

Abstract
Claudins form size- and charge-selective pores in the tight junction that control the paracellular flux of inorganic ions and small molecules. However, the structural basis for ion selectivity of paracellular pores is poorly understood. Here we applied cysteine scanning to map the paracellular pathway of ion permeation across claudin-2-transfected Madin-Darby canine kidney type I cells. Four potential pore-lining amino acid residues in the first extracellular loop were mutated to cysteine and screened for their accessibility to thiol-reactive reagents. All mutants were functional except D65C, which formed dimers by intermolecular disulfide bonding, leading to a loss of charge and size selectivity. This suggests that claudin-2 pores are multimeric and that Asp65 lies close to a protein-protein interface. Methanethiosulfonate reagents of different size and charge and the organic mercury derivate, p-(chloromercuri)benzenesulfonic acid, significantly decreased paracellular ion permeation across I66C-transfected cells by a mechanism that suggests steric blocking of the pore. The conductance of wild-type claudin-2 and the other cysteine mutants was only weakly affected. The rate of reaction with I66C decreased dramatically with increasing size of the reagent, suggesting that Ile66 is buried deep within a narrow segment of the pore with its side group facing into the lumen. Furthermore, labeling with N-biotinoylaminoethyl methanethiosulfonate showed that I66C was weakly reactive, whereas Y35C was strongly reactive, suggesting that Tyr35 is located at the protein surface outside of the pore.
Sheets of polarized epithelia constitute barriers that separate fluid compartments of different chemical composition and mediate exchange of solutes and ions via transcellular and paracellular pathways. A large body of evidence suggests that transport via the paracellular pathway occurs through pores in the tight junctions that are formed by tetraspan membrane proteins, known as claudins (1–3).
Our current understanding of paracellular pores is that they are size- and charge-selective water-filled channels that, in contrast to channels for transmembrane transport, are oriented parallel instead of perpendicular to the lipid layer of the cell membrane. Size exclusion experiments suggest a pore diameter of 6.4–8 Å (4, 5). Furthermore, site-directed mutagenesis and overexpression of claudins in epithelial cells identified the first extracellular domain as playing an important role in the charge selectivity of paracellular transport (6–8). The first extracellular domain of claudins contains various basic and acidic amino acids, some of which are conserved in different claudin isoforms, and these could be involved in the mechanism of ion permeation. Several studies have demonstrated homo- or heterotypic interaction of claudins, suggesting that paracellular pores are formed by oligomers of claudins (9–11). Taken together, significant progress has been made in uncovering the nature of the paracellular pathway and mechanisms of selectivity of paracellular ion permeation. However, it is unknown how the extracellular domains of claudins fold to form paracellular pores and which amino acid residues line the pathway of ion diffusion.
Epithelia in vivo and epithelial cell lines express characteristic sets of different claudin isoforms that determine paracellular permeability and permselectivity. Claudin-2 is expressed in epithelia with a high capacity for passive paracellular cation transport, such as the epithelium lining the proximal renal tubules (12). The transfection of claudin-2 into high resistance Madin-Darby canine kidney (MDCK)2 type I cells converts the tight junction from a “tight” into a “leaky” paracellular barrier by selectively increasing Na+ permeability (13, 14), suggesting a physiologic role of claudin-2 in creating paracellular Na+ channels. Because of the high signal/noise ratio of the claudin-2-induced permeability, this isoform provides an excellent model to study paracellular transport. We have recently generated a stable expression system of claudin-2 in MDCK I cells under the control of a TetOff promoter. This inducible system allows us to specifically determine the macroscopic conductance and permeability of claudin-2 pores by subtracting background measurements of uninduced cells. Using this expression system, we could recently demonstrate that the cation selectivity of claudin-2 cells is mediated by electrostatic interaction of partially dehydrated permeating cations with aspartate 65 (5). However, further investigations are necessary to study the position and function of this and other residues of the first extracellular domain and to elucidate their role in the transport mechanism of paracellular pores.
The substituted cysteine accessibility method (SCAM), developed by the Karlin group, has proved to be a powerful tool in the mapping of the structures of membrane ion channels and transport proteins (15, 16). In SCAM, thiol-reactive reagents are used to covalently modify endogenous cysteines, or cysteines introduced into a protein by site-directed mutagenesis. SCAM can be used to study channel-lining amino acid side chains, the secondary structures of membrane-spanning segments, and the localization of selectivity filters, channel gates, and inhibitor binding sites.
Here, we used SCAM to analyze the paracellular pathway of ion permeation across claudin-2 transfected MDCK I cells. Our data show that thiol-reactive reagents strongly block ion transport in at least one of the cysteine mutants that we have generated and, thus, provide a tool to map residues that line the paracellular pore.
EXPERIMENTAL PROCEDURES
Generation and Screening of MDCK I TetOff Claudin-2 Cell Lines
MDCK I TetOff cells expressing wild-type (WT) and mutant claudin-2 were generated by methods described previously (5, 17). In brief, cysteine mutants of mouse claudin-2 were generated by site-directed mutagenesis using the QuikChange kit (Stratagene). cDNA of WT mouse claudin-2 and cysteine mutants were ligated into the retroviral Tet response vector, pRevTREP, and lipofected into the viral packaging line, PT67. Stable transfected cells were selected with hygromycin. Retrovirus encoding mouse claudin-2 was produced and transduced into MDCK I Tet-Off cells, and stable clonal cell lines were selected in hygromycin using cloning cylinders. To induce claudin-2 expression, doxycycline was omitted from the culture medium starting from the day of plating (Dox−), whereas in control plates, 20 ng/ml doxycycline was added to the culture medium to suppress expression (Dox+). Electrophysiological studies were generally performed after 7 days. The expression and localization of claudin-2 was tested by immunofluorescence staining by methods described before (18) using a 1:200 dilution of claudin-2 antibody (Zymed Laboratories Inc.). Double labeling with anti-ZO-1 (Zymed Laboratories Inc.) was used to confirm the correct localization of claudin-2 to the tight junction.
Western Blotting
Claudin-2 protein expression and oligomerization was tested by immunoblotting. Confluent cells, grown on tissue culture dishes, were homogenized in buffer (pH 7) containing 0.25 m sucrose, 30 mm histidine, 1 mm EDTA, and protease inhibitors (Complete Mini; Roche Applied Science). Cell lysates were mixed as indicated with non-reducing loading buffer (pH 6.8; containing 2% (w/v) SDS) or reducing loading buffer (1% (v/v) 2-mercaptoethanol added to non-reducing buffer) and heated for 10 min at 80 °C. The samples were loaded on a 15% denaturing polyacrylamide gel in aliquots of 10 μg of protein and analyzed as described before (18) using claudin-2 antibody (1:1000; Invitrogen). The effect of claudin-2 on the expression of endogenous claudins was assessed by immunoblotting with antibodies to claudin-1, -3, -4, and -7 (Invitrogen).
Affinity Purification of Biotinylated Claudin-2
The accessibility of WT and mutant claudin-2 to MTSEA-biotin was tested as follows. Cells were grown in 6-well plates (10 cm2) for 6 days. Cells were washed with phosphate-buffered saline and incubated on the apical side with 0.5 mg/ml MTSEA-biotin (Toronto Research Chemicals) in phosphate-buffered saline containing 1 mm CaCl2 and MgCl2 for 5 min at 4 °C. The reaction was stopped by washing the cells with phosphate-buffered saline. Cells were harvested and homogenized in radioimmune precipitation assay buffer (pH 8; 50 mm Tris-HCl, 150 mm NaCl, 1% (v/v) Nonidet P-40, 0.5% (w/v) deoxycholic acid, 0.1% (w/v) SDS). Insoluble material was sedimented by brief centrifugation at 16,000 × g, and biotinylated protein was precipitated from the supernatant using streptavidin-coated agarose beads (Pierce) for 1 h at 4 °C. The beads were washed with radioimmune precipitation assay buffer and mixed with reducing loading buffer, and attached protein was released by heating the beads to 80 °C for 10 min. The samples were analyzed by SDS-PAGE as described above using claudin-2 antibody (Invitrogen).
