

Abstract
Schistosomiasis is a parasitic disease affecting over 200 million people currently treated with one drug, praziquantel. A possible drug target is the seleno-protein thioredoxin-glutathione reductase (TGR), a key enzyme in the pathway of the parasite for detoxification of reactive oxygen species. The enzyme is a unique fusion of a glutaredoxin domain with a thioredoxin reductase domain, which contains a selenocysteine (Sec) as the penultimate amino acid. Auranofin (AF), a gold-containing compound already in clinical use as an anti-arthritic drug, has been shown to inhibit TGR and to substantially reduce worm burden in mice. Using x-ray crystallography we solved (at 2.5 Å resolution) the structure of wild type TGR incubated with AF. The electron density maps show that the actual inhibitor is gold, released from AF. Gold is bound at three different sites not directly involving the C-terminal Sec residue; however, because the C terminus in the electron density maps is disordered, we cannot exclude the possibility that gold may also bind to Sec. To investigate the possible role of Sec in the inactivation kinetics, we tested the effect of AF on a model enzyme of the same superfamily, i.e. the naturally Sec-lacking glutathione reductase, and on truncated TGR. We demonstrate that the role of selenium in the onset of inhibition by AF is catalytic and can be mimicked by an external source of selenium (benzeneselenol). Therefore, we propose that Sec mediates the transfer of gold from its ligands in AF to the redox-active Cys couples of TGR.
Schistosomiasis is one of the most important human parasitic infections in the world, affecting over 200 million people in developing countries with 280,000 deaths/year in sub-Saharan Africa alone (1). The therapy for schistosomiasis is based on a single drug, praziquantel, that is administered to millions of people yearly (2, 3). A reliable alternative to praziquantel does not exist at the moment. Oxamniquine, the only other drug commercially available, is expensive and is active against only one of the three schistosome species capable of infecting humans, namely Schistosoma mansoni. Artemisinins, although safe, are active only against the immature stages of the parasites. A treatment strategy that relies on just a single drug is at risk given the high probability that drug-resistant parasites will emerge (4).
Recently, a possible solution to this urgent problem has been proposed, i.e. targeting the antioxidant pathway of the parasite. In platyhelminths, like Schistosoma and Echinococcus, thiol redox homeostasis is completely dependent on the enzyme thioredoxin-glutathione reductase (TGR),4 which directs the NADPH reducing equivalents to both GSH and thioredoxin (4–6). On the other hand, in humans and other vertebrates the same function is fulfilled by two distinct enzymes that initiate two parallel pathways, namely thioredoxin reductase (TR) and glutathione reductase (GR).
TGR is a natural chimeric flavo-enzyme whose structure results from the fusion of a TR domain with a glutaredoxin domain (7–9). We recently solved the crystal structure of TGR from S. mansoni (10). The redox activity of the enzyme relies on at least three redox sites communicating with one another: (i) the FAD site, composed by the isoalloxazine ring of the flavin and the Cys154–Cys159 couple (characteristic of all the enzymes of the TR/GR family); (ii) the C terminus, constituted by the Gly-Cys-Sec-Gly sequence shared with the majority of TRs but not with GRs; and (iii) the glutaredoxin redox site represented by Cys28–Cys31 at the N-terminal portion of the protein.
The presence of this peculiar enzyme in schistosomes was exploited in the search of new schistosomicidal drugs (11, 12). The significance of S. mansoni (Sm)TGR as a putative drug target was first demonstrated using an RNA interference approach, which killed 90% of treated parasites in vitro. Moreover, it was demonstrated that SmTGR activity is inhibited by two schistosomicidal drugs used in the past to fight the infection, antimonyl potassium tartrate and oltipraz, suggesting that the enzyme is the main target of these compounds (6). Based on these promising results, a quantitative high throughput screening of ∼71000 compounds was carried out to isolate new leads targeting the S. mansoni redox pathway (13).
A potentially interesting inhibitor of TGR is auranofin (AF; Au(I); 3,4,5-triacetyloxy-6-(acetyloxymethyl) oxane-2-thiolate; triethyl phosphanium; C20H34O9S-gold-P(C2H5)3; see the formula in Scheme 1). In vitro, 10 μm AF causes unpairing of male and female worms after 1 h and results in 100% mortality after 9 h of exposure (6). In the same study it was found that TR and GR activities of TGR in worm homogenates were nearly 100% inhibited after 1 h and that the GSH:GSSG ratio decreased from 18:1 in control worms to 2.6:1 (85% decrease) after 6 h of treatment. Larval, juvenile, and adult stages of the parasite are killed by 5 μm AF in 24 h, which acquires significance given that this concentration of the drug is well tolerated by mammalian cells. Indeed, in preliminary experiments, AF administered to infected mice at dosages well tolerated by the host killed 60% of adult schistosomes. This reduction of worm burden would lead to a significant decrease in the pathology and morbidity associated with schistosomiasis (6). Moreover, it has recently been demonstrated that AF also inhibits the TGRs from the related parasites Echinococcus granulosus (14) and Taenia crassiceps (15).
SCHEME 1.

Possible reaction path for AF and SmTGR. One molecule of AF reacts with wild type reduced SmTGR (species 1) to yield a transient intermediate bearing an Au(I) coordinated with Sec597 and triethylphosphine (species 2); we hypothesize that acetoxythioglucose is released first (see text). The intermediate species releases triethylphosphine to form a Sec-gold-Cys complex with any of the available cysteinyl residues; three reaction products are possible (species 3, 4, and 6). The chemical species in which the C-terminal arm is cross-linked via the gold ion to a distant Cys residue (species 4 and 6) may rearrange to yield the more stable short distance Cys-gold-Cys complexes (species 5 and 7). When equilibrium is reached, a mixture of species 3, 5, and 7 is expected to be present, dynamically interchanging via the less stable intermediates 4 and 6. Additional molecules of AF may be introduced, leading to SmTGR molecules bearing two or three gold ions. The gold-binding site 3, in which the metal ion is not coordinated by Cys or Sec, is not considered in the scheme.
