

Abstract
The human holocarboxylase synthetase (HCS) catalyzes transfer of biotin to biotin-dependent carboxylases, and the enzyme is therefore of fundamental importance for many physiological processes, including fatty acid synthesis, gluconeogenesis, and amino acid catabolism. In addition, the enzyme functions in regulating transcription initiation at several genes that code for proteins involved in biotin metabolism. Two major forms of HCS exist in humans, which differ at the amino terminus by 57 amino acids. In this work, the two proteins were expressed in Escherichia coli, purified, and subjected to biochemical characterization. Equilibrium sedimentation indicates that the two proteins are monomers both in their apo-forms and when bound to the enzymatic intermediate biotinyl 5′-AMP. Steady state kinetic analyses as a function of biotin, ATP, or a minimal biotin-accepting substrate concentration indicate similar behaviors for both isoforms. However, pre-steady state analysis of biotin transfer reveals that the full-length HCS associates with the minimal biotin acceptor substrate with a rate twice as fast as that of the truncated isoform. These results are consistent with a role for the HCS amino terminus in biotin acceptor substrate recognition.
Introduction
Biotin protein ligases are enzymes that are required for viability of all organisms. In metabolism these enzymes catalyze covalent linkage of biotin to biotin-dependent carboxylases as indicated in Reactions 1 and 2,
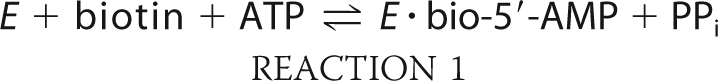 |
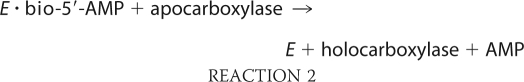 |
in which the adenylated derivative of biotin, biotinyl 5′-AMP (bio-5′-AMP), is first synthesized from the substrates biotin and ATP (1). In the second step, the enzyme·adenylate complex interacts with the biotin acceptor domain or subunit of a carboxylase, and the biotin is covalently linked to the ϵ-amino group of a specific lysine residue on the acceptor. In humans the ligase is referred to as holocarboxylase synthetase (HCS).2
Five biotin-dependent carboxylases, including acetyl-CoA carboxylases 1 and 2, 3-methylcrotonyl-CoA carboxylase, pyruvate carboxylase, and propionyl-CoA carboxylase, are the HCS substrates in humans. The biotin moiety serves as the transient carboxylate carrier as it is transferred from a donor to an acceptor in the reaction catalyzed by each enzyme. Biotin-dependent carboxylase-catalyzed reactions contribute to fatty acid synthesis and oxidation, gluconeogenesis, and amino acid catabolism. Mutations in the HCS gene cause multiple carboxylase deficiency, a potentially fatal disease characterized by the concurrence of symptoms associated with each individual carboxylase deficiency (2, 3).
In response to changes in biotin availability, HCS has been demonstrated to function in regulating transcription of the gene that encodes HCS itself as well as those that code for propionyl-CoA carboxylase, pyruvate carboxylase, acetyl-CoA carboxylase 1, and the sodium-dependent multivitamin transporter (4, 5). This transcriptional regulatory process is associated with the soluble guanylyl cyclase signal transduction pathway (6). It has also been suggested that HCS exerts its transcriptional regulatory role by catalyzing biotin linkage to histones (7) (Fig. 1).
FIGURE 1.

HCS function in metabolism and transcription regulation. The enzyme utilizes substrates biotin and ATP to catalyze synthesis of bio-5′-AMP. The HCS·intermediate complex interacts with carboxylases to transfer biotin or functions in transcription, perhaps through histone biotinylation.