Ussing Chamber Electrophysiological Studies
Ussing chamber diffusion potential measurements to determine ion permeabilities were performed as described recently (5). Cells were plated at confluent density on Snapwell filters (Corning Glass) and cultured for 7 days, which is sufficient to achieve steady-state transepithelial resistance (19). The filters were mounted in Ussing chambers and stirred with gas lifts in 100% O2 at 37 °C. A pair of voltage-sensing electrodes made of Ag/AgCl pellets and current-passing electrodes of Ag wire were immersed into the apical and basolateral medium via agar bridges containing 3 m KCl. The electrodes were connected via head stage amplifiers to a microcomputer-controlled voltage/current clamp (VCC-MC6) and data were acquired at 1-s intervals using Acquire and Analyze software (Physiologic Instruments, San Diego, CA). Data shown in time-independent experiments are usually averages of 3–6 data points.
The standard Ringer solution used at base line contained 150 mm NaCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm glucose, 10 mm Tris-HEPES, at pH 7.4. For measurements of the NaCl dilution potential the medium of the basolateral chamber was replaced by Ringer solution containing 75 mm NaCl (osmolarity adjusted with mannitol). For alkali metal bi-ionic potentials, the basolateral medium was switched to a solution containing a 150 mm concentration of the alkali metal cation-chloride salt. For organic cation diffusion potentials, we used a solution containing 75 mm of the organic cation-chloride salt and 75 mm NaCl. Diffusion potentials were determined as the apical voltage with respect to the basolateral side. Liquid junction potentials at the tip of the agar bridges were estimated with blank filters in place and used to correct the data, as recently described (5). Ion permeability ratios were calculated by the Goldman-Hodgkin-Katz voltage equation. In all calculations, activities rather than concentrations were used, assuming that the activity coefficient of each XCl salt is the same as that of NaCl. The absolute permeabilities, PNa and PCl, were derived by the method of Kimizuka and Koketsu (20) using the equation,
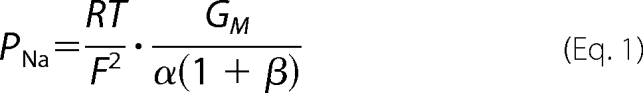 |
where GM represents the monolayer conductance per unit of surface area, α is the ratio of NaCl activity in the apical and the basolateral compartment (aNaCl,ap/aNaCl,bl), and β is the ratio of the permeability of Cl− and Na+ (PCl/PNa).
Unless indicated differently, the conductance and permeability of uninduced (Dox+) were subtracted from measurements of induced (Dox−) cells to determine changes in the transepithelial conductance and permeability attributable to the introduction of claudin-2 and to obtain a quantitative measure of the macroscopic conductance and permeability of claudin-2 paracellular pores.
SCAM Studies
For SCAM studies, methanethiosulfonate (MTS) and p-(chloromercuri)benzenesulfonic acid (pCMBS) solutions were prepared fresh as 100-fold concentrated stock solutions directly before starting the experiment to avoid hydrolysis (16). Unless indicated differently, reagents were added simultaneously to the medium of the apical and basolateral compartment of the Ussing chamber and rapidly mixed by gas lifts. The concentrations of MTSEA and MTSET were adjusted to compensate for differences in their reactivity with thiols in free solution (21). All thiol-reactive reagents were applied in concentrations sufficient to obtain maximum effects on conductance, which were tested in initial experiments by consecutive readdition of reagents. The rate of reaction of MTS and pCMBS with cysteine was derived from the rate of the inhibition of the conductance of claudin-2 pores, assuming that the concentration of MTS remains nearly constant during the reaction so that we can determine a pseudo-first order rate constant by fitting conductance, G, to an exponential decay equation,
with G0 representing the initial conductance and G∞ representing conductance at maximum inhibition. The second order rate constant, k, was determined by dividing the pseudo-first order rate constant k′ by the concentration of the applied reagent.
The dimensions of thiol-reactive reagents were estimated by drawing the molecules in ChemSketch 11.0 (ACD Labs) and measuring molecular distances and radii with the three-dimensional viewer function.
Ca2+ Permeability Measurements
Ca2+ permeability was determined in unidirectional tracer flux experiments across cells grown on Transwell filters. 3 μCi of 45Ca2+ was added to the cis side, and after different time intervals, samples were taken from the basolateral (trans) compartment and analyzed by liquid scintillation counting. The initial rate of flux was used to determine the permeability of filters with cell monolayers (PC = flux/substrate concentration in cis compartment). This was then corrected for the permeability of blank filters, PB, to obtain the true transepithelial permeability (PT), using the following equation.
Statistical Analysis
The data are presented as mean ± S.E. Statistical significance was determined using the unpaired two-tailed Student's t test.
RESULTS
Characterization of Stable Transfected MDCK I TetOff Cells Expressing Cysteine Mutants of Claudin-2
Using site-directed mutagenesis, cysteines were introduced at four different positions of the first extracellular domain of claudin-2 (Table 1). These positions were chosen because mutation of the homologous residues in other claudin isoforms was found either to regulate charge selectivity or to cause familial hypomagnesemia with hypercalciuria and nephrocalcinosis, a genetic disease that affects renal paracellular permeability (22). Each cysteine mutant and WT claudin-2 was transfected into MDCK I TetOff cells, and stable transfected clones were selected. Protein expression and localization of WT and mutant claudin-2 were determined by immunofluorescence staining, together with the tight junction protein ZO-1, and studied by confocal microscopy (Fig. 1; see also supplemental Fig. S1 for magnified orthogonal sections). As reported previously (5), WT claudin-2 was mostly expressed at the tight junction, colocalizing with ZO-1, and also found in other cell compartments as an artifact of overexpression. The subcellular distribution of cysteine mutants of claudin-2 was indistinguishable from WT claudin-2, showing that the substitution of the selected amino acids with cysteine does not affect protein targeting and localization. The inducible expression of WT and mutant claudin-2 was verified by Western blots (Fig. 2A), which showed a characteristic band for claudin-2 in the absence of doxycycline (Dox−) at the expected size of about 22 kDa. Importantly, the induced expression of WT and mutant claudin-2 did not affect the expression of other claudin isoforms endogenously expressed by the host cell line (claudin-1, -3, -4, and -7; supplemental Fig. S2), so that changes in the paracellular transport of induced cells are most likely attributable to claudin-2. For subsequent electrophysiological studies, clones were selected that showed similar levels of claudin-2 expression.
TABLE 1.
Amino acid residues of the first extracellular domain of claudin-2 that were selected for cysteine mutagenesis
| Residue | Rationale for selection (based on observations on homologous residues in other claudin isoforms) | Reference |
|---|---|---|
| Tyr35 | R32D in claudin-10a decreases anion selectivity | Ref. 7 |
| D104S in claudin-16 partially inhibits cation permeation | Ref. 27 | |
| His57 | D55K in claudin-15 converts from cation to anion selectivity | Ref. 6 |
| Q57E in claudin-19 causes familial hypomagnesemia with hypercalciuria and nephrocalcinosis | Ref. 22 | |
| Asp65 | K65D of cation-blocking claudin-4 eliminates discrimination against Na+ | Ref. 6 |
| D65N in claudin-2 abolishes a cation-binding site within the pore lumen | Ref. 5 | |
| Ile66 | Asp65 and Glu63, respectively, in claudin-10b and claudin-16, which are both highly cation-selective, are both negatively charged | |
| E64K in claudin-15 converts from cation to anion selectivity | Ref. 6 |
FIGURE 1.