Compared with other newly identified leads (11), AF has an important advantage as a schistosomicidal drug, because it has been in clinical use to cure rheumatoid arthritis for 25 years and thus presents a well known and quite safe toxicity profile (16, 17). Finding new uses for clinically established drugs represents a clever strategy of drug discovery, considering the drastic reduction of time and cost relative to developing novel drugs (18). These considerations are of extreme importance for neglected diseases, such as schistosomiasis, which are not of primary interest to the pharmaceutical industry.
AF has been demonstrated to be a nanomolar inhibitor of Sec-containing enzymes such as TR and TGR (6), whereas 1000-fold higher concentrations appear to be required to inhibit GR, which lacks the C-terminal Sec (19). These observations suggest that Sec is either the binding site of AF or is essential in displacing the gold from its ligands given that its nucleophilic power is greater than sulfur. Other gold(I) compounds are known to irreversibly inhibit human glutathione reductase by the formation of a covalent adduct between catalytic cysteines and the metal (Cys-gold-Cys); a similar mechanism for AF has been previously hypothesized but not proven. It was suggested that AF might lose both the thioglucose and the triethylphosphine ligands by sequential exchange reactions with cellular thiols, leading to final gold-protein thiol adducts similar to those produced with gold compounds in GR (20).
Using x-ray crystallography, we hereby demonstrate for the first time the formation of Cys-gold-Cys adducts in two different sites of the three-dimensional structure of wild type SmTGR incubated with AF. Unfortunately we cannot determine the exact position of the C terminus in any of the solved structures, and therefore we do not know whether it is bound to AF derivatives or gold. We have also determined the effect of AF on the activity of yeast GR (a protein lacking Sec) and on a Sec-lacking truncated form of SmTGR. Interestingly we found that the addition of benzeneselenol increases considerably the velocity of inactivation of GR by AF. Taken together, kinetic and structural data allow us to assign a catalytic role to the C-terminal Sec, suggesting a possible mechanism whereby AF inhibits TGRs and TRs.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of SmTGR Forms
The truncated SmTGR lacking the last two amino acids, i.e. Sec597 and Gly598, and the Sec-containing form of SmTGR were prepared as described previously (6, 10).
Crystallization
9 μm wild type SmTGR, reduced with a saturating amount of NADPH (300 μm) was stirred overnight in 50 mm Tris, pH 7.4, 100 mm NaCl at 4 °C in presence of 100 μm auranofin. AF was kindly provided by Dr. Frank Shaw (Normal, IL). The mixture was then dialyzed (1:27000) against 20 mm Tris, pH 7.4, 100 mm NaCl and reconcentrated to 5 mg/ml before crystallization trials were set up. The protein solution was mixed with an equal amount of the reservoir solution containing 0.1 m Hepes at pH 7.0, 20% polyethylene glycol 3350, 0.2 m potassium iodide, and 5 mm GSH. Crystals grew in 3–4 weeks to 0.4 × 0.2 × 0.2 mm3 and were cryoprotected with the same reservoir solution plus 30% polyethylene glycol 200.
X-ray Data Collection
The data were collected at 100 K. The best data set has been collected as 1.0° oscillation frames using the MAR CCD detector on the BL14-1 Beamline at BESSY (Berlin, Germany) at a wavelength of 0.98 Å. Data analysis performed with HKL2000 (21) indicated that the crystals belongs to the C2 space group with the following unit cell dimensions: a = 147.51 Å; b = 102.16 Å; c = 60.57 Å; and β = 114.51°. The data were scaled and are 99.7% complete at 2.55 Å resolution, with an Rmerge of 12% and a χ2 of 1.2. The crystal contains 1 subunit/asymmetric unit, with a VM of 3.2 Å3 Da−1 and a solvent content of 61.3%.
Structure Refinement and Analysis
The structure was solved by molecular replacement using the program PHASER (22) and the structure of truncated SmTGR (Protein Data Bank code 2v6o) as the search model. Refinement of the atomic coordinates and displacement parameters was carried out by means of Refmac5 (23). Model building was performed using the program package COOT (24). The structure of the complex was refined to 2.55 Å resolution with an overall temperature factor of 35.4 Å2 (for other data collection and refinement statistics, see Table 1). The configuration was verified by PROCHECK (25). The locations of gold atoms were defined using the anomalous Fourier maps. Atomic coordinates and structure factors have been deposited in the Protein Data Bank with the accession code 3H4K.
TABLE 1.
Summary of crystallographic data
| Wild type SmTGR | |
|---|---|
| Data collection | |
| Space group | C2 |
| Cell dimensions | |
| a, b, c (Å) | 147.51, 102.16, 60.57 |
| α, β, γ (°) | 90.00, 114.15, 90.00 |
| Resolution (Å) | 40.0-2.55 (2.59-2.55)a |
| Rmerge | 0.12 (0.48) |
| I/σI | 14.7 (2.4) |
| Completeness (%) | 99.7% (99.9%) |
| Redundancy | 5.2 (5.2) |
| Refinement | |
| Resolution (Å) | 40.0-2.55 |
| No. reflections | 25362 |
| Rwork/Rfree | 0.231/0.256 |
| No. of atoms (1 subunit/au) | 4745 |
| Protein | 4540 |
| Ligand/ion | 104 |
| Water | 101 |
| B-factors (overall) | 35.42 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.006 |
| Bond angles (°) | 0.91 |
| Ramachandran plot | |
| Most preferred | 90.4% |
| Allowed | 9.2% |
| Generously allowed | 0.4% |
Highest resolution shell is shown in parentheses.