Full-length HCS is a 726-amino acid polypeptide, characterized by a molecular mass of 81 kDa. Although residues 448–701 are homologous to the catalytic region of the Escherichia coli ligase, BirA (8), the sequence of the amino-terminal 447 residues of HCS bears homology only to the amino-terminal sequences of other mammalian holocarboxylase synthetases. Although only one copy of the HCS gene is present per haploid genome, analysis of the 5′ termini of cDNAs (9), elucidation of the structure of the HCS gene (10), and purification of HCS from human tissue (11) indicate the existence of more than one form of the protein. Characterization of HCS cDNAs revealed an mRNA in which the initiator codon corresponds to methionine 58 in the full-length coding sequence (9). In addition, Western blot analysis of partially purified HCS from human placenta revealed three species that were assigned to the full-length HCS protein (FL-HCS) and species that initiated translation at methionine 7 or methionine 58 (58-HCS) (11).
The significance of these distinct HCS forms for functional biology is not known. It is possible that the two isoforms are characterized by different catalytic activities. Alternatively, the different forms may play distinct roles in the metabolic versus transcriptional functions of the enzyme. To investigate the enzymatic properties of the two isozymes, we have overexpressed and purified FL-HCS and 58-HCS to homogeneity. Sedimentation equilibrium measurements of the two proteins indicate that each is monomeric in both the unliganded state and when saturated with the intermediate in biotin transfer, biotinyl 5′-AMP. Steady state parameters governing the overall biotin transfer reaction using the model substrate p67, a fragment of propionyl-CoA carboxylase, show that they are very similar. However, pre-steady state kinetic analysis of the second half-reaction revealed that FL-HCS associates with p67 at a faster rate than does 58-HCS. The results indicate that the two isoforms are functionally distinct and that the amino terminus influences the kinetics of interaction of the enzyme with the biotin acceptor protein.
EXPERIMENTAL PROCEDURES
Chemicals and Biochemicals
All chemicals used were at least reagent grade. The stock ATP solutions were prepared by dissolving ATP disodium salt (Sigma) into water and adjusting the pH to 7.5. The nucleotide concentration was determined by UV spectroscopy using an extinction coefficient at 259 nm of 15,400 m cm−1. The d-[carbonyl-14C]biotin (GE Healthcare) was stored desiccated under nitrogen at −20 °C. Biotin d-2,3,4,6-3H was purchased from American Radiolabeled Chemicals, Inc., and stored at −20 °C. The bio-5′-AMP was synthesized and purified as described previously (1, 12). Protein extinction coefficients were calculated according to Gill and von Hippel (13).
Expression Plasmid Construction
The strategy developed to overexpress FL-HCS and 58-HCS involved constructing plasmids that encode amino-terminal His6-SUMO fusions of each protein using the linearized pSUMOpro (LifeSensors Inc.) expression vector (14). Primers containing an Esp3I restriction site were used to amplify the HCS coding sequences by PCR. The coding sequence for 58-HCS was amplified from a pGEX construct provided by Dr. Roy Gravel. The FL-HCS cDNA sequence (GenBankTM accession number NM_000411.4) was purchased from OriGene Technologies, Inc. After digestion of the PCR product with Esp3I (Fermentas), the resulting two coding sequence fragments were ligated into the linearized pSUMO vector using T4 ligase (Roche Applied Science), and the ligation mixtures were transformed into the E. coli top10 strain. Plasmids were verified by sequencing the entire insert.