Characterization of MDCK I TetOff cells, stably transfected with WT claudin-2 and the cysteine mutants Y35C, H57C, D65C, and I66C. The expression and distribution of claudin-2 was tested by double immunofluorescence staining with antibodies to claudin-2 and to the tight junction marker, ZO-1. Confocal en face images of induced cells (Dox−) show that WT and mutant claudin-2 colocalize predominantly with ZO-1 to the tight junction but are also found in the cell bodies and at the apical membrane and, to a lesser extent, along the basolateral membrane (see also supplemental Fig. S1). Scale bar, 10 μm.
FIGURE 2.

Western blot analysis of claudin-2 in cell lysates isolated from cell lines expressing WT claudin-2 and cysteine mutants. A, claudin-2 appears as a band at ∼22 kDa in induced cells (Dox−), whereas expression is suppressed in the presence of doxycycline (Dox+). An additional band at between 40 and 45 kDa was observed with cells induced to express the D65C mutant of claudin-2. (The band at ∼35 kDa present in all lanes represents nonspecific antibody binding.) All samples were prepared with reducing sample buffer, containing 1% (v/v) 2-mercaptoethanol. B, testing the identity of the high molecular mass band at 40–45 kDa. Lysates of D65C and WT cells without pretreatment were prepared for SDS-PAGE using non-reducing sample buffer (lane 1) or reducing sample buffer (lane 2). Alternatively, intact cell monolayers were pretreated with 10 mm dithiothreitol (lane 3) or 110 mm Cu(II)-phenanthroline (lane 4) for 5 min to reduce and oxidize cysteines, respectively. The reagents were vigorously washed off prior to cell lysis, and samples were prepared with non-reducing buffer. The blot shows that preparation of the sample under reducing conditions or pretreatment of the cells with reducing reagent decreases the high molecular mass band.
Interestingly, in addition to the 22 kDa band, a high molecular mass band between 40 and 45 kDa consistently appeared in Western blots of lysates isolated from the cysteine mutant D65C (Fig. 2A). This observation is specific for the substitution of aspartate with cysteine, since such a high molecular mass band was not reported for a recently generated mutant of claudin-2 in which this aspartate residue was replaced by asparagine (5). We therefore tested the identity of this band (Fig. 2B). Interestingly, the intensity of the high molecular mass band was increased, and the signal of the 22 kDa claudin-2 band decreased when cell lysates were prepared under non-reducing conditions (lane 1) compared with reducing conditions (lane 2). Similarly, the pretreatment of cells with 10 mm dithiothreitol led to a shift of claudin-2 from the high molecular mass band to the 22 kDa band (lane 3), whereas the opposite observation was made for cells pretreated with the oxidative reagent Cu(II)-phenanthroline (lane 4). As a control, lysates of WT claudin-2-expressing cells were prepared under the same conditions, and a high molecular mass band was never detected. The data indicate that the introduction of cysteine at position 65 enables individual claudin-2 units to form intermolecular disulfide bonds that could lead to the formation of claudin dimers.
Base-line Permeability and Selectivity of Cysteine Mutants of Claudin-2
The effect of cysteine mutagenesis on the electrophysiological characteristics of claudin-2 was determined in Ussing chamber measurements of conductance and dilution potentials. Fig. 3 shows representative results of two clones of WT claudin-2 and each cysteine mutant, expressed in MDCK I TetOff cells. The expression of WT claudin-2 led to a ∼10-fold increase in conductance (Fig. 3A), which was due to an increase in Na+ permeability (PNa) (Fig. 3B), in agreement with previous observations (5, 13, 14). Cl− permeability (PCl) was also increased (Fig. 3C) but to a lesser extent than PNa, yielding a ratio of PNa/PCl of about 8 (Fig. 3D), consistent with a role of claudin-2 to form cation-selective pores within the tight junction. PNa, PCl, and PNa/PCl of the cysteine mutants, except for D65C, were similar to WT claudin-2, with only minor or inconsistent differences; one of the Y35C clones had a lower permeability, whereas both I66C clones had a slightly higher PNa and conductance compared with WT claudin-2. The most striking differences, however, were observed for D65C, which consistently showed a low conductance, which was due to a low PNa, whereas PCl was increased. Accordingly, PNa/PCl was drastically decreased (Fig. 3D), showing that D65C is much less cation-selective than WT claudin-2. Interestingly, these results are different from data obtained with the D65N mutant of claudin-2, which also showed a decrease in PNa but remained highly cation-selective (5).
FIGURE 3.

Characterization of the electrophysiological properties of claudin-2 by Ussing chamber measurements of conductance and the transepithelial NaCl dilution potential. The figure shows representative data from two independent clones of MDCK I TetOff cells for each WT or cysteine mutant claudin-2. The results were corrected for the base-line conductance and permeability obtained with uninduced (Dox+) cells and represent the macroscopic conductance and permeability of claudin-2 pores. A, conductance of MDCK I TetOff cells induced to express WT and mutant claudin-2. The conductance of uninduced cells was typically <1 millisiemens (not shown). B, Na+ permeability (PNa). C, Cl− permeability (PCl) of claudin-2 pores, calculated from the dilution potential. D, permeability ratio of Na+ to Cl− (PNa/PCl). The diagram shows that D65C is much less cation-selective than WT claudin-2 and the other cysteine mutants. Data points represent means of 3–4 filters ± S.E. p values refer to statistically significant differences in comparison with WT claudin-2. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Next, we measured bi-ionic diffusion potentials to test whether mutation of claudin-2 affected the relative permeabilities of different alkali metal ions (PM+) and hence the Eisenman sequence (23). The Eisenman sequence can be used as a measure of the strength of the electrostatic interaction between charged binding site(s) within a channel or pore and permeating ions. WT claudin-2 exhibits a relatively high permeability for small cations, such as Li+, that have a high charge density and, consequently, large hydration shell, and low permeability for large, poorly hydrated cations, such as Cs+. We interpret this as indicating that the claudin-2 pore is narrow so that cations permeate in a partially dehydrated form and that there is a strong negatively charged intrapore binding site that is able to offset the high energetic cost of dehydration for small cations (5). The relative permeabilities of alkali metal ions (normalized to PNa; Fig. 4A) in Y35C, H57C, and I66C were identical to WT claudin-2. In contrast, the relative permeabilities of larger cations (K+, Rb+, and Cs+) were increased in D65C compared with WT, whereas that of Li+ was slightly decreased (Fig. 4B) so that the permeability sequence was similar to the sequence of free solution mobilities of these ions (24). We know that this is not solely due to loss of the negatively charged binding site at Asp65, because neutralizing this site by mutating it to asparagine (D65N) only decreases relative Li+ permeability but does not affect the permeability to larger cations, as we have previously shown (5). The data suggest that cysteine mutagenesis of Asp65 widens the pore, allowing the relatively unhindered diffusion of hydrated ions.
FIGURE 4.

