an EC50 of ∼2 μm after both 4-h and 24-h incubations. However, 4-h incubation with EGF-AB did not affect the cellular incorporation of diI-LDL in the absence of exogenous PCSK9 (Fig. 2a, open squares). In contrast, after a 24-h incubation of naïve HepG2 cells (secreting ∼4 nm PCSK9, as estimated from cells expressing a nonsecretable PCSK9-H226A form; see Fig. 4) with an increasing amount of EGF-AB we observed a ≤20% increase in LDLR activity (Fig. 2b, open squares). In agreement, EGF-AB only slightly affected total LDLR protein levels in naïve HepG2 cells after incubation with as much as 12.5 μm for 24 h (Fig. 2c). We also noted that exogenously added EGF-AB peptide was not internalized by HepG2 cells (Fig. 2c). This excludes a possible interference with the PCSK9 intracellular LDLR degradation pathway. We conclude that inhibition of endogenously secreted PCSK9 by EGF-AB is only seen after a 24-h incubation, suggesting that the dosage and/or incubation time is an important factor for extracellular PCSK9 activity on LDLR degradation.
FIGURE 2.

Extracellular EGF-AB does not affect endogenous PCSK9 effect on LDLR. a and b, HepG2 cells were preincubated for either 4 h (a) or 24 h (b) with 0.01, 0.1, 0.3, 1, 3, and 10 μm EGF-AB peptide together with 0, 67, 135 or 200 nm pure human PCSK9. Then, 5 μg/ml diI-LDL was added, and the incubation continued for another 2 h. Cells were washed and analyzed for fluorescence incorporation. Normalized data are presented. d, Western blot analyses of HepG2 cells incubated for 24 h with 0, 1, 5, and 12.5 μm EGF-AB are shown. These data are representative of three independent experiments.
FIGURE 4.

The gain-of-function mutant PCSK9-D374Y, but not WT PCSK9, decreases cell surface LDLR on adjacent cells. a, immunocytochemistry of cell surface LDLR (blue) in HepG2 transiently overexpressing a bicistronic vector (pIRES) encoding WT PCSK9 and EGFP (green) is shown. b–d, at 24 h after transfection, cells overexpressing a nonsecreted catalytically inactive PCSK9 (H226A), WT, or the gain-of-function mutant (D374Y) were sorted for EGFP and LDLR. Graphs were derived from FACS analyses (supplemental Fig. S1), and values were normalized to those of the H226A-expressing cells. Secreted human PCSK9 levels were measured using an ELISA (b). Error bars represent three separate experiments. Scale bar, 20 μm.
Secreted PCSK9 from WT Hepatocytes Does Not Affect LDLR in PCSK9-deficient Ones in 24-Hour Incubations
In agreement with whole liver data (20, 25), LDLR levels increased by ∼2.5-fold in primary hepatocytes from Pcsk9−/− (knock-out (KO)) versus Pcsk9+/+ (WT) mice (Fig. 3). Thus, we tested the effect of secreted PCSK9 from primary hepatocytes of WT(+/+) mice (48-h spent media) on LDLR levels in hepatocytes of KO(−/−) mice. No significant effect was observed on LDLR levels after a 24-h incubation (Fig. 3 and supplemental Fig. S1). In contrast, incubation with 100 nm purified human PCSK9 resulted in its internalization and a ∼70% reduction in total LDLR. Accordingly, the accumulated PCSK9 in the 48-h spent media of primary hepatocytes of WT mice (supplemental Fig. S1) was unable to influence the levels of LDLR in KO(m−/−) hepatocytes.
FIGURE 3.

Secreted PCSK9 does not reduce LDLR levels in PCSK9 KO hepatocytes. Primary hepatocytes of Pcsk9−/− (−/−) mice were incubated for 24 h in duplicate with either 100 nm PCSK9 or with 48-h conditioned media from PCSK9 KO or WT hepatocytes (from two mice, WT1 and WT2). Note the lower LDLR levels in Pcsk9+/+ (+/+) hepatocytes, the source of WT media, versus (−/−) hepatocytes. Western blot analyses of LDLR, human PCSK9, and β-actin are shown. Note the level of cell-associated human PCSK9 (hPCSK9) after the 100 nm incubation using a human PCSK9 antibody that does not recognize mouse PCSK9 on Western blots (20). Similar data were obtained in two separate experiments.