Purification of 58-HCS and FL-HCS
The plasmids encoding His6-SUMO-FL-HCS and His6-SUMO-58-HCS were transformed into E. coli RosettaTM(DE3). Once the culture had reached an A600 of 0.6, induction was achieved by the addition of lactose (Difco) to a final concentration of 0.5% (w/v) and allowed to proceed for 18–20 h at 25 °C. Cells were harvested, resuspended in 30 ml of lysis buffer/liter of culture (50 mm sodium phosphate buffer, pH 8, 500 mm sodium chloride, 1 mm 2-mercaptoethanol, 1 mm phenylmethylsulfonyl fluoride, and 5% (v/v) glycerol containing 10 mm imidazole), and lysed with three French press passes at 800–1000 p.s.i. The cellular debris was pelleted by centrifugation, and the supernatant was loaded onto a Ni2+-Sepharose column (GE Healthcare). After washing extensively, 20 ml of lysis buffer containing 1 mm ATP, 0.5 mm magnesium chloride, and 1 μm of the biotin acceptor protein p67 was loaded on the column to remove residual biotin and bio-5′-AMP that was associated with the enzyme. After a wash step, the HCS protein was eluted with lysis buffer containing 400 mm imidazole. Sumo protease-1 (15) that was purified in this laboratory using an overexpression plasmid provided by Dr. C. Lima was added at a 1:100 (w/w) ratio to the HCS, and the mixture was dialyzed into buffer containing 10 mm sodium phosphate, pH 8.0, 60 mm NaCl, 5% (v/v) glycerol, 5 mm 2-mercaptoethanol. Following overnight incubation, the imidazole concentration was adjusted to 40 mm, and the sample was loaded again onto Ni2+-Sepharose resin to remove the His6-SUMO tag and the His6-tagged protease. The flow-through was collected and dialyzed against buffer containing 10 mm sodium phosphate, pH 8.0, 30 mm NaCl, 1 mm 2-mercaptoethanol, 5% glycerol. The resulting sample was loaded onto a Toyopearl anion exchange column (Tosoh Bioscience) and eluted with a linear gradient to 0.3 m NaCl. HCS was concentrated and stored in 10 mm Tris-HCl, pH 8, 200 mm sodium chloride, 10% glycerol at −80 °C. The yield was 2 mg of protein/liter of bacterial culture and the concentration of each protein was determined spectrophotometrically using an extinction coefficient at 280 nm of 56,610 m cm−1 (13). The proteins were at least 97% pure as judged by Coomassie Brilliant Blue staining of samples subjected to SDS-PAGE.
Purification of p67
The pDest17 plasmid coding for His6-p67 was a generous gift from Dr. Roy Gravel. Purification of His6-p67 was carried out as described previously (16), except for the use of an additional SP-Sepharose Fast Flow column (GE Healthcare). This column was run with a linear KCl gradient in 50 mm Tris-HCl, pH 7.5, at 4 °C, 5% glycerol, 1 mm 2-mercaptoethanol. The concentration was determined spectrophotometrically using an extinction coefficient at 276 nm of 4350 m cm−1. The presence of biotinylated p67 in the preparations was undetectable, as determined by MALDI-TOF mass spectrometry using the α-cyano-4-hydroxycinnamic acid matrix (17). The same method was used to determine that all of the p67 preparation is active in accepting biotin.
Equilibrium Sedimentation Measurements
Equilibrium sedimentation measurements were carried out in a Beckman Optima XL-I analytical centrifuge equipped with a 4-hole An60-Ti rotor (Beckman Coulter). Double sector 12-mm path length cells with charcoal-filled Epon centerpieces and sapphire windows were used. Samples (VTOT = 140 μl) were prepared at final concentrations ranging from 2 to 20 μm from protein that had been extensively dialyzed against reaction buffer (10 mm Tris-HCl, pH 7.50 ± 0.02 at 20.0 ± 0.1 °C, 200 mm KCl, 2.5 mm MgCl2). Samples that contained HCS, FL- or 58-, and bio-5′-AMP were prepared with 1.5-fold molar excess of the ligand over the protein. Centrifugation was carried out at speeds ranging from 14,000 to 22,000 rpm for 12 h at each speed (18), and absorbance scans were acquired at 280 or 295 nm, if the bio-5′-AMP contribution needed to be avoided, with a step size of 0.001 cm and five averages per step. The data obtained for the samples run at the multiple speeds were subjected to a global single species analysis using the program WinNonLin (19), and the best fit value of σ obtained from the analysis was converted to molecular mass using Equation 1,
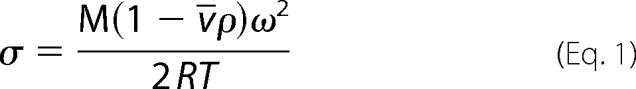 |
in which M is the molecular mass; v̄ is the partial specific volume of the protein, ρ is the buffer density; ω is the angular velocity; R is the gas constant; and T is the temperature. The value of ρ (1.007 g/ml) was determined pycnometrically (20), and v̄ was calculated, based on the protein sequences, to be 0.7389 ml/g for 58-HCS or 0.7392 ml/g for FL-HCS using the program SedenTerp.