Ussing chamber measurements of bi-ionic diffusion potential to determine the permeabilities of WT and mutant claudin-2 for alkali metal ions (Eisenman sequence) and nitrogenous cations. A, the permeabilities of WT claudin-2, Y35C, H57C, and I66C to alkali metal ions are plotted as relative data compared with Na+ permeability. B, relative permeabilities of D65C and D65N to alkali metal ions in comparison with WT claudin-2, showing an increase in the relative permeabilities of D65C to larger cations. C, relative permeabilities of the nitrogenous cations, methylamine (MA), ethylamine (EA), tetramethylammonium (TMA), tetraethylammonium (TEA), and N-methyl-d-glucamine (NMDG). All were increased for D65C compared with WT claudin-2. The permeabilities of MA, EA, and TMA were fitted to a Renkin equation by non-linear regression to obtain the pore diameter, which was found to be as follow (±S.E.): 7.0 ± 0.8 Å (WT), 6.9 ± 0.1 Å (Y35C), 6.7 ± 0.1 Å (H57C), 10.2 ± 1.2 Å (D65C), 6.6 ± 0.3 Å (I66C). Data points represent means of four filters ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001. p values refer to statistically significant differences in comparison with WT claudin-2.
This hypothesis was tested by measurements of the permeabilities (PM+) to larger organic cations. The relative permeabilities (PM+/PNa) of five nitrogenous cations, methylamine, ethylamine, tetramethylammonium, tetraethylammonium, and N-methyl-d-glucamine, were similar between the cysteine mutants, Y35C, H57C, and I66C, and WT claudin-2 (Fig. 4C). However, D65C had a higher permeability to nitrogenous cations with a selectivity sequence that was similar to their relative mobilities in free solution (25). The diameters of WT and mutant claudin-2 pores were estimated by fitting the permeabilities of methylamine, ethylamine, and tetramethylammonium to a Renkin equation, as described previously (5). The pore diameters obtained for WT claudin-2 and Y35C, H57C, and I66C fell within a narrow range of 6.6–7 Å, similar to previous estimates for WT claudin-2 (4, 5). However, D65C had a significantly larger pore diameter of 10.2 ± 1.2 Å. In summary, the results show that the base-line permeability characteristics of cysteine mutants of claudin-2 are similar to those of WT claudin-2, with the exception of D65C, which shows a dramatic loss in charge selectivity, change in alkali metal Eisenman sequence, and widening of the pore.
Screening for the Accessibility of Engineered and Endogenous Cysteines to Thiol-reactive Reagents
In the next set of experiments, we analyzed the structure of the first extracellular domain of claudin-2 by testing the accessibility of engineered cysteines to membrane-impermeant thiol-reactive reagents that covalently bind to free cysteines. The thiol-reactive reagents that we used in this study were MTS molecules of different size and charge and the mercury derivative, pCMBS. The chemical structure, size, and charge of each reagent are listed in Table 2. A basic assumption is that only reduced cysteines that are accessible from the extracellular aqueous phase can react with these, mostly lipid-insoluble, reagents. Claudin-2 also contains two conserved endogenous cysteines in the first extracellular domain of unknown function that could potentially undergo reaction.
TABLE 2.
Structure, size, charge, and working concentration of thiol-reactive reagents used in this study*

* Reagents are listed in order of net charge and increasing size.
† Determined by reaction with β-mercaptoethanol, from Table II in Ref. (16). n/a, not available.
First, we tested whether thiol-reactive reagents modify the conductance of WT and mutant claudin-2 pores. Representative data of time-dependent changes in conductance upon the addition of MTS or pCMBS in induced or non-induced cells are shown in Fig. 5, and the quantitation of these effects is summarized in Fig. 6A. The positively charged molecule, MTSET, induced a rapid decrease in the conductance of I66C (∼25%) and, to a much lesser extent, of H57C but had only very small effects on WT claudin-2 or the other cysteine mutants (Fig. 5A). Similarly, the smaller positively charged reagent, MTSEA, strongly decreased the conductance of I66C but induced only a slight decrease in the conductance of WT and the other mutants (Fig. 5B). All of the reagents we tested had little or no effect on the conductance of uninduced (Dox+) cells, thus confirming that these reagents modulate the conductance of claudin-2 and not of endogenous paracellular or transcellular transport proteins. Moreover, the lack of effect of these reagents on WT claudin-2 indicates that their effects on I66C were not due to their reaction with the endogenous cysteines in the first extracellular domain but were specific for the engineered cysteine at position 66.
FIGURE 5.

Time-dependent measurements of conductance of WT and mutant claudin-2 and effects of thiol-reactive reagents. MTS and pCMBS were added to both sides of the cell monolayers mounted in Ussing chambers. The graphs show representative data of induced and uninduced cells incubated with MTSET (1 mm) (A), MTSEA (2.5 mm) (B), MTSES (5 mm, only WT and I66C shown) (C), and pCMBS (0.5 mm, only WT and I66C shown) (D). In all measurements, the effects on uninduced cells were very small.
FIGURE 6.

A, quantification of SCAM conductance measurements. The diagram shows the relative changes in conductance of WT and mutant claudin-2 induced by different MTS reagents. The data are means of three filters ± S.E. of representative experiments, measured 3 min after the addition of MTS. B, testing the reaction of MTSES and MTSACE with I66C. Cells expressing I66C were preincubated with either MTSES or MTSACE for 5 min prior to the addition of MTSEA. Control cells were not preincubated. Preincubation with either MTSES or MTSACE largely prevented the inhibition of conductance by MTSEA. C, Western blot of claudin-2 to test the reaction of endogenous and engineered cysteines with MTSEA-biotin. Intact cell layers were incubated with MTSEA-biotin, and cells were homogenized with radioimmune precipitation buffer. Samples were taken from the whole cell lysate to test overall claudin-2 expression (bottom). Biotin-labeled proteins were affinity-purified with streptavidin-coated agarose beads, and biotinylated claudin-2 was detected using a claudin-2-specific antibody (top).
We also tested thiol-reactive reagents that were negatively charged or uncharged. Like MTSET and MTSEA, the negatively charged reagent, pCMBS, strongly decreased the conductance of I66C but did not affect WT claudin-2 (Fig. 5D). In contrast, another negatively charged reagent, MTSES, initially increased the conductance of I66C slightly, followed by a modest decrease. The initial slight increase in conductance was also observed with WT and the other claudin-2 mutants (Fig. 5C, only WT and I66C shown). The neutral reagent, MTSEA-biotin, reduced the conductance of I66C by 27%, whereas the neutral MTSACE had minimal effects on I66C that were no different from WT (Fig. 6A).
The lack of effect of MTSACE and MTSES on I66C could be because the reagents were unable to gain access to the cysteine at position 66. Alternatively, they could gain access and react covalently with this cysteine but nevertheless fail to inhibit conductance through the pore. To distinguish between these two possibilities, we tested whether MTSACE or MTSES protected the cysteine from MTSEA. I66C-expressing cells were preincubated with MTSACE or MTSES for 5 min prior to the addition of MTSEA (Fig. 6B). As shown before, MTSEA induced a rapid and large decrease in conductance of control cells that had not been preincubated. By contrast, pretreatment with either MTSACE or MTSES largely prevented the subsequent inhibition of conductance by MTSEA. This suggests that MTSES and MTSACE do react with the cysteine at position 66 and that their presence does not affect base-line conductance but does protect this cysteine from subsequent reaction with MTSEA.
Apart from I66C, the thiol-reactive reagents had minimal effects on the other claudin-2 cysteine mutants. Again, this could be explained by two possible hypotheses; either these cysteines are inaccessible to the reagents, or covalent binding of the reagents does not influence conductance (e.g. if the cysteine does not line the permeation pathway). Because none of the thiol-reactive reagents had functional effects on conductance of these other mutants, we could not use a cysteine protection experiment. Thus, to distinguish between these two hypotheses we used a biochemical approach in which MTSEA-biotin was used to label accessible cysteines in intact cell layers. The reaction was followed by immunoprecipitation of biotinylated proteins from the cell lysate with streptavidin beads and analysis of biotinylated claudin-2 by Western blots. In parallel, whole cell lysates were prepared to quantify the overall expression of claudin-2 in different clones. As expected, I66C was biotinylated, whereas WT claudin-2 showed minimal biotinylation Fig. 6C). Interestingly, the other mutants were also biotinylated. Remarkably, the strongest signal of biotinylated claudin-2 was obtained with Y35C. Normalized to the overall amount of claudin-2 expression, the signal for Y35C was almost 50 times stronger than for WT and 6 times stronger than for I66C, respectively. This result contrasts with the conductance measurements in which the response of Y35C to various MTS reagents was minimal (Figs. 5 and 6A). The data suggest that engineered Cys35 is located outside of the pore or at the pore entrance and able to react with large molecules, such as MTSEA-biotin, without affecting ion conductance.