Local Effect of Overexpressed PCSK9 on Cell Surface LDLR
PCSK9 was shown to specifically degrade the LDLR and two other family members, i.e. VLDLR and apo-ER2, but not angiotensin-converting enzyme 2 (26) nor the high density lipoprotein receptor SR-BI, hepatocyte growth factor receptor HGFR, or transferrin receptor TfR (27). It was also shown previously that overexpression of PCSK9 in a rat hepatic cell line reduces the levels of LDLR mostly in transfected cells (15). Here, a similar observation was also made in hepatic human HepG2 cells, transiently transfected with a bicistronic vector (pIRES-EGFP) that encodes separately WT hPCSK9 and EGFP proteins. At 24 h after transfection, immunocytochemistry revealed that cell surface LDLR protein levels (Fig. 4a, blue) were appreciably decreased in PCSK9-positive cells (Fig. 4a, green) relative to nontransfected adjacent cells. In contrast, HepG2 cells overexpressing the gain-of-function mutant D374Y resulted in a decrease of LDLR levels in both transfected and neighboring cells (data not shown). To quantify these observations, the cells were transfected with a nonsecreted catalytically inactive PCSK9 mutant (H226A) (5), WT, or the D374Y variant (28). At 24 h after transfection, cells were sorted for EGFP and LDLR (supplemental Fig. S2). Compared with PCSK9-H226A-expressing cells, the number of LDLR-negative cells increased by 4- and 6-fold in cells overexpressing WT or D374Y PCSK9, respectively (GFP+, Fig. 4c). Although the LDLR levels of adjacent, nontransfected cells were not affected by WT PCSK9, the number of LDLR-negative cells increased ∼3-fold by the D374Y mutant (GFP−, Fig. 4d). Secreted PCSK9 from the ∼20% transfected cells (quantified by an ELISA (29) were ∼3-fold those of endogenous levels (H226A media; Fig. 4b).
Compared with HepG2 cells, HEK293 cells exhibit a higher transfection efficiency (∼75%) leading to the secretion of up to 1 μg/ml per 24 h (20 nm) of PCSK9. Similar to HepG2 cells, overexpression of WT PCSK9 in HEK293 cells resulted in a ∼50% reduction of cell surface LDLR (GFP+; supplemental Fig. S3). As evidenced by FACS analysis, mixing of these cells with naïve ones, at various ratios, did not modify LDLR levels in the latter (GFP−; supplemental Fig. S3). Thus, after a 24-h incubation, in both HepG2 and HEK293 cells, overexpression of WT PCSK9 mainly reduces local cell surface LDLR levels, with little influence on adjacent cells. This suggests that at 24 h after transfection, overexpressed WT PCSK9 efficiently down-regulates local LDLR levels, either intracellularly or by an autocrine fashion.
Disruption of Intracellular Trafficking Increases Endogenous LDLR Levels Only in PCSK9-expressing Cells
Clathrin light chains CLCa and CLCb are not required for clathrin-mediated endocytosis but are critical for clathrin-mediated trafficking between the trans-Golgi network and the endosomal system (19). Accordingly, KD of both isoforms in HepG2 cells (KDCLCs) resulted in the clustering of the cation-independent mannose 6-phosphate receptor, characteristic of disruption of the intracellular Golgi-lysosomal pathway (Fig. 5a, see arrows), as reported in other cell lines (19). At 72 h after transfection, HepG2 and HEK293 cell lysates were analyzed by Western blotting. The level of endogenous mature N- and O-glycosylated ∼160-kDa LDLR protein (30) was increased by ∼2.9-fold in HepG2 cells, endogenously expressing PCSK9, but not in HEK293 cells (PCSK9-negative) (5) (Fig. 5, b and c). As controls, we also show that the levels of the transferrin receptor TfR (Fig. 5b) or PCSK9 (data not shown) are not affected by KDCLCs. Analysis of HepG2 cells by immunocytochemistry revealed that the levels of both cell surface (Fig. 5d) and intracellular (supplemental Fig. S4a) LDLR were heavily increased. Although KDCLCs resulted in higher LDLR levels, it did not significantly affect its overall subcellular localization (data not shown). Furthermore, upon KDCLCs, LDLR endocytosis is functional as we observed an enhanced diI-LDL uptake (supplemental Fig. S4b), which correlates with the observed increased cell surface LDLR (Fig. 5d). The observed effect on LDLR seems to be relatively rapid because at 72 h after KDCLCs we can already see an increased rate of de novo biosynthesized LDLR in a 4-h pulse with [35S]Met/Cys (7), whereas nontarget control siRNAs (siCtl) are ineffective (supplemental Fig. S5).