Steady State Kinetic Measurements
Kinetic measurements of the overall HCS-catalyzed reaction as a function of ATP or p67 concentration were performed by monitoring the incorporation of [14C]biotin into p67. The reactions were carried out in reaction buffer at 37.0 ± 0.1 °C and initiated by the addition of enzyme to a final concentration between 50 and 150 nm. At designated time points, a 17-μl aliquot was quenched into a solution containing 5.1 μl of loading dye and 3.4 μl of 1% trifluoroacetic acid (in H2O). The loading dye was composed of 50 mm Tris-HCl, pH 6.8, 4% SDS, 30% glycerol, 0.67 mg/ml Coomassie Brilliant Blue G-250, 735 mm 2-mercaptoethanol. Unincorporated [14C]biotin was separated from the 14C-biotinylated p67 by electrophoresis on a 16% acrylamide-SDS-Tricine gel (21). The gels were dried on Whatman 3MM paper and exposed to a phosphor screen (GE Healthcare) for 3 days. The phosphor screen image was scanned using a Storm PhosphorImager, and the volumes of the bands corresponding to [14C]biotin-labeled p67 were quantified using the ImageQuant software (GE Healthcare). Initial velocities were obtained from the slope of counts versus time plot. The initial velocity versus substrate concentration data were subjected to nonlinear least squares analysis using the Michaelis-Menten equation with GraphPad Prism to obtain Vmax and Km. The Vmax and kcat values were converted from counts/min to micromolars of biotin using the relation between counts/min and micromolars obtained from a reaction containing 50 μm p67, 150 nm HCS, 10 μm biotin, and 1 mm ATP in standard reaction buffer incubated for 1 h to allow complete incorporation of the biotin into the acceptor protein. In experiments in which the Michaelis-Menten constants for p67 were determined, the [14C]biotin and ATP concentrations were set at saturating values of 10 μm and 1 mm, respectively. The Km and kcat values for ATP were determined in a similar fashion, with the exception that the reactions contained 50 μm p67, 10 μm [14C]biotin, and variable ATP concentrations.
The biotin concentration dependences of the reactions catalyzed by FL- and 58-HCS were determined by quantitating the incorporation of [3H]biotin into p67. Each reaction contained 16.67 nm [3H]biotin and a supplement of unlabeled biotin to obtain the desired final total biotin concentration. The p67 and ATP concentrations were maintained at 150 μm and 1 mm, respectively. Reactions were initiated by addition of enzyme to 5 nm final concentration, and aliquots were quenched with trifluoroacetic acid added to a final concentration of 0.3% (v/v). Quenched reactions were filtered though BA 85 Protran nitrocellulose membranes (Whatman), and the retained radioactivity was quantified in ProteinReady+ scintillation fluid (Beckman) using a LS6500 Beckman counter. The measured disintegrations/min were converted to molar quantities of biotin incorporated by multiplying by total biotin concentration/dpm of unfiltered sample. The correction for filter retention was achieved by using the disintegrations/min associated with a filtered and unfiltered reaction in which 10 μm cold biotin and 16.67 nm [3H]biotin were fully incorporated into p67.
Steady State Fluorescence Spectra
Steady state fluorescence spectra were acquired at 20 °C in reaction buffer using an ISS PC1 instrument. The excitation wavelength was 295 or 300 nm, and emission was monitored from 310 to 450 nm. The excitation slit width was set to 4 or 8 nm, and the emission slit width was 8 nm. Spectra were corrected for contributions from buffer and ligand and for dilution.