GR Activity Assay
Glutathione reductase from baker's yeast (Sigma-Aldrich) was washed four times at 3500 × g with 50 mm Tris/HCl, 0.1 m NaCl, pH 7.4, using Amicon Ultra 15 centrifugal filter devices (Millipore, Cork, Ireland) with a molecular mass cut-off of 30 kDa to change buffer and remove trace amounts of dithiothreitol. GR activity was assayed as a NADPH-dependent reduction of GSSG (26). In all of the assays described below, 0.5-ml reaction mixtures contained 30 μl of the stock GR solution (0.5 μm, final concentration 30 nm) plus 200 μm NADPH and 500 μm GSSG.
To assess the effect of AF on GR, incubation mixtures were prepared containing 0.5 μm GR and 1, 4, 10, and 50 μm AF, all containing 100 μm NADPH in 50 mm Tris, pH 7.4, and 100 mm NaCl. The measurements were performed at 0, 15, 30, 60, 120, and 240 min after the aerobic incubation was started. The reaction was initiated by the addition of 500 μm GSSG and followed by the decrease in absorbance at 340 nm. Controls containing no AF were measured at each time point, and the activity was expressed as a percentage of each sample against the control.
SmTGR Inhibition Assay
To assess the effect of AF on truncated TGR, incubation mixtures were prepared containing 3.0 μm SmTGR (truncated form, lacking the last two amino acids) with and without 8 μm AF, eventually with 3 μm BzSe. All of the samples, under aerobic conditions, contained 100 μm NADPH in 50 mm Tris, pH 7.4, and 100 mm NaCl. Aliquots of 85 μl were tested for activity by the 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) assay in a 0.5-ml cuvette. The measurements were performed at 0, 30, 60, 120, and 240 min after the incubation was started. The reaction was initiated by the addition of 1 mm DTNB and followed by the increase in absorbance at 412 nm. Controls containing no AF were measured at each time point, and the activity was expressed as a percentage of each sample against the control.
RESULTS
Structural Analysis
The structure of wild type SmTGR treated with AF was solved by molecular replacement. After the refinement processes and geometry optimization of the model, an anomalous difference Fourier map was calculated from 40 to 2.5 Å and contoured at 8 σ. Its superimposition on the difference density map (Fo − Fc) contoured at the same σ and calculated on the refined SmTGR structure without the metal (10), clearly shows the position of the gold atoms in the asymmetric unit. AF itself was never detected in our structures, but three gold-binding sites were identified in each subunit, as shown in Fig. 1A. As already reported for the truncated form of the enzyme (10), the C terminus was not visible in the electron density maps.
FIGURE 1.

The three gold-binding sites of wild type SmTGR. A, the three-dimensional model of one subunit is shown with the three gold-binding sites. Site 1 shows the gold in between Cys154 and Cys159; site 2 shows the gold in between Cys520 and Cys574; site 3 shows the gold in the putative NADPH-binding pocket. The glutaredoxin domain of TGR is shown in green, whereas the thioredoxin domain is shown in red. The bound flavin is also highlighted. B, site 1. The linear geometry of the Cys154-gold-Cys159 adduct is shown. The occupancy of the gold atom is about 50%. Both distances of the sulfur-gold bond are 2.3 Å, as expected for this type of coordination moiety. C, site 2. The electron density map (2Fo − Fc) contoured at 1 σ shows the possible charge transfer complex between the gold and Phe505. D, site 2. The gold atom between Cys574 and Cys520 is shown together with the other residues that surround the metal, i.e. Phe505, Pro507, and Pro542. E, site 3. Gold in the putative NADPH-binding site of SmTGR. Tyr296 is known to swing upon NADPH binding in thiol reductase enzymes (30). Ser295 is the residue closest to the gold (Ser295(OG)-gold: 3.2 Å). Other van der Waals' contacts are with the main chain atoms of the polypeptide (Ala390, Val391, Gly392, and Arg393).
Site 1
The metal is bound between the catalytic cysteines (Cys154–Cys159) over the FAD-binding site. The geometry of the Cys-gold-Cys array is linear, and the distance between the sulfurs and the gold is 2.3 Å. The metal occupancy is ∼50%. Cys154 is found in double conformation (Fig. 1B); in the gold-containing site, this residue coordinated to the metal points toward Cys159, whereas in the gold-free site Cys154 is not involved in a disulfide bridge with Cys159 and points away from the FAD and Cys159. At this resolution, the orientation of Cys159 is almost independent of the presence of gold.
Site 2
Also in this case, the gold was found between two cysteines, namely Cys520 and Cys574, with an occupancy of ∼50%. As in site 1, the geometry is linear, but the sulfur-gold distances are longer than the canonical values (Fig. 1, C and D); this might indicate a fluxional behavior of the gold between the two cysteines (27). Thus the geometry calculated from the electron density maps should be taken as an average.
The gold atom in site 2 is surrounded by hydrophobic residues, i.e. Phe505, Pro507, and Pro542 (Fig. 1, C and D). The aromatic ring of Phe505 is 2.6 Å away from the gold atom (Fig. 1D), this distance being smaller than the sum of the van der Waals' radii of the sp2 carbon and the metal. It is known from gas phase studies that benzene and gold(I) interact to form a charge transfer complex (28). The stability of the complex was calculated to be 70 kcal/mol, in the range of a covalent bond (29). Therefore, the position of the gold atom between Cys520 and Cys574 might be also stabilized by a charge transfer complex that forces the metal into a peculiar position.
Because no functional role has been assigned as yet to the couple Cys520–Cys574, we cannot infer the role of site 2 in the inhibition of TGR by AF, but we point out that: (i) site 2 is invariant in all the TGRs and mammalian TRs that have a Sec at the penultimate position, whereas it is absent in GRs or in TRs lacking the penultimate Sec (sequence alignment not shown); and (ii) site 2 of one subunit is in close proximity to the C terminus of the same subunit and to the FAD-binding site of the other subunit (Cys574/SG-Cys154/SG = 14.5 Å).