FIGURE 5.

KD of CLCs increases endogenous LDLR levels only in PCSK9-expressing cells. HepG2 or HEK293 cells were transiently transfected with either a nonsilencing siRNA (siCtl) or with siRNAs against both CLC a and b isoforms (siCLCa+b). a, immunocytochemistry analyses confirmed that KDCLCs (green) results in the clustering of the cation-independent mannose 6-phosphate receptor, characteristic of disruption of the intracellular Golgi-lysosomal pathway (blue; see arrows). b and c, at 72 h after transfection, HepG2 and HEK293 cells were analyzed by Western blotting. d, immunocytochemistry under nonpermeabilizing conditions of cell surface LDLR (green) upon KDCLCs (siCLCa+b) compared with nonsilencing siRNA transfected cells (siCtl) is shown. e, at 48 h after transfection, addition of 40 nm exogenous PCSK9 enhances the degradation of LDLR irrespective of KDCLCs. (f) Effect of KDCLCs on LDLR protein levels in HepG2 cells with (shNT) or without (shPCSK9*) endogenous PCSK9 is shown. The 160 kDa band representing mature LDLR was quantified and normalized with those of actin. These data are representative of 3–6 independent experiments. Scale bars, 20 μm.
It was reported that KDCLCs can reduce the levels of endosomal/lysosomal degrading enzymes by preventing their normal trafficking and thus delaying their zymogen activation (19). Accordingly, KDCLCs may affect both intra- and extracellular pathways that presumably depend on similar lysosomal degrading enzymes. However, incubating HepG2 cells with 40 nm purified PCSK9 added 48 h after KDCLCs and then allowing the cells to grow for another 24 h did not affect its ability to degrade the LDLR (Fig. 5e). Thus, KDCLCs does not affect the extracellular LDLR degradation pathway induced by PCSK9.
To substantiate further the critical importance of PCSK9 in the regulation of LDLR levels upon KDCLCs, we generated stable pools of HepG2 cells expressing either a nontargeting or a PCSK9-specific shRNA. Quantitative PCR analyses revealed that one of the shPCSK9-expressing cellular pools (shPCSK9-2b) exhibits ∼90% reduction in PCSK9 mRNA (data not shown). Western blot analysis of the latter pool shows a ∼60% reduction in secreted PCSK9 protein levels, concomitant with a 1.6-fold increase in LDLR levels but without any effect on the level of the control TfR (supplemental Fig. S6). To enrich cells that express the highest level of shPCSK9, we further sorted the shPCSK9-expressing cells by FACS and selected for those expressing the highest level of LDLR. Such an LDLR-enriched shPCSK9* cellular pool exhibits virtually no PCSK9 (Fig. 5f). The increased LDLR levels observed upon KDCLCs was PCSK9-dependent because it is no longer seen in shPCSK9* (Fig. 5f). We conclude that in HepG2 cells, a relatively rapid, endogenous intracellular pathway exists for PCSK9-enhanced LDLR degradation.
Additive Effects of KDCLCs and LPDS Treatment on LDLR Levels
Exposure of cells to LPDS media, which are poor in cholesterol, results in an up-regulation of both LDLR and PCSK9 mRNAs via a common SREBP-2 pathway (31). Accordingly, at 48 h after transfection of HepG2 cells with either control siRNA (−) or KDCLCs (+), the cells were incubated overnight with complete media (FBS) or LPDS. Western blot analyses revealed that LPDS media resulted in a ∼2- and ∼1.5-fold up-regulation of the endogenous protein levels of LDLR and PCSK9, respectively in the presence of control siRNA (−; Fig. 6). However, only LDLR levels were further increased by another ∼2-fold upon KDCLCs in LPDS versus FBS media, but not those of PCSK9 or TfR (+; Fig. 6). We conclude that incubation of cells in LPDS media coupled to a blockade of the intracellular trafficking by KDCLCs results in an additive ∼4-fold up-regulation of LDLR levels.
FIGURE 6.