Kinetics of Reaction of MTS with I66C
In the following experiments, we analyzed the rate at which modification of I66C by MTS reagents leads to the observed changes in conductance. We focused on I66C due to its large response to MTS compared with other cysteine mutants.
The overall rate of reaction between accessible cysteines and MTS is dependent on the rate of diffusion of the MTS reagent through the unstirred layers at the cell surface, the rate of its diffusion through the aqueous paracellular pore to the site of reaction, and its rate of covalent reaction with the thiol group of the substituted cysteine. Fig. 7A shows the time course of inhibition of the conductance of I66C by four MTS reagents differing in size and charge: MTSEA, MTSET, MTSPTrEA, and MTSEA-biotin. For reagents for which the rate constant for reaction with free thiol groups was known, the concentration was adjusted accordingly. Thus, MTSEA was added in a 2.5-fold higher concentration than MTSET to compensate for its 2.5-fold lower second order rate constant (Table 2) (16). From these data, the pseudo-first order rate constant of inhibition, k′, was determined by non-linear regression. The second order rate constant, k, was obtained by dividing k′ by the concentration of the reagent (Fig. 7B). The data showed that with increasing size of the reagent, the maximum amount of inhibition tended to increase (with the exception of MTSEA-biotin), whereas the reaction rate constant decreased (k, mean ± S.E. from n = 3): 105 ± 21 m−1 s1 (MTSEA), 53.5 ± 8.7 m−1 s1 (MTSET), 40.1 ± 3.6 m−1 s1 (MTSPTrEA), and 10.8 ± 1.6 m−1 s1 (MTSEA-biotin).
FIGURE 7.

Kinetics of the inhibition of conductance of I66C by different MTS reagents. Cationic MTS reagents of increasing size, MTSEA, MTSET, and MTSPTrEA, and the uncharged reagent, MTSEA-biotin, were used. A, raw data showing change in normalized conductance with time after adding the MTS reagent. B, relationship between the molecular mass of the reagent and the second order rate constant, k.
Modification of Paracellular Pores of I66C by Thiol-reactive Reagents
The next set of experiments was designed to investigate the mechanism by which the conductance of I66C is inhibited by thiol-reactive reagents. This could be due either to a partial or complete steric blocking of the pore or to electrostatic effects inhibiting ion permeation. To distinguish between these possibilities, we performed four different experiments.
First, the effect of thiol-reactive reagents of different size and charge on PNa and PCl was determined by measuring dilution potentials. Fig. 8 summarizes the effects of MTSET, MTSEA, MTSEA-biotin, and pCMBS on the conductance, PNa, and PCl of I66C. The reagents caused a 20% (MTSET) to 40% (pCMBS) decrease in conductance (Fig. 8A), which correlated with similar changes in both PNa (Fig. 8B) and PCl (Fig. 8C). The observation that the decrease in ion permeability was similar regardless of the net charge on the thiol-reactive reagent and that none of the reagents altered the charge selectivity of claudin-2 pores argues strongly against an electrostatic effect of the covalently bound reagent on permeant ions.
FIGURE 8.

Ussing chamber experiments with I66C to determine the effect of thiol-reactive reagents of different size and charge, MTSEA, MTSET, and MTSEA-biotin pCMBS, on conductance (A), Na+ permeability (PNa) (B), and Cl− permeability (PCl) (C) of claudin-2 pores. The MTS- and pCMBS-induced decreases in conductance correlate with similar changes in PNa and PCl. Data points represent means of four filters ± S.E. The data are corrected for measurements with uninduced cells. *, p < 0.05; **, p < 0.01; ***, p < 0.001. p values refer to statistically significant differences compared with control cells.
Second, the Ca2+ permeability of MTSET-treated and -untreated cells was determined by radiotracer flux assays. Electrostatic changes would be expected to exert disproportionately larger effects on divalent ions compared with monovalent ions. MTSET induced a decrease in PCa by about 50% (Fig. 9A), which is approximately double that observed for Na+. This is a relatively small difference (compare the effect of abolishing a single negatively charged binding site at Asp65, which decreased PCa by 95% (5)) that can be attributed to the slightly larger radius of Ca2+ (0.99 Å, compared with 0.97 Å of the dehydrated Na+ cation).
FIGURE 9.

Testing electrostatic effects created by positively charged MTS after reaction with I66C. A, measurements of Ca2+ permeability (PCa) of control and MTSET-treated cells, grown on Transwell filters, by tracer flux experiments (n = 4). B, to unmask surface charge effects, the dependence of conductance on ionic strength was determined in Ussing chambers. The NaCl activity of the medium of control and MTSEA-treated cells was gradually increased from 0 to 90 mm, replacing with mannitol as needed to maintain osmolality (n = 3). ***, p < 0.001, compared with control.
Third, electrostatic interactions at the entrance of a pore or channel between charged amino acid residues and ions, so-called surface charge effects, can be identified by decreasing the ionic strength of the solution and become detectable at low ion concentrations as deviations from the Michaelis-Menten relationship between conductance and ion concentration (26). We performed measurements of the conductance-concentration relationship of I66C at varying NaCl concentrations to identify potential surface charge effects created by the headgroup of MTSEA. Fig. 9B shows that inhibition of conductance by MTSEA did not affect the relationship between conductance and the NaCl activity of the medium, which is normally linear within physiologic range (5).
Finally, we measured the permeability of different alkali metal cations to test if covalent modification of I66C by MTS or pCMBS affects the Eisenman sequence of claudin-2 pores. We previously showed that cation permeation through claudin-2 pores requires a negatively charged binding site at Asp65 (5). If the positively charged MTS reagent neutralized the charge at this binding site, one would expect that the reduction in cation conductance would be accompanied by a disproportionate reduction in the smallest and most heavily hydrated cations (Li+ and Na+) and a shift in the Eisenman sequence to a lower number (24). Instead, treatment with MTSET actually led to a disproportionately larger decrease in the relative permeability of larger cations, K+, Rb+, and Cs+, and less inhibition of Li+ and Na+ permeability (Fig. 10A). Similar results were obtained with MTSEA (not shown). Taken together, these four experiments exclude electrostatic interaction between covalently bound thiol-reactive reagents and permeant ions as the mechanism of inhibition.
FIGURE 10.

Bi-ionic diffusion potential measurements to determine the effect of different MTS reagents and pCMBS on the relative cation permeability properties of I66C. The data were plotted as relative data compared with PNa. A, effect of MTSET on the relative permeabilities of alkali metal cations (Eisenman sequence) and nitrogenous cations of different diameter. Shown are the effects of MTSEA-biotin (B) and pCMBS (C) on the Eisenman sequence of I66C. Data points represent means of four filters ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001. p values refer to statistically significant differences compared with untreated control cells.
The most plausible interpretation of the effect of MTSEA and MTSET on the alkali cation permeabilities is that these reagents partially block the permeation pathway for the dehydrated cation in a charge-independent manner. Thus, cations with larger unhydrated ionic radius are disproportionately inhibited. Consistent with this, MTSET also caused a disproportionate inhibition of the large organic cation, methylamine (radius 1.89 Å), and there was a trend toward a similar effect for ethylamine (radius 2.29 Å) (Fig. 10A).