Site 3
The third gold atom is located in a pocket on the side of the FAD opposite to Cys154–Cys159, with an occupancy of ∼40% (Fig. 1E). The cavity is surrounded by the amino acid side chains of Ser295, Tyr296, and Val297 and the main chains of Ala390, Val391, Gly392, and Arg393. No atoms have the correct distance to form coordination bonds, the closest atom being the oxygen of the Ser296 at 3.2 Å; however, all of the above residues can be engaged in van der Waals' contacts. Superposition of the structures of SmTGR and mouse TR bound to NADPH (Protein Data Bank code 1zdl) (30) suggests that site 3 may correspond to the NADPH-binding site (Fig. 2), and thus it might be relevant to the mechanism of inactivation by AF.
FIGURE 2.

The SmTGR crystal structure in complex with gold ions (in green) is superimposed to the mouse TR with NADPH bound (in magenta; Protein Data Bank code 1zdl (30)). The root mean square deviation is 0.82 Å over the 462 aligned residues. The residues surrounding NADPH in mouse TR are conserved in SmTGR (sequence alignment not shown). The structural comparison shows the change in conformation of the loop 293–296 and in particular of Tyr296 and Ser295, highlighted for the two enzymes as balls and sticks (the other amino acid side chains are omitted for clarity). The OG atom of Ser295 is the closest contact with the gold ion in the SmTGR crystal structure (see “Results” and Fig. 1). In the mouse TR structure, Ser295 shifts in position to make room for the bound NADPH; the clash between the metal in site 3 and the phosphate of the cofactor in SmTGR is self-evident.
Reaction of a Sec-lacking, Truncated SmTGR Variant with AF
Because AF behaves as a gold carrier and the final complex contains the bare metal bound either to Cys residues or to a hydrophobic pocket (site 3), we decided to test whether Sec is indeed essential for inactivation of TGR by AF, as is usually thought. X-ray diffraction maps were thus collected for a Sec-lacking artificial variant of the enzyme, namely the truncated SmTGR, a construct lacking the last two C-terminal residues, Sec597 and Gly598 (10). This variant, when crystallized in the presence of a slight molar excess of AF (2–3-fold) plus a catalytic source of selenium (see below), incorporates gold at sites 1 and 3 (results not shown); we are at present unable to explain why no gold was observed in site 2. The time courses of inactivation of truncated and wild type SmTGR by AF are reported in Fig. 3C. The Sec-lacking variant, which is 15-fold less active than the wild type in the DTNB assay (10), is inhibited by AF 200-fold more slowly than the wild type enzyme (which is inactivated with a rate constant of 0.21/min).
FIGURE 3.

A, baker's yeast GR is inactivated by AF: time courses obtained at concentrations of 1 μm (squares), 4 μm (closed circles), 10 μm (open circles), and 50 μm (open triangles). B, effect of BzSe on baker's yeast GR inactivation by AF. Time course in the presence of 4 μm AF (circles) or 4 μm AF plus 2 μm BzSe (triangles). GR exposed to a mixture in which AF 4 μm and BzSe 2 μm were preincubated for 2 h before the assay (pentagons). C, inactivation by AF of truncated SmTGR and wild type SmTGR, and the effect of BzSe. Time courses of truncated SmTGR in the presence of 8 μm AF (circles) or 8 μm AF plus 3 μm BzSe (triangles). Truncated SmTGR exposed to a mixture in which AF 8 μm and BzSe 3 μm were preincubated for 2 h before the assay (pentagons); time course of wild type SmTGR 20 nm incubated with 50 nm AF (open circles).
GR Inactivation Assays
The observation that the gold-binding sites identified by x-ray crystallography do not involve the penultimate Sec challenges the currently accepted mechanism of inhibition of TGR/TR by AF (14, 19). To investigate this point, we carried out functional experiments using GR as a model enzyme. GR has three crucial features that make it suitable for this purpose: (i) it lacks the Sec residue and the C-terminal tail, thus serving as a negative control for the role of selenium; (ii) it has only one of the gold-binding sites identified in SmTGR, i.e. that homologous to site 1 (20) (the Cys couple of site 2 of TGRs and TRs is lacking, and most of the side chains that surround the gold in site 3 are not conserved, e.g. Ser295 is replaced by a Gly in the baker's yeast GR; sequence alignment not shown); and (iii) it has much higher catalytic activity than truncated SmTGR, thus simplifying the assays. A spectrophotometric enzymatic assay was carried out using 0.5 μm GR incubated with 1, 4, 10, and 50 μm AF over a period of 4 h; activity was followed as the NADPH-dependent (200 μm) reduction of GSSG (500 μm), monitoring the reaction at 340 nm (26). When GR was incubated with AF in the absence of NADPH, no inhibition was detected, a finding consistent with the entrapment of gold between cysteines (as for site 1, a reaction dependent on the enzyme being reduced). The concentrations of GR, NADPH, and AF in the assay mixture were chosen following Gromer et al. (19). The results of these experiments (Fig. 3A) show that AF inactivates GR at every concentration from 1 to 50 μm, the time course being in all cases complex and the rate slowing down progressively. The experiment shows that AF is capable of inhibiting an enzyme that lacks Sec, as expected on the basis of the structural considerations made on SmTGR (see above).
Given the complex time course and the very slow inactivation rate, quantitative analysis is not easy, and thus we resorted to estimate the initial rate of inactivation of GR by AF, at the different concentrations. Initial first order rate constants plotted as a function of AF concentration, reported in Fig. 4, shows that inactivation rate is AF concentration-dependent but tends to a plateau. Nevertheless we have direct evidence that the inactivation is faster at higher AF concentrations and that the reaction is essentially irreversible (because activity is not recovered even after long dialysis). This is consistent with the high stability of the sulfur-gold-sulfur complex observed in site 1 of SmTGR and presumably formed also in the homologous site of GR.
FIGURE 4.