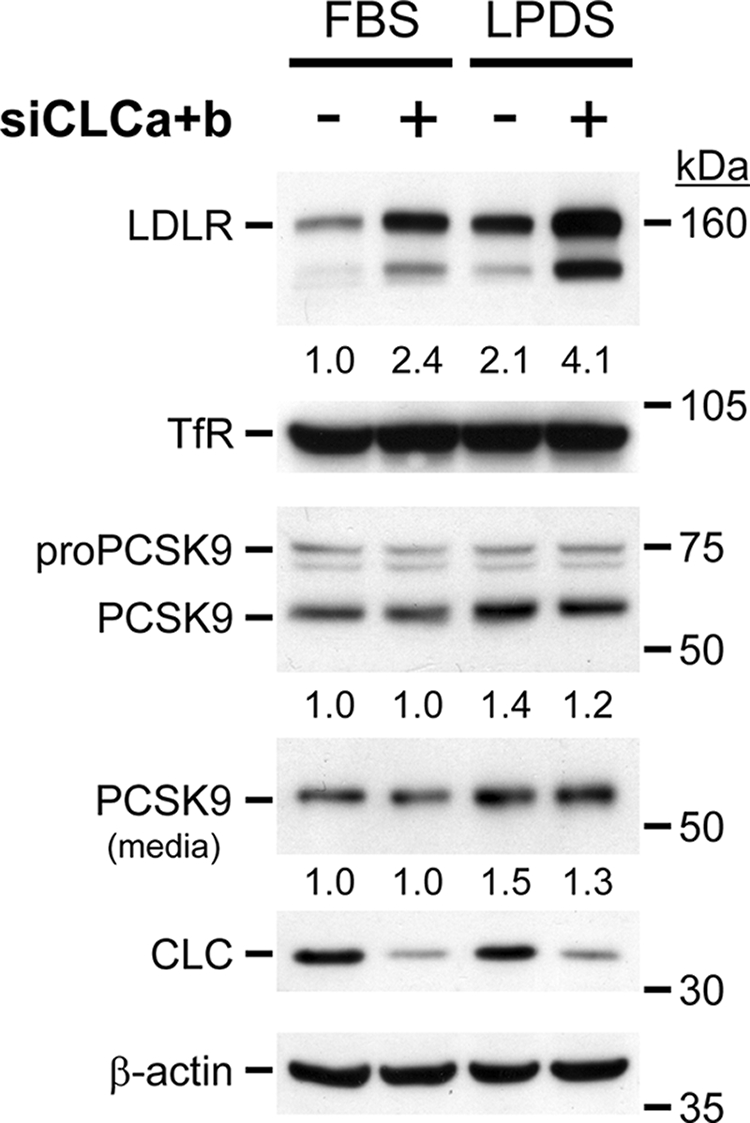
Synergic effect of KDCLCs with LPDS treatment. HepG2 cells were transiently transfected with either a nonsilencing siRNA or with siRNAs against both CLC a and b isoforms (siCLCa+b). At 48 h after transfection, cells were washed twice with DMEM and incubated with either complete (10% FBS) or with 5% LPDS medium. After a 24-h incubation, cells were washed and incubated for 6 h in DMEM, and protein extracts and media were analyzed by Western blotting. These data are representative of three independent experiments.
DISCUSSION
The cell surface internalization of PCSK9 depends primarily on the presence of LDLR and the adaptor protein ARH that binds the cytosolic tail of the LDLR (16, 32, 33). In addition, we showed that LDLR and PCSK9 interact early in the secretory pathway (10) and questioned whether some PCSK9·LDLR complex or PCSK9 alone can be targeted directly from the trans-Golgi network to LE/L, without cycling through the cell surface. Aside from the extracellular pathway, the existence of an intracellular one is supported by the ability of PCSK9 to degrade the LDLR in vivo in the absence of ARH (9) and on the capability of PCSK9-Lamp1 chimeras, which directly traffic to LE/L, to enhance the degradation of the LDLR, VLDLR and apo-ER2 efficiently (26). In liver, ARH seems to be critical for the endocytosis of the extracellular PCSK9·LDLR complex (16) but does not seem to play an important role in regulating total levels of endogenous LDLR (Fig. 1d) (17, 34).