A prediction of this model is that if we use a reagent sufficiently large to completely block the pore, then permeation of cations would be inhibited equally, regardless of their size. Consistent with this, although the larger neutral reagent, MTSEA-biotin, also disproportionately inhibited the larger cations more than the smaller ones (Fig. 10B), the effect was less than for MTSET. Finally, pCMBS, which is also large but much more rigid than the MTS reagents and showed the greatest inhibition of Na+ conductance, showed no size selectivity at all (Fig. 10C). Our results are consistent with a model in which thiol-reactive reagents act to inhibit the I66C claudin-2 pore conductance by steric block of the permeation pathway for dehydrated ions.
DISCUSSION
Effect of Cysteine Substitutions on Base-line Properties of Claudin-2
Here, we describe for the first time the use of cysteine scanning to investigate the structure of paracellular pores. The introduction of cysteines at various positions of the first extracellular domain did not affect the subcellular localization of claudin-2 compared with the WT protein, and the expression of exogenous claudin-2 did not alter the expression of endogenous claudin isoforms. Recent studies from our group and others have shown that claudin pores are charge-selective (PNa/PCl ∼ 8) and small (∼6.5–8-Å diameter) (4, 5). Our data show that the substitution of Tyr35, His57, and Ile66 with cysteine does not affect these properties, suggesting that none of these residues in claudin-2 is directly involved in the mechanism of ion permeation and that their substitution does not lead to changes in protein conformation. The conductance of some clones differed from that observed for WT claudin-2, which was most likely an artifact of clone-dependent variations in claudin-2 expression. In contrast to our results, the substitution of homologous residues in other claudin isoforms, including claudin-4, -10a, -10b, -15, and -16 (6, 7, 27) (Table 1), led to drastic changes in ion permeability and, in some cases, even to reversal of charge selectivity. This could reflect isoform-dependent differences in the localization and role of individual residues between different claudins.
Asp65 Is Located near an Intermolecular Interface
In contrast to the other cysteine mutants, D65C showed a striking loss in both charge and size selectivity and an increase in the pore diameter. The unusual properties of D65C pores can be explained by two mechanisms. First, Asp65 is a site for electrostatic interaction that binds to permeating dehydrated cations. Accordingly, the substitution of Asp65 with cysteine or, as described previously, asparagine (5), would cause a decrease in the paracellular permeability of cations and reduction in charge selectivity. Second, the increase in the pore diameter permits passage of larger ions, perhaps even in their hydrated form. Our data show that the cysteine at position 65 forms intermolecular disulfide bonds. We suggest that this causes a conformational change in the extracellular fold, leading to the observed increase in paracellular pore diameter.
The spontaneous or chemically induced formation of disulfide bridges between engineered cysteines has been widely used to identify protein oligomerization and sites of protein-protein interaction (28, 29). There is already accumulating evidence for oligomeric protein assembly of claudins. Some claudins, for instance, appear as oligomers, up to hexamers, in perfluorooctanoic acid-PAGE (30, 31). Blasig et al. (9) used fluorescence resonance energy transfer to demonstrate the self-association of claudin-5, which seems to be mediated by trans-dimerization of the second extracellular domain (11). Our data support a model in which individual claudin units are in sufficiently close proximity to interact with their extracellular domains to form dimers. For a disulfide bond to form, the Cys65 β-carbons of two adjacent claudin-2 molecules, which are predicted to face into the pore lumen, must be separated by less than 7 Å (32). However, it is possible that they could be further apart if there is sufficient flexibility and motion in the region of the protein where the cysteines are located. Indeed, the fact that disulfide bonding leads to conformational changes supports this idea. It is not clear at this point whether D65C forms disulfide bridges with another claudin-2 molecule (homodimerization) or if it interacts with endogenous cysteines of other claudin isoforms to form heterodimers and if this interaction occurs within the same membrane (cis) or between adjacent membranes (trans). These questions will be addressed in future experiments by co-immunoprecipitation and co-cultures of cells expressing differently tagged claudins.
Endogenous Extracellular Cysteines Are Unavailable for Modification by Thiol-reactive Reagents
SCAM was used to study the structure of the first extracellular domain of claudin-2. This method was developed for structural and functional analysis of channels and transporters of the transcellular pathway (15). We assume that only cysteines that are located at the water-exposed surface of the protein and accessible from the bulk solution can react with hydrophilic thiol-reactive reagents. Lack of reaction indicates that cysteines are buried in the protein interior, sterically blocked, or in an oxidized state (16). Claudin-2 contains two endogenous cysteines in the first extracellular domain that are conserved among all claudin isoforms. It has been suggested that the conserved cysteines form an intramolecular disulfide bond (3), although this has not been supported yet by experimental evidence. MTSEA slightly increased the conductance of WT claudin-2 (Fig. 5). This result should be regarded somewhat skeptically, since a considerable fraction of MTSEA is deprotonated and, hence, membrane-permeable at physiologic pH, which enables MTSEA to react with cysteines from the cytosolic side. MTSES slightly increased the conductance of WT claudin-2. This could indicate a reaction with the endogenous cysteines and increase of cation permeation by electrostatic interaction with the negatively charged headgroup of MTSES. On the other hand, WT claudin-2 was only very weakly labeled by MTSEA-biotin, which argues against accessibility of the endogenous cysteines (Fig. 6C). We conclude that SCAM studies on claudin-2 do not require elimination of endogenous cysteines, since the effects of MTS on WT claudin-2 are only minor.
Tyr35 Is Located outside the Pore and Exposed at the Surface
Three of four cysteine mutants, Y35C, H57C, and D65C, showed only small to moderate responses to the addition of MTS in conductance measurements. However, Y35C strongly reacted with MTSEA-biotin, proving that engineered C35 is easily accessible to this bulky reagent. The results suggest that Tyr35 in claudin-2 does not line the lumen of the pore but is most likely located outside of the pore or at the pore entrance. In the claudin-2 sequence, Tyr35 is much more N-terminally located than the other residues that we tested, suggesting that pore-lining residues, including Asp65 and Ile66 (see below), are more C-terminally located. This is also consistent with observations by the Anderson laboratory (6, 7) that residues that seemed to influence the charge selectivity of ion permeation are mostly located in the C-terminal half of the first extracellular domain. H57C was inhibited slightly by MTSET but to a lesser extent than I66C and was also labeled with MTSEA-biotin but less than Y35C. Furthermore, MTSET decreased the conductance of H57C more than the smaller MTSEA, which had minimal effect. These observations suggest that, unlike Tyr35, His57 is located in the pore but in a wider part than Ile66, perhaps in a vestibule. Because D65C formed disulfide bonds, its MTS reactivity was not informative. However, our previous observations already suggest that it is located in a narrow part of the pore (5).
Ile66 Is Buried in a Narrow Part of the Pore
The most striking effects on conductance by thiol-reactive reagents were observed with I66C, so these are discussed in detail. The majority of MTS reagents that we tested, as well as pCMBS, significantly inhibited the conductance of I66C. This strongly suggests that the engineered Cys66 lines the aqueous lumen of the paracellular pore so that covalent binding of various thiol-reactive reagents impedes ion permeation. Alternatively, the reaction of MTS and pCMBS with Cys66 may lead to changes in gating of claudin-2 pores and a decrease in the macroscopic conductance. The latter explanation seems less likely for three reasons. First, gating mechanisms such as ligand-controlled gating have not been reported for the paracellular pathway. Second, we show that modification of Cys66 by MTS reagents alters the size selectivity of the pore. Third, Ile66 lies adjacent to Asp65, which has already been identified to be a binding site for permeating cations located inside the claudin-2 pore. Accordingly, Ile66 is much more likely to also be located in a pore-lining segment of the first extracellular domain rather than in a gating-controlling segment, if such a domain should exist.