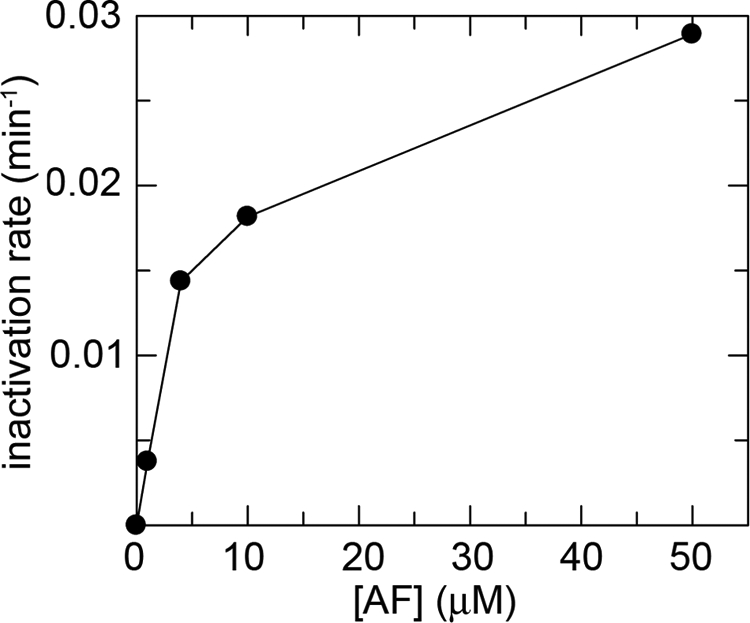
Rate constant of inactivation (min−1) of yeast GR (0.5 μm) as a function of AF concentration (1, 4, 10, and 50 μm), as derived from linearization of the first points of the time courses reported for Fig. 3A.
The Role of Selenium
The limited level of inhibition of GR by AF reported in the literature (19) is most probably due to the insufficient time allowed for complex formation (i.e. ≤20 min); the more effective inhibition seen with TR/TGR may be due to the faster reactivity of Sec-containing reductases rather than to higher affinity for AF or gold. To test this hypothesis, GR was incubated with AF in the presence of an external source of selenium, BzSe. Because BzSe is unstable, being easily oxidized to diphenyl diselenide, it was reduced with NaBH4 before use; consequently, the concentrations reported in our experiments are probably overestimated, the actual concentration of the reduced form probably falling under aerobic conditions. The assay was performed on aliquots of an incubation mixture containing 0.5 μm GR, 4 μm AF, and 2 μm BzSe. Parallel control experiments (not shown) demonstrated that in the absence of AF, BzSe is not an inhibitor of GR, at least in the low micromolar concentration range. Moreover, a third condition was tested, where AF and BzSe were preincubated at 25 °C for 2 h before the addition of the enzyme. The results in Fig. 3B show that the kinetics of inhibition by AF is significantly affected by the addition 2 μm BzSe to the assay mixtures, the onset of inactivaion being clearly accelerated. GR approaches 0% activity within 4 h, whereas the addition of 4 μm AF alone yields only 50% inhibition over the same time interval. Fig. 3B also shows that incubating AF and BzSe together before addition to the assay mixtures increases substantially the rate of the reaction. These experiments confirm that although selenium is not absolutely required for inactivation of the enzyme by AF, it has a significant effect on the rate of onset of inactivation.
The effect of the external source of selenium was also tested for the truncated SmTGR variant that lacks the last two amino acids (Sec-Gly). Also in this case the onset of enzyme inactivation is accelerated in the presence of BzSe (Fig. 3C).
DISCUSSION
Structural Basis of the Inhibition of Thiol-dependent Reductases by Gold
Despite the extensive use of AF as an antiarthritic drug, the rationale for its mechanism of action in relation with its chemistry is not yet established. In general, gold-containing compounds are thiol/selenol reactive species; indeed AF and other gold compounds are strong inhibitors of mammalian TR and GR.
Our data indicate that the actual inhibitor is gold(I) rather than the whole AF molecule. The phosphine and thioglucose ligands cannot be seen in any of our electron density maps, confirming that AF is actually a pro-drug whose function is to carry and deliver gold to its final target(s) with the metal bound in between Cys couples of the enzyme.
As to the geometry and the presumable relative affinities of the gold-binding sites observed in our maps, we note that in site 1 the gold atom is bound between the catalytic cysteines Cys154–Cys159 in a linear two-coordination geometry, typical of the gold(I)-sulfur complexes (Fig. 1). Site 2 also shows the gold bound with an almost linear geometry between two cysteines, Cys520–Cys574, close to the C terminus of the same subunit and the FAD-binding site of the other subunit (Fig. 1); a charge transfer complex with Phe505 is probably also involved. Because the functional role of this site is not clear, the consequences of gold binding to Cys520–Cys574 are at present uncertain. Site 3 is different from the other two because it contains no Cys, the metal being located in a cavity with no atoms at the correct distance to form coordination bonds (Fig. 1); however, it probably overlaps with the NADPH-binding site (Fig. 2).
Our results suggest a possible mechanism of inhibition of thiol-dependent reductases by AF, namely the transfer of gold(I) to one or more redox-active Cys (or Sec-Cys) couples of the enzyme (Scheme 1 and below). Our proposal is consistent with what is known about the electron flow in TR and GR (29, 30); the FAD cofactor is reduced by NADPH and donates its electrons to the first Cys couple (Cys154–Cys159 in SmTGR). Reduction of GSSG in GR is presumed to occur at this site. In TRs and TGRs the path of electrons is more complex, because they are transferred from the first Cys couple to the C-terminal Cys596–Sec597 couple and from here to thioredoxin, or, in the case of TGR, to the additional Grx domain. TRs and TGRs possess an additional conserved Cys couple (Cys520–Cys574 in SmTGR), whose functional role is unknown.