Endogenous PCSK9 secreted from primary hepatocytes of WT mice has no effect on those derived from Pcsk9−/− mice, whereas incubation with 100 nm PCSK9 clearly decreased LDLR levels (Fig. 3). Thus, after 24-h incubations, only high, nonphysiological concentrations of WT PCSK9 can reduce LDLR levels in primary hepatocytes (Fig. 3) and in HepG2 cells (Figs. 1 and 5). Interestingly, we observed that in the plasma of Ldlr−/− mice the levels of circulating PCSK9 and its Furin-cleaved form (20) are at least 10-fold higher than in WT mice (supplemental Fig. S1b), and yet it was reported that sharing the blood circulation (parabiosis) of Ldlr−/− mice with that of WT mice did not affect the levels of liver LDLR in the latter nor plasma LDL-cholesterol (16). Accordingly, the physiological role of circulating WT PCSK9 and its Furin-truncated form remains undefined. Although hepatocyte-specific transgenic mice weakly overexpressing PCSK9 (3-fold) exhibit no significant change in circulating LDL-cholesterol, a ∼30-fold overexpression increased it by ∼5-fold (20). Data from transgenic mice expressing very high levels of human PCSK9 in mouse kidney (35) or liver (16), or continuous infusions of recipient WT mice with recombinant PCSK9 (36) indicated that >100 nm amounts of circulating PCSK9 are required to affect liver LDLR protein levels significantly, without affecting extrahepatic LDLR. In that context, we recently showed that annexin A2 inhibits the effect of extracellular PCSK9 on LDLR, and in view of its absence from hepatocytes in vivo, it may exert its inhibitory effect in extrahepatic tissues (14). The available compiled data suggest that only supraphysiological levels of circulating PCSK9 have an impact on liver LDLR. Several ELISAs evaluated the level of circulating human PCSK9 to be ∼2–4 nm (16, 29, 37). However, because the local concentration of PCSK9 secreted from hepatocytes into the extracellular microenvironment is expected to be much higher than in plasma, PCSK9 may well have an autocrine/paracrine effect in vivo.
Our data revealed that a 4-h incubation of HepG2 cells with an EGF-AB peptide readily inhibits the LDLR-degrading activity of exogenously added PCSK9 but had little effect on endogenous secreted PCSK9 (Fig. 2a). Interestingly, it took at least 24 h to detect an effect of EGF-AB on LDLR levels and activity (Fig. 2, b and c). Thus, it is possible that the extracellular PCSK9 effect is a relatively slow process and may require a long incubation time and/or a critical threshold concentration. This may be related to the lower binding affinity of PCSK9 to LDLR at neutral versus acidic pH values (11).
A very recent report revealed that a monoclonal antibody (mAb) that neutralizes the interaction of PCSK9 and LDLR is able to reduce the extracellular activity of PCSK9 both in naïve HepG2 cells and in vivo in mouse and cynomolgus monkey (38). Similar to our data in HepG2 cells using EGF-AB as a neutralizing agent (Fig. 2), the mAb effect was only reported for times ≥24-h incubation. The latter seems to be due to a conformational change as the mAb does not completely mask the PCSK9-LDLR interaction surface (38). Because no evidence was provided for antibody-antigen cellular internalization, we presume that the mAb blocks the extracellular PCSK9 effect on LDLR.
Here, we showed that overexpression of PCSK9 or its D374Y mutant in HepG2 cells effectively reduces cell surface LDLR levels within 24 h. In contrast, only the D374Y mutant affects the levels of LDLR in nontransfected neighboring cells (Fig. 4), in agreement with its reported enhanced LDLR-binding affinity at neutral pH (11, 18, 23). Concentrations of secreted PCSK9, up to 12 nm (HepG2 cells; ∼3× endogenous levels; Fig. 4) or 20 nm (HEK293 cells; supplemental Fig. S3), were unable to enhance the degradation of the LDLR on neighboring cells in 24 h. This suggests that within 24 h and at levels close to endogenous ones, PCSK9 exerts its function mostly intracellularly and/or in an autocrine fashion.