The rate of reaction of MTS reagents of different size with Cys66 sheds some light on its position within the pore. The overall rate of reaction is dependent on three steps: 1) diffusion of the MTS reagent through unstirred layers to the cell surface, 2) diffusion through the paracellular pore to the site of the target cysteine, and 3) covalent reaction with the thiol group. The rates of chemical reaction of MTSEA and MTSET with free thiol groups (step 3) (16) have been determined by Karlin and Akabas and are 700 and 4000 times faster, respectively, than the overall reaction rates we observed (Table 2). We have estimated the unstirred layer thickness and, from this, estimated that the rate of diffusion of MTS regents through this layer (step 1) is substantially faster than the overall rate of reaction that we observed (supplemental material). Furthermore, the decrease in rate of reaction that we observed with increasing molecular size was much greater than would be expected for simple diffusion through an aqueous unstirred layer. We therefore conclude that the rate-determining step is diffusion of the MTS reagents within the claudin-2 pore (step 2). Thus, the steep dependence of the overall rate on the molecular size of the reagent (Fig. 7A) suggests that Cys66 is located within a relatively narrow portion of the pore so that access to it by the larger MTS reagents is sterically constrained.
If Cys66 is indeed buried in the narrow part of the pore, the size and shape of the molecules that can react with it may reveal information about the geometry of the access pathway. Molecules like MTSEA, MTSET, and pCMBS are small enough to fit into the narrowest part of claudin-2 pores (diameter 6.5–7 Å) (5) unless permeation is restricted by other factors (e.g. charge selectivity). However, Cys66 also reacts with larger reagents, MTSPTrEA and MTSEA-biotin, whose dimensions exceed the pore diameter. The dimensions of MTSPTrEA are about 13.7 × 8 Å, with the triethylammonium group being the widest part of the molecule and about 6 Å away from the reactive disulfide. MTSEA-biotin consists of the flexible MTS-derivate chain with dimensions of about 15 × 4.8 Å connected to a biotin group of about 9 × 7 Å. We therefore hypothesize that the claudin-2 pore has a conical shape. We envision that Ile66 in WT claudin-2 is located in the narrow region of the pore accessible through an opening of at least 4.8 Å in diameter, which opens up to at least 8–9 Å to incorporate the bulky triethylammonium group of MTSPTrEA and the biotin of MTSEA-biotin.
Thiol-reactive Reagents Inhibit I66C by Steric Blocking
Our data suggest that covalent modification of Cys66 leads to a steric blocking of the pore and that changes in the electrostatic interactions of the thiol-reactive reagent with permeant ions are of minor importance. This is supported by our findings that 1) cationic, neutral, and anionic thiol-reactive reagents decrease the conductance of I66C without affecting the charge selectivity of the pore, and 2) we could not find evidence for surface charge effects by charged MTS. It is not clear at this point why MTSES and MTSACE fail to significantly block ion permeation, although these molecules have dimensions comparable with those of MTSEA and MTSET. Our data show that both reagents block the access of MTSEA, but it is unclear if they directly react with Cys66 or if accessibility is blocked by another mechanism (e.g. by reaction with other proteins of the tight junction complex). Such differences in the effect of thiol-reactive reagents of similar size, charge, and/or chemical properties have also been observed by many investigators in other SCAM experiments and are probably related to differences in their shape and orientation. For example, Xu and Akabas (33) measured strong inhibition of conductance of multiple pore-lining cysteine mutants of the γ-aminobutyric acid type A receptor α1 unit with pCMBS but not with MTSES. They postulated that this difference might exist because MTSES is quite flexible, whereas the benzene ring in pCMBS makes it rigid. Thus, in MTSES the sulfonate group on one end can potentially rotate to a position adjacent to the reactive thiosulfonate on the other end and inhibit its reactivity with cysteine thiol groups.
Interestingly, thiol-reactive reagents do not completely inhibit claudin-2 pores but decrease conductance only by 25% (MTSET and MTSEA) to 45% (pCMBS). One possible explanation is that only a subpopulation of all pores (25% in the case of MTSET) is inhibited. Such a scenario could be possible if claudin-2 participated in the formation of different subpopulations of pores that differ in structure and hence accessibility to MTS (e.g. homotypic and heterotypic pores formed between claudin-2 and other claudin isoforms). It is also possible that all pores are equally inhibited, but for each one the unitary conductance is reduced by only 25%. Also, a combination of both scenarios could lead to the macroscopic observations. Zhou et al. investigated a similar problem when studying channel-lining residues in hemichannels of gap junctions, expressed in Xenopus oocytes (34). Using a large maleimide derivate, maleimido-butyryl biocytin, they observed ∼40% inhibition of membrane conductance and inhibition of the flux of a large but not of a small tracer molecule. The authors concluded that maleimido-butyryl biocytin partially blocks the hemichannels, leading to an incomplete inhibition of the conductance of each channel. Similarly, we observed that MTSET and MTSEA-biotin disproportionately affect the permeability of claudin-2 pores to larger cations (Fig. 10, A and B), which supports the idea of a partial block of each pore rather than the possibility that just a subpopulation of pores is inhibited. Interestingly, such a disproportionate effect on larger cations was not observed with pCMBS. This reagent differs substantially from MTS derivates in terms of molecular structure and flexibility. Therefore, pCMBS might inhibit the conductance of claudin-2 pores by another mechanism. Further studies, including experiments that address the activation energy of ion permeation, may be needed to clarify how MTS reagents modify paracellular pores. A partial blocking of the pore should reveal itself by an increase in the activation energy of ion permeation. However, showing this will be experimentally challenging, since this degree of decrease in conductance (e.g. by 25%) is predicted to be caused by a very small increase in activation energy (RT ln(1.25) = 0.14 kcal/mol).
Further Evidence That Claudin Pores Are Oligomeric
Our biotinylation results also suggest that the claudin pore is oligomeric. Although I66C was clearly labeled by MTSEA-biotin, a much stronger signal was obtained with Y35C (Fig. 6C). This result at first sight seems puzzling, since our conductance measurements showed a robust reaction between MTSEA-biotin and I66C. These observations can be explained by a multimeric model of the claudin-2 pore. In this model, MTSEA-biotin has unhindered access to engineered Cys35, so multiple molecules can bind to a single pore complex. By contrast, Cys66 is located in a narrow part of the pore, so binding of only one bulky MTSEA-biotin molecule may be allowed. This would be sufficient to sterically block access to the other Cys66 residues inside of the pore, and hence, less biotin-labeled claudin-2 would be detected in the streptavidin pull-down assay. Fig. 11 summarizes our data in a model depicting the putative location of each residue relative to the claudin-2 pore.
FIGURE 11.

Model of claudin-2 showing the putative location of the mutated residues in the first extracellular domain. The pore is hypothesized to be a homomultimer (residues from 2–3 subunits are shown). Wild-type claudin-2 (WT) is depicted in the left panel, and the consequences of cysteine mutagenesis are shown in the other panels. D65 is located in the narrowest part of the pore facing the lumen and close to an intersubunit interface, so that the D65C mutation leads to dimerization by disulfide bonding. Ile66 is also within the pore facing the lumen, but residues from neighboring subunits are further apart, so that disulfide bonding in I66C is precluded. Thiol-reactive MTS reagents (green) enter the pore to react with I66C, partially blocking the pore to ion permeation. Only a single MTS molecule is accommodated in each pore. Tyr35 is outside the pore facing extracellularly. Thus, reaction of MTS reagents with Y35C does not block the pore. Furthermore, because there is no steric restriction, every Y35C residue can react with an MTS molecule, so that multiple MTS molecules are associated with each pore multimer.