We observe gold binding to two of these redox sites (sites 1 and 2), plus to the putative NADPH-binding site (site 3), and a further site may be present at the C terminus (Cys596–Sec597) that is disordered in our structures. Because binding to any of these may in principle be sufficient to inhibit the enzyme, one may ask which is the most relevant binding site in vivo. We assume that gold is redistributed at equilibrium among the three redox couples of SmTGR (Scheme 1) and that in the presence of enough AF all the three redox couples would be fully occupied, provided that enough time is allowed and that the enzyme is kept reduced. Based on what is known about TRs and TGRs (31), we also assume that any derivative of SmTGR bearing gold at the C-terminal binding site would be incapable of reducing thioredoxin or the glutaredoxin domain, whereas any species bearing gold at site 1 would be inactive toward any substrate, including the artificial DTNB. Site 2 participates in this exchange of gold, but its functional role is unclear, whereas site 3 is probably relevant to the inhibition of SmTGR (see above), but we cannot see how it could participate to the gold exchange with the other sites.
Our hypothesis is consistent with data by other researchers; e.g. irreversible inhibition of human GR by gold (released by GoPi) has been shown by x-ray crystallography to be due to the formation of a Cys-gold-Cys adduct with the Cys couple lying over the FAD (Cys58 and Cys63, homologous to Cys154–Cys159 in SmTGR) (20, 32). Moreover, the clinically used plasma levels of AF are in the order of 20 μm (19); these values are similar to our reaction conditions that require 10 μm SmTGR and 100 μm AF (i.e. a stoichiometric excess of only 2–3-fold, assuming four binding sites/enzyme subunit); thus significant occupancy of all the binding sites of SmTGR may be expected if AF therapy of schistosomiasis were to become a standard practice.
Affinity of the Sulfur-Gold-Sulfur Complex
The gold-enzyme complex has very high affinity, because inhibition is insensitive to both dilution in the final assay mixture and extensive dialysis. An adduct analogous to that observed for site 1 of SmTGR has been found in human GR treated with the gold compound GoPi (20), and also in this case the inhibition was demonstrated to be irreversible. What is the affinity of GR or TGR for gold? Our experiments demonstrate that complete inhibition is obtained using micromolar concentrations of AF, provided that enough time is allowed for the reaction to go to completion; in our hands, complete inhibition of GR and truncated SmTGR is only achieved when BzSe is added to accelerate the reaction. Thus our best estimate suggests IC50 < 10−7 m, consistent with our previous estimate of IC50 ≈ 10−8 m for wild type SmTGR (6) and much lower than the concentrations needed to kill the worms “in vitro” (10 μm).
This estimate must be taken with caution, because in such a complex reaction as that depicted in Scheme 1, the affinity of the enzyme for its inhibitor (the gold(I) ion) must be considered from at least two different view points, i.e. the true thermodynamic reversibility and the (possible) release of free gold. If the mechanism of inhibition is transfer of gold from AF to a Cys couple, as revealed by x-ray crystallography, true thermodynamic reversibility would imply not only the dissociation of the metal from the enzyme but simultaneous resynthesis of AF from gold, phosphine, and thioglucose. Resynthesis of AF, besides being unlikely, has a paradoxical consequence: given the stoichiometry of the reaction (TGR-gold + ATG + P(Et)3 ↔ TGR + AF, where ATG stands for acetoxy-thioglucose, and P(Et)3 stands for triethylphosphine), dilution is expected to increase inhibition, rather than vice versa. Moreover, free triethyl-phosphine in solution is prone to oxidation; thus its concentration decreases with time (16) and makes it impossible to accurately determine a precise value of Ki. For all practical purposes, the true IC50 of AF must be indistinguishable from zero, and enzyme activity may probably be restored only by the addition of compounds capable of chelating gold with affinity higher than Cys couples (e.g. 2,3 dimercapto-propanol; see Ref. 19).
If, on the other hand, one considers the affinity of gold for its binding sites in GR, TR, or TGR (i.e. the tendency of the complex to dissociate releasing the free metal ion, rather than AF), we may only recall that the energy of the gold-sulfur bond has been estimated at ∼ 55 kcal/mol (33).
Given such a high affinity of gold for sulfur pairs, the incomplete (50%) occupancy of sites 1 and 2 in our structures is puzzling, and we can offer two, nonmutually alternative hypotheses, whose testing requires further experimental work. Incomplete occupancy of sites 1 and 2 might be due to the slow redistribution of gold between its protein-binding sites (see Scheme 1). Moreover, negative kinetic cooperativity between the two subunits of the dimer or between the two sites of the same protomer cannot be excluded; binding of gold to one site could slow down the reaction at the other.
Because the same Cys couple exists in TR, TGR, and GR and because binding of gold to this couple is essentially irreversible, the 1000-fold lower apparent affinity of AF for GRs reported by Gromer et al. (19) seemed puzzling. Our data solve this issue by demonstrating that the incubation of GR with AF in these experiments (20 min) was too short to allow the formation of the GR-gold complex. Indeed if we analyze the time courses of Fig. 3A up to 15 min., we reproduce similar results of Gromer et al. (19).
The time courses of inactivation indicate a complex reaction mechanism whose order decreases from 2 at low concentration of AF to lower values. This behavior (Fig. 4), which is typical of reactions in which an intermediate populated in the course of a bimolecular reaction decays monomolecularly to more stable chemical species, is not inconsistent with the first two reactions of Scheme 1 (i.e. the bimolecular formation of the Cys- or Sec-gold-Pt(Et)3 complex, followed by the irreversible monomolecular entrapment of the metal in the Cys-gold-Cys complex).
Selectivity of AF for TR/TGR: the Role of Selenium
Two strongly correlated issues remain to be discussed, namely the kinetics of gold incorporation and the possible reason for the strong preference of AF for TRs and TGRs over GRs. Our data show that Sec is not the only binding site of the drug, and indeed we demonstrate that (i) GR and Sec-free SmTGR are significantly inhibited by the addition of AF stoichiometric with Cys couples, provided enough time is allowed for the slow transfer of gold to the protein, and (ii) external selenium (in the form of benzeneselenol) plays a significant and in some way surprising catalytic role and facilitates the transfer of gold from AF to the redox active Cys couples, both in GR and in truncated SmTGR.