Our previous work demonstrated the importance of clathrin heavy chain for the PCSK9-dependent degradation of the LDLR (10). However, because clathrin-mediated membrane trafficking is needed for both direct sorting from the trans-Golgi network to LE/L and internalization of cargo by endocytosis, the route taken by the PCSK9·LDLR complex remained unresolved. In the present work, our data support the presence of a relatively rapid intracellular route for the PCSK9-induced degradation of LDLR at endogenous levels because blockade of the trans-Golgi network to lysosome vesicular trafficking by KDCLCs clearly up-regulated LDLR levels already after 4 h (supplemental Fig. S5), without affecting endocytosis (Fig. 5 and supplemental Fig. S4b). Even though the KDCLCs slows down the activation of procathepsin D (19), a lysosomal hydrolase that is targeted to LE/L, our results also showed that after KDCLCs, the cognate hydrolases retained their capacity to degrade the LDLR (Fig. 5e). In addition, we also demonstrated that the effect of KDCLCs is PCSK9-dependent because LDLR levels are no longer affected when PCSK9 is absent (Fig. 5f). Although the level of LDLR is increased, its intracellular and surface localization remained comparable in cells lacking CLCs (supplemental Fig. S4a). Similarly, intracellular and secreted levels as well as the localization of PCSK9 were not significantly altered by KDCLCs (data not shown). Thus, although the underlying mechanism of CLCs effect is presently not fully unraveled, it may rely on another factor that is essential for the direct intracellular trafficking of PCSK9 to the LDLR degradation compartment(s).
PCSK9 has emerged as a viable attractive target to reduce coronary heart disease through control of dyslipidemias (39). The choice of the best approach to diminish the effect of PCSK9 on LDLR degradation in vivo effectively may be influenced by the relative contribution of both cellular pathways under physiological and pathological conditions. The combination of a statin with an agent that blocks either the extracellular pathway (38) or the intracellular one (present work) or both would constitute a powerful novel approach to control hypercholesterolemia. With the rapid pace of discoveries in the field, it is hoped that in a few years lead molecules reducing the level and/or activity of PCSK9 will be uncovered and that these will find their way in clinical trials to assess their potency and safety.
Supplementary Material
Acknowledgments
We thank R. Essalmani, S. Benjannet, M. Dupuis, E. Massicotte, D. Meyers, and G. Duke for excellent technical advice and help. We also thank J. Davignon, G. Dubuc, and M. Tremblay for the ELISAs and V. Franklin and Y. Marcel for advice on primary hepatocytes. Many thanks to all members of the Seidah laboratory for helpful discussions and to Brigitte Mary for editorial assistance.
This work was supported in part by Canadian Institutes of Health Research Grants MOP-36496 and CTP-82946 (to N. G. S.) and MOP-15396 (to P. S. M.), a Strauss Foundation grant (to N. G. S.), a Bristol-Myers Squibb research grant, and Canada Chair 201652 (to N. G. S.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S6.
- LDL
- low density lipoprotein
- ARH
- autosomal recessive hypercholesterolemia
- CLC
- clathrin light chain
- diI-LDL
- LDL coupled with 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate
- DMEM
- Dulbecco's modified Eagle's medium
- EGF-A
- epidermal growth factor domain A
- EGF-AB
- epidermal growth factor domain AB
- EGFP
- enhanced green fluorescent protein
- ELISA
- enzyme-linked immunosorbent assay
- FACS
- fluorescence-activated cell sorting
- FBS
- fetal bovine serum
- KD
- knockdown
- KO
- knock-out
- LDLR
- low density lipoprotein receptor
- LE/L
- late endosomes/lyososomes
- LPDS
- lipoprotein-deficient serum
- mAb
- monoclonal antibody
- PBS
- phosphate-buffered saline
- PCSK9
- proprotein convertases subtilisin kexin 9
- shRNA
- small hairpin RNA
- siRNA
- small interfering RNA
- TfR
- transferrin receptor
- WT
- wild type.