In conclusion, we have, for the first time, applied SCAM to study paracellular transport and started to map the structure of claudin pores. Our data suggest that claudin pores are oligomeric and probably conical in shape, that Ile66 faces the lumen in a narrow region of the pore, and that covalent modification of engineered Cys66 with thiol-reactive reagents leads to steric blocking of the pore.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants DK062283 (to A. Y.) and DK48522 (to the USC Center for Liver Diseases, for the Confocal Microscopy Sub-Core). This work was also supported by a postdoctoral research grant by the National Kidney Foundation (to S. A.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
- MDCK
- Madin-Darby canine kidney
- SCAM
- substituted cysteine accessibility method
- WT
- wild-type
- MTS
- methanethiosulfonate
- pCMBS
- p-(chloromercuri)benzenesulfonic acid
- MTSEA
- (2-aminoethyl) methanethiosulfonate
- MTSET
- [2-(trimethylammonium)ethyl]methanethiosulfonate
- MTSES
- (2-sulfonateoethyl)methanethiosulfonate
- MTSPTrEA
- [3-(triethylammonium)propyl]methanethiosulfonate
- MTSACE
- [2-(aminocarbonyl)ethyl]methanethiosulfonate
- MTSEA-biotin
- N-biotinylaminoethyl methanethiosulfonate.
REFERENCES
- 1.Furuse M., Fujita K., Hiiragi T., Fujimoto K., Tsukita S. (1998) J. Cell Biol. 141, 1539–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsukita S., Furuse M. (2000) J. Cell Biol. 149, 13–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Itallie C. M., Anderson J. M. (2006) Annu. Rev. Physiol. 68, 403–429 [DOI] [PubMed] [Google Scholar]
- 4.Van Itallie C. M., Holmes J., Bridges A., Gookin J. L., Coccaro M. R., Proctor W., Colegio O. R., Anderson J. M. (2008) J. Cell Sci. 121, 298–305 [DOI] [PubMed] [Google Scholar]
- 5.Yu A. S., Cheng M. H., Angelow S., Günzel D., Kanzawa S. A., Schneeberger E. E., Fromm M., Coalson R. D. (2009) J. Gen. Physiol. 133, 111–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colegio O. R., Van Itallie C. M., McCrea H. J., Rahner C., Anderson J. M. (2002) Am. J. Physiol. Cell Physiol. 283, C142–C147 [DOI] [PubMed] [Google Scholar]
- 7.Van Itallie C. M., Rogan S., Yu A., Vidal L. S., Holmes J., Anderson J. M. (2006) Am. J. Physiol. Renal Physiol. 291, F1288–F1299 [DOI] [PubMed] [Google Scholar]
- 8.Alexandre M. D., Jeansonne B. G., Renegar R. H., Tatum R., Chen Y. H. (2007) Biochem. Biophys. Res. Commun. 357, 87–91 [DOI] [PubMed] [Google Scholar]
- 9.Blasig I. E., Winkler L., Lassowski B., Mueller S. L., Zuleger N., Krause E., Krause G., Gast K., Kolbe M., Piontek J. (2006) Cell Mol. Life Sci. 63, 505–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daugherty B. L., Ward C., Smith T., Ritzenthaler J. D., Koval M. (2007) J. Biol. Chem. 282, 30005–30013 [DOI] [PubMed] [Google Scholar]
- 11.Piontek J., Winkler L., Wolburg H., Müller S. L., Zuleger N., Piehl C., Wiesner B., Krause G., Blasig I. E. (2008) FASEB J. 22, 146–158 [DOI] [PubMed] [Google Scholar]
- 12.Enck A. H., Berger U. V., Yu A. S. (2001) Am. J. Physiol. Renal Physiol. 281, F966–F974 [DOI] [PubMed] [Google Scholar]
- 13.Furuse M., Furuse K., Sasaki H., Tsukita S. (2001) J. Cell Biol. 153, 263–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amasheh S., Meiri N., Gitter A. H., Schöneberg T., Mankertz J., Schulzke J. D., Fromm M. (2002) J. Cell Sci. 115, 4969–4976 [DOI] [PubMed] [Google Scholar]
- 15.Akabas M. H., Stauffer D. A., Xu M., Karlin A. (1992) Science 258, 307–310 [DOI] [PubMed] [Google Scholar]
- 16.Karlin A., Akabas M. H. (1998) Methods Enzymol. 293, 123–145 [DOI] [PubMed] [Google Scholar]
- 17.Yu A. S., Enck A. H., Lencer W. I., Schneeberger E. E. (2003) J. Biol. Chem. 278, 17350–17359 [DOI] [PubMed] [Google Scholar]
- 18.Angelow S., El-Husseini R., Kanzawa S. A., Yu A. S. (2007) Am. J. Physiol. Renal. Physiol. 293, F166–77 [DOI] [PubMed] [Google Scholar]
- 19.Angelow S., Kim K. J., Yu A. S. (2006) J. Physiol. 571, 15–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kimizuka H., Koketsu K. (1964) J. Theor. Biol. 6, 290–305 [DOI] [PubMed] [Google Scholar]
- 21.Stauffer D. A., Karlin A. (1994) Biochemistry 33, 6840–6849 [DOI] [PubMed] [Google Scholar]
- 22.Konrad M., Schaller A., Seelow D., Pandey A. V., Waldegger S., Lesslauer A., Vitzthum H., Suzuki Y., Luk J. M., Becker C., Schlingmann K. P., Schmid M., Rodriguez-Soriano J., Ariceta G., Cano F., Enriquez R., Juppner H., Bakkaloglu S. A., Hediger M. A., Gallati S., Neuhauss S. C., Nurnberg P., Weber S. (2006) Am. J. Hum. Genet. 79, 949–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eisenman G., Horn R. (1983) J. Membr. Biol. 76, 197–225 [DOI] [PubMed] [Google Scholar]
- 24.Diamond J. M., Wright E. M. (1969) Annu. Rev. Physiol. 31, 581–646 [DOI] [PubMed] [Google Scholar]
- 25.Moreno J. H., Diamond J. M. (1975) J. Membr. Biol. 21, 197–259 [DOI] [PubMed] [Google Scholar]
- 26.Green W. N., Andersen O. S. (1991) Annu. Rev. Physiol. 53, 341–359 [DOI] [PubMed] [Google Scholar]
- 27.Hou J., Paul D. L., Goodenough D. A. (2005) J. Cell Sci. 118, 5109–5118 [DOI] [PubMed] [Google Scholar]
- 28.Nagamori S., Nishiyama K., Tokuda H. (2000) J. Biochem. 128, 129–137 [DOI] [PubMed] [Google Scholar]
- 29.Guo W., Shi L., Javitch J. A. (2003) J. Biol. Chem. 278, 4385–4388 [DOI] [PubMed] [Google Scholar]
- 30.Coyne C. B., Gambling T. M., Boucher R. C., Carson J. L., Johnson L. G. (2003) Am. J. Physiol. Lung Cell Mol. Physiol. 285, L1166–L1178 [DOI] [PubMed] [Google Scholar]
- 31.Mitic L. L., Unger V. M., Anderson J. M. (2003) Protein Sci. 12, 218–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Careaga C. L., Falke J. J. (1992) J. Mol. Biol. 226, 1219–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu M., Akabas M. H. (1996) J. Gen. Physiol. 107, 195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou X. W., Pfahnl A., Werner R., Hudder A., Llanes A., Luebke A., Dahl G. (1997) Biophys. J. 72, 1946–1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.