If we accept that TRs/TGRs and GRs all have very high affinity for gold, our best hypothesis to explain the selectivity of AF for the former is that TRs and TGRs react faster. Indeed we demonstrated in this paper that yeast GR reacts slowly with AF and that the incubation times used in previous experiments (e.g. 19) were too short to allow extensive inactivation, resulting in an overestimate of the Ki (in the micromolar range). Our data demonstrate that (i) wild type SmTGR is inactivated by AF faster than both its truncated (i.e. Sec lacking) variant and yeast GR and (ii) an external source of selenium greatly speeds up the reactivity of Sec-free thiol reductases. These observations point to a catalytic role of Sec that would promote the displacement of the first gold ligand of AF, probably thioglucose (in analogy with the reaction of AF with the human serum albumin) (16) (Scheme 1). The intermediate adduct Sec-gold-triethylphosphine thus formed would transfer the metal to its final protein ligands. We hypothesize that BzSe is able to accelerate the rate of inhibition of GR or Sec-free TGR with AF because it displaces thioglucose and helps transfer of the metal to Cys residues. This ability would be due to the fact that BzSe shares some properties of Sec, e.g. it has a similar pKa (4.6 for BzSe versus 5.2 for Sec (5, 34)) and has high nucleophilic character when ionized.
There are two possible and mutually nonexclusive reasons why Sec reacts with and activates AF faster than the Cys residues, i.e. (i) a more reactive electronic structure and (ii) a better exposure to the solvent of the C terminus that by-passes the slow diffusion to site 1 of the bulky molecule of AF. The first point is in apparent contrast with the fact that selenium has a greater affinity than sulfur for gold (16). Nevertheless, studies have shown that derivatives of AF in which the thioglucose moiety has been substituted by a selenol glucose or a cyanide ion (both high affinity ligands for gold) are more apt to loose the P(Et)3 ligand when they react with human albumin (16, 35). The labilization of phosphine in the albumin sulfur-gold-PEt3 adduct is attributed to a strong trans-effect of the albumin thiolate of Cys34 (36). In TGRs or TRs, this first labilization of the gold-P(Et)3 bond is probably followed by the formation of the Sec-gold-Cys species (see Scheme 1). In turn, this adduct could be easily transformed into the final Cys-gold-Cys given that Sec or in general selenium compounds (e.g. BzSe) are better leaving groups than sulfur (Ref. 37 and Scheme 1). The second hypothesis, which considers a steric effect, is consistent with the crystal structures of TRs and SmTGR in which it is clear that the C terminus is more exposed to the solvent than the buried FAD catalytic site (10, 30).
Consistent with the literature and based on our own results, the mechanism of action of AF is envisaged as a series of ligand displacement reactions (Scheme 1). Indeed it is known that when administered in vivo, AF partially releases the gold atom to protein thiols and to free thiols; through ligand displacement reactions, gold is then transferred from serum albumin (16) to other proteins or thiols in the body, until it reaches a final acceptor that traps the gold in a stable linear two-coordination geometry (20, 38). The peculiarity of TRs and TGRs would be their own ability to carry out the undressing and stable coordination of gold without the help of other proteins or free thiols, because of the C-terminal Sec. This reaction intermediate would further transfer the gold atom to its final acceptor within the same polypeptide chain (Scheme 1). This mechanism is similar to that proposed for mercuric ion reductase, a homodimeric enzyme belonging to the same family as TGR (39).
Acknowledgments
We are grateful to Angela Kuntz for preparation of the Sec-containing wild type S. mansoni TGR and to Fulvio Saccoccia and Loredana Sposato for preparation of the truncated form of the enzyme. We thank the synchrotron facilities Berliner Elektronenspeicherring BESSY II (BESSY; Berlin, Germany) and ELETTRA (Trieste, Italy).
This work was supported, in whole or in part, by National Institutes of Health Grant AI065622. This work was also supported by the Fondazione Roma Project “Rational Approach to the Specific Inhibition of Plasmodium falciparum and Schistosoma mansoni,” the “Sapienza” University of Rome Progetto Università 2006 and 2007 and Progetto Ateneo Federato 2006 and 2007, the Ministero dell'Università e della Ricerca Italy Fondo per gli Investimenti della Ricerca di Base/Proteomica 2007-protRBRN07BMCT, and the European Community Research Infrastructure Action under FP6 “Structuring the European Research Area” Contract R II 3-CT-2004-506008.
The atomic coordinates and structure factors (code 3H4K) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- TGR
- thioredoxin-glutathione reductase
- SmTGR
- TGR from Schistosoma mansoni
- TR
- thioredoxin reductase
- GR
- glutathione reductase
- GoPi
- [{1-phenyl-2,5-di(2-pyridyl)-phosphole}AuCl]
- BzSe
- benzeneselenol
- Sec
- selenocysteine
- AF
- auranofin
- DTNB
- 5,5′-dithiobis-(2-nitrobenzoic acid)
- GSSG
- glutathione disulfide.