REFERENCES
- 1.Lloyd-Jones D., Adams R., Carnethon M., De Simone G., Ferguson T. B., Flegal K., Ford E., Furie K., Go A., Greenlund K., Haase N., Hailpern S., Ho M., Howard V., Kissela B., Kittner S., Lackland D., Lisabeth L., Marelli A., McDermott M., Meigs J., Mozaffarian D., Nichol G., O'Donnell C., Roger V., Rosamond W., Sacco R., Sorlie P., Stafford R., Steinberger J., Thom T., Wasserthiel-Smoller S., Wong N., Wylie-Rosett J., Hong Y. (2009) Circulation 119, e21–e181 [DOI] [PubMed] [Google Scholar]
- 2.Brown M. S., Goldstein J. L. (1986) Science 232, 34–47 [DOI] [PubMed] [Google Scholar]
- 3.Varret M., Abifadel M., Rabès J. P., Boileau C. (2008) Clin. Genet. 73, 1–13 [DOI] [PubMed] [Google Scholar]
- 4.Seidah N. G., Prat A. (2007) J. Mol. Med. 85, 685–696 [DOI] [PubMed] [Google Scholar]
- 5.Seidah N. G., Benjannet S., Wickham L., Marcinkiewicz J., Jasmin S. B., Stifani S., Basak A., Prat A., Chretien M. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 928–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abifadel M., Varret M., Rabès J. P., Allard D., Ouguerram K., Devillers M., Cruaud C., Benjannet S., Wickham L., Erlich D., Derré A., Villéger L., Farnier M., Beucler I., Bruckert E., Chambaz J., Chanu B., Lecerf J. M., Luc G., Moulin P., Weissenbach J., Prat A., Krempf M., Junien C., Seidah N. G., Boileau C. (2003) Nat. Genet. 34, 154–156 [DOI] [PubMed] [Google Scholar]
- 7.Benjannet S., Rhainds D., Essalmani R., Mayne J., Wickham L., Jin W., Asselin M. C., Hamelin J., Varret M., Allard D., Trillard M., Abifadel M., Tebon A., Attie A. D., Rader D. J., Boileau C., Brissette L., Chrétien M., Prat A., Seidah N. G. (2004) J. Biol. Chem. 279, 48865–48875 [DOI] [PubMed] [Google Scholar]
- 8.Maxwell K. N., Fisher E. A., Breslow J. L. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 2069–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park S. W., Moon Y. A., Horton J. D. (2004) J. Biol. Chem. 279, 50630–50638 [DOI] [PubMed] [Google Scholar]
- 10.Nassoury N., Blasiole D. A., Tebon O. A., Benjannet S., Hamelin J., Poupon V., McPherson P. S., Attie A. D., Prat A., Seidah N. G. (2007) Traffic 8, 718–732 [DOI] [PubMed] [Google Scholar]
- 11.Cunningham D., Danley D. E., Geoghegan K. F., Griffor M. C., Hawkins J. L., Subashi T. A., Varghese A. H., Ammirati M. J., Culp J. S., Hoth L. R., Mansour M. N., McGrath K. M., Seddon A. P., Shenolikar S., Stutzman-Engwall K. J., Warren L. C., Xia D., Qiu X. (2007) Nat. Struct. Mol. Biol. 14, 413–419 [DOI] [PubMed] [Google Scholar]
- 12.Kwon H. J., Lagace T. A., McNutt M. C., Horton J. D., Deisenhofer J. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 1820–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang D. W., Garuti R., Tang W. J., Cohen J. C., Hobbs H. H. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 13045–13050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mayer G., Poirier S., Seidah N. G. (2008) J. Biol. Chem. 283, 31791–31801 [DOI] [PubMed] [Google Scholar]
- 15.Maxwell K. N., Breslow J. L. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 7100–7105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lagace T. A., Curtis D. E., Garuti R., McNutt M. C., Park S. W., Prather H. B., Anderson N. N., Ho Y. K., Hammer R. E., Horton J. D. (2006) J. Clin. Invest. 116, 2995–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones C., Hammer R. E., Li W. P., Cohen J. C., Hobbs H. H., Herz J. (2003) J. Biol. Chem. 278, 29024–29030 [DOI] [PubMed] [Google Scholar]
- 18.McNutt M. C., Kwon H. J., Chen C., Chen J. R., Horton J. D., Lagace T. A. (2009) J. Biol. Chem. 284, 10561–10570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poupon V., Girard M., Legendre-Guillemin V., Thomas S., Bourbonniere L., Philie J., Bright N. A., McPherson P. S. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 168–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zaid A., Roubtsova A., Essalmani R., Marcinkiewicz J., Chamberland A., Hamelin J., Tremblay M., Jacques H., Jin W., Davignon J., Seidah N. G., Prat A. (2008) Hepatology 48, 646–654 [DOI] [PubMed] [Google Scholar]