REFERENCES
- 1.Hotez P. J., Molyneux D. H., Fenwick A., Kumaresan J., Sachs S. E., Sachs J. D., Savioli L. (2007) N. Engl. J. Med. 357, 1018–1027 [DOI] [PubMed] [Google Scholar]
- 2.Doenhoff M. J., Kusel J. R., Coles G. C., Cioli D. (2002) Trans. R. Soc. Trop. Med. Hyg. 96, 465–469 [DOI] [PubMed] [Google Scholar]
- 3.Angelucci F., Basso A., Bellelli A., Brunori M., Pica-Mattoccia L., Valle C. (2007) Parasitology 134, 1215–1221 [DOI] [PubMed] [Google Scholar]
- 4.Cioli D., Valle C., Angelucci F., Miele A. E. (2008) Trends Parasitol. 24, 379–382 [DOI] [PubMed] [Google Scholar]
- 5.Salinas G., Selkirk M. E., Chalar C., Maizels R. M., Fernández C. (2004) Trends Parasitol. 20, 340–346 [DOI] [PubMed] [Google Scholar]
- 6.Kuntz A. N., Davioud-Charvet E., Sayed A. A., Califf L. L., Dessolin J., Arnér E. S., Williams D. L. (2007) PLoS Med. 4, e206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun Q. A., Kirnarsky L., Sherman S., Gladyshev V. N. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 3673–3678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alger H. M., Williams D. L. (2002) Mol. Biochem. Parasitol. 121, 129–139 [DOI] [PubMed] [Google Scholar]
- 9.Sharma M., Khanna S., Bulusu G., Mitra A. (2009) J. Mol. Graph Model 27, 665–675 [DOI] [PubMed] [Google Scholar]
- 10.Angelucci F., Miele A. E., Boumis G., Dimastrogiovanni D., Brunori M., Bellelli A. (2008) Proteins 72, 936–945 [DOI] [PubMed] [Google Scholar]
- 11.Sayed A. A., Simeonov A., Thomas C. J., Inglese J., Austin C. P., Williams D. L. (2008) Nat. Med. 14, 407–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loukas A., Bethony J. M. (2008) Nat. Med. 14, 365–367 [DOI] [PubMed] [Google Scholar]
- 13.Simeonov A., Jadhav A., Sayed A. A., Wang Y., Nelson M. E., Thomas C. J., Inglese J., Williams D. L., Austin C. P. (2008) PLoS Negl. Trop. Dis. 2, e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonilla M., Denicola A., Novoselov S. V., Turanov A. A., Protasio A., Izmendi D., Gladyshev V. N., Salinas G. (2008). J. Biol. Chem. 283, 17898–17907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rendón J. L., del Arenal I. P., Guevara-Flores A., Uribe A., Plancarte A., Mendoza-Hernández G. (2004) Mol. Biochem. Parasitol. 133, 61–69 [DOI] [PubMed] [Google Scholar]
- 16.Shaw C. F., 3rd (1999). Chem. Rev. 99, 2589–2600 [DOI] [PubMed] [Google Scholar]
- 17.Becker K., Gromer S., Schirmer R. H., Müller S. (2000) Eur. J. Biochem. 267, 6118–6125 [DOI] [PubMed] [Google Scholar]
- 18.Chong C. R., Sullivan D. J., Jr. (2007) Nature 448, 645–646 [DOI] [PubMed] [Google Scholar]
- 19.Gromer S., Arscott L. D., Williams C. H., Jr., Schirmer R. H., Becker K. (1998). J. Biol. Chem. 273, 20096–20101 [DOI] [PubMed] [Google Scholar]
- 20.Urig S., Fritz-Wolf K., Réau R., Herold-Mende C., Tóth K., Davioud-Charvet E., Becker K. (2006). Angew. Chem. Int. Ed. 45, 1881–1886 [DOI] [PubMed] [Google Scholar]
- 21.Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 22.Read R. J. (2001) Acta Crystallogr. Sect. D Biol. Crystallogr. 57, 1373–1382 [DOI] [PubMed] [Google Scholar]
- 23.Murshudov G. N., Vagin A. A., Lebedev A., Wilson K. S., Dodson E. J. (1999). Acta Crystallogr. Sect. D Biol. Crystallogr. 55, 247–255 [DOI] [PubMed] [Google Scholar]
- 24.Emsley P., Cowtan K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 25.Laskowsky R. A., MacArthur M. W., Moss D. S., Thorntorn J. (1993) J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 26.Carlberg I., Mannervik B. (1985) Methods Enzymol. 113, 484–490 [DOI] [PubMed] [Google Scholar]
- 27.Mézailles N., Avarvari N., Maigrot N., Ricard L., Mathey F., Le Floch P., Cataldo L., Berclaz T., Geoffroy M. (1999). Angew. Chem. Int. Ed. Engl. 38, 3194–3197 [DOI] [PubMed] [Google Scholar]
- 28.Ho Y.-P., Dunbar R. C. (1999) Int. J. Mass Spectrosc. 182–183, 175–184 [Google Scholar]
- 29.Schröder D., Hrusak J., Hertwig R. H., Wolfram K., Schwerdtfeger P., Schwarz H. (1995) Organometallics 14, 312–316 [Google Scholar]
- 30.Biterova E. I., Turanov A. A., Gladyshev V. N., Barycki J. J. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 15018–15023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arnér E. S. (2009) Biochim. Biophys. Acta 1790, 495–526 [DOI] [PubMed] [Google Scholar]
- 32.Deponte M., Urig S., Arscott L. D., Fritz-Wolf K. R. R., Herold-Mende C., Koncarevic S., Meyer M., Davioud-Charvet E., Ballou D. P., Williams C. H., Jr., Becker K. (2005). J. Biol. Chem. 280, 20628–20637 [DOI] [PubMed] [Google Scholar]
- 33.Schröder D., Schwarz H., Hrušák J., Pyykkö P. (1998) Inorg. Chem. 37, 624–632 [Google Scholar]
- 34.Filipovska A., Kelso G. F., Brown S. E., Beer S. M., Smith R. A., Murphy M. P. (2005) J. Biol. Chem. 280, 24113–24126 [DOI] [PubMed] [Google Scholar]
- 35.Isab A. A., Hormann A. L., Coffer M. T., Shaw C. F. (1988) J. Am. Chem. Soc. 110, 3278–3284 [Google Scholar]
- 36.Coffer M. T., Shaw C. F., 3rd, Hormann A. L., Mirabelli C. K., Crooke S. T. (1987) J. Inorg. Biochem. 30, 177–187 [DOI] [PubMed] [Google Scholar]
- 37.Lothrop A. P., Ruggles E. L., Hondal R. J. (2009) Biochemistry 48, 6213–6223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Viry E., Battaglia E., Deborde V., Müller T., Réau R., Davioud-Charvet E., Bagrel D. (2008) ChemMedChem. 3, 1667–1670 [DOI] [PubMed] [Google Scholar]
- 39.Engst S., Miller S. M. (1999) Biochemistry 38, 3519–3529 [DOI] [PubMed] [Google Scholar]