- 21.Wang M. D., Franklin V., Sundaram M., Kiss R. S., Ho K., Gallant M., Marcel Y. L. (2007) J. Biol. Chem. 282, 22525–22533 [DOI] [PubMed] [Google Scholar]
- 22.Macia E., Ehrlich M., Massol R., Boucrot E., Brunner C., Kirchhausen T. (2006) Dev. Cell 10, 839–850 [DOI] [PubMed] [Google Scholar]
- 23.Bottomley M. J., Cirillo A., Orsatti L., Ruggeri L., Fisher T. S., Santoro J. C., Cummings R. T., Cubbon R. M., Lo S. P., Calzetta A., Noto A., Baysarowich J., Mattu M., Talamo F., De Francesco R., Sparrow C. P., Sitlani A., Carfi A. (2009) J. Biol. Chem. 284, 1313–1323 [DOI] [PubMed] [Google Scholar]
- 24.Shan L., Pang L., Zhang R., Murgolo N. J., Lan H., Hedrick J. A. (2008) Biochem. Biophys. Res. Commun. 375, 69–73 [DOI] [PubMed] [Google Scholar]
- 25.Rashid S., Curtis D. E., Garuti R., Anderson N. N., Bashmakov Y., Ho Y. K., Hammer R. E., Moon Y. A., Horton J. D. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 5374–5379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poirier S., Mayer G., Benjannet S., Bergeron E., Marcinkiewicz J., Nassoury N., Mayer H., Nimpf J., Prat A., Seidah N. G. (2008) J. Biol. Chem. 283, 2363–2372 [DOI] [PubMed] [Google Scholar]
- 27.Labonté P., Begley S., Guévin C., Asselin M. C., Nassoury N., Mayer G., Prat A., Seidah N. G. (2009) Hepatology 50, 17–24 [DOI] [PubMed] [Google Scholar]
- 28.Timms K. M., Wagner S., Samuels M. E., Forbey K., Goldfine H., Jammulapati S., Skolnick M. H., Hopkins P. N., Hunt S. C., Shattuck D. M. (2004) Hum. Genet. 114, 349–353 [DOI] [PubMed] [Google Scholar]
- 29.Dubuc G., Tremblay M., Pare G., Jacques H., Hamelin J., Benjannet S., Boulet L., Genest J., Bernier L., Seidah N. G., Davignon J. (2009) J. Lipid Res., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cummings R. D., Kornfeld S., Schneider W. J., Hobgood K. K., Tolleshaug H., Brown M. S., Goldstein J. L. (1983) J. Biol. Chem. 258, 15261–15273 [PubMed] [Google Scholar]
- 31.Dubuc G., Chamberland A., Wassef H., Davignon J., Seidah N. G., Bernier L., Prat A. (2004) Arterioscler. Thromb. Vasc. Biol. 24, 1454–1459 [DOI] [PubMed] [Google Scholar]
- 32.Cameron J., Holla Ø. L., Ranheim T., Kulseth M. A., Berge K. E., Leren T. P. (2006) Hum. Mol. Genet. 15, 1551–1558 [DOI] [PubMed] [Google Scholar]
- 33.Qian Y. W., Schmidt R. J., Zhang Y., Chu S., Lin A., Wang H., Wang X., Beyer T. P., Bensch W. R., Li W., Ehsani M. E., Lu D., Konrad R. J., Eacho P. I., Moller D. E., Karathanasis S. K., Cao G. (2007) J. Lipid Res. 48, 1488–1498 [DOI] [PubMed] [Google Scholar]
- 34.Harada-Shiba M., Takagi A., Marutsuka K., Moriguchi S., Yagyu H., Ishibashi S., Asada Y., Yokoyama S. (2004) Circ. Res. 95, 945–952 [DOI] [PubMed] [Google Scholar]
- 35.Luo Y., Warren L., Xia D., Jensen H., Sand T., Petras S., Qin W., Miller K. S., Hawkins J. (2009) J. Lipid Res. 50, 1581–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grefhorst A., McNutt M. C., Lagace T. A., Horton J. D. (2008) J. Lipid Res. 49, 1303–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alborn W. E., Cao G., Careskey H. E., Qian Y. W., Subramaniam D. R., Davies J., Conner E. M., Konrad R. J. (2007) Clin. Chem. 53, 1814–1819 [DOI] [PubMed] [Google Scholar]
- 38.Chan J. C., Piper D. E., Cao Q., Liu D., King C., Wang W., Tang J., Liu Q., Higbee J., Xia Z., Di Y., Shetterly S., Arimura Z., Salomonis H., Romanow W. G., Thibault S. T., Zhang R., Cao P., Yang X. P., Yu T., Lu M., Retter M. W., Kwon G., Henne K., Pan O., Tsai M. M., Fuchslocher B., Yang E., Zhou L., Lee K. J., Daris M., Sheng J., Wang Y., Shen W. D., Yeh W. C., Emery M., Walker N. P., Shan B., Schwarz M., Jackson S. M. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 9820–9825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seidah N. G. (2009) Expert. Opin. Ther. Targets. 13, 19–28 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.