Initial Rate of Bio-5′-AMP Synthesis
The rate of bio-5′-AMP synthesis was measured at 20 °C by monitoring the intrinsic protein fluorescence decrease that accompanies synthesis of the intermediate. The experiments were performed in a Kintek 2001 stopped flow instrument using an excitation wavelength of 295 or 300 nm and monitoring the emission through a 340-nm cutoff filter (Corion Corp.). Several ATP solutions prepared at concentrations ranging from 25 to 800 μm were rapidly mixed in a 1:1 ratio with a solution of a fixed concentration of HCS containing 30 μm biotin. Initial HCS concentrations varied from 2 to 6 μm, and all solutions were prepared in degassed reaction buffer. A minimum of five traces that spanned at least eight half-lives of the process was collected at each ATP concentration. Transients were analyzed using a double exponential function with the Kintek software. The dependences of the apparent rates of the two phases on ATP concentration are discussed under “Results.”
Measurement of the Bimolecular Association Rate Constant of HCS with p67
The bimolecular rate of association of HCS with p67 was obtained by monitoring the intrinsic fluorescence increase of HCS upon depletion of the bio-5′-AMP·HCS complex. The instrument and experimental setup were as described above, with the exception that one syringe contained varying concentrations of p67, and the second syringe contained a fixed concentration of the HCS·bio-5′-AMP complex. The HCS·bio-5′-AMP complex concentration before mixing ranged from 1 to 3 μm in different experiments. This solution was prepared by combining HCS with biotin at half of the enzyme concentration and 100 μm ATP and incubating for 10 min to allow completion of bio-5′-AMP synthesis. This solution was rapidly mixed with p67, and the resulting time-dependent increase in intrinsic HCS fluorescence was monitored. At least five traces acquired at each [p67] were fit to single exponential to obtain apparent rates. The observed rates as a function of p67 concentration were subjected to further analysis as described under “Results.”
RESULTS
Purification of HCS Isoforms
The two HCS isoforms were expressed in E. coli as His6-SUMO fusion proteins, and the purification protocol, which included three chromatography steps, yielded preparations that were >97% pure (Fig. 2). Because SUMO-protease-1 leaves no exogenous amino acids after cleavage, the final proteins correspond to native 58-HCS and FL-HCS. In the purification, it was necessary to incorporate steps to remove bio-5′-AMP from the preparations. This contamination was revealed by the ability to detect biotinylation of p67 in the absence of added biotin. Biotinylation was assayed as a shift in the mass of a fraction of the p67 using MALDI-TOF mass spectrometry. Both the E. coli and Pyroccocus horikoshii enzymes have been purified in this laboratory, and in contrast to the Homo sapiens enzyme, no contaminating substrate or intermediate has ever been detected in those preparations.
FIGURE 2.

SDS-polyacrylamide gel of 58-HCS purification fractions. A, lane 1, low range molecular weight markers (Bio-Rad). Lane 2, crude extract after induction. Lane 3, sample obtained from the first Ni2+-Sepharose column. Lane 4, sample after SUMO protease 1 cleavage. Lane 5, flow-through from the second Ni2+-Sepharose column. Lane 6, material obtained after Toyopearl anion exchange column. B, 12% SDS-polyacrylamide gel of the pure proteins. Lane 1, low range molecular weight markers. Lane 2, FL-HCS; lane 3, 58-HCS.
Sedimentation Equilibrium Analysis of FL-HCS and 58-HCS Oligomeric State
Sedimentation equilibrium measurements were used to determine the oligomeric state of the two HCS enzymes in reaction buffer at 20 °C. Scans obtained for unliganded 58-HCS are shown in Fig. 3, along with the results of nonlinear least squares analysis of the data. Global analysis of data obtained for samples prepared at two protein concentrations and centrifuged at two rotor speeds using a single species model indicates a molecular weight consistent with the monomer. The experimentally determined molecular masses were 74 ± 4 and 83 ± 3 kDa for 58-HCS and FL-HCS, respectively. These values agree within error with the analytical molecular masses of 74 kDa for 58-HCS and 81 kDa for FL-HCS calculated from the amino acid sequence.
FIGURE 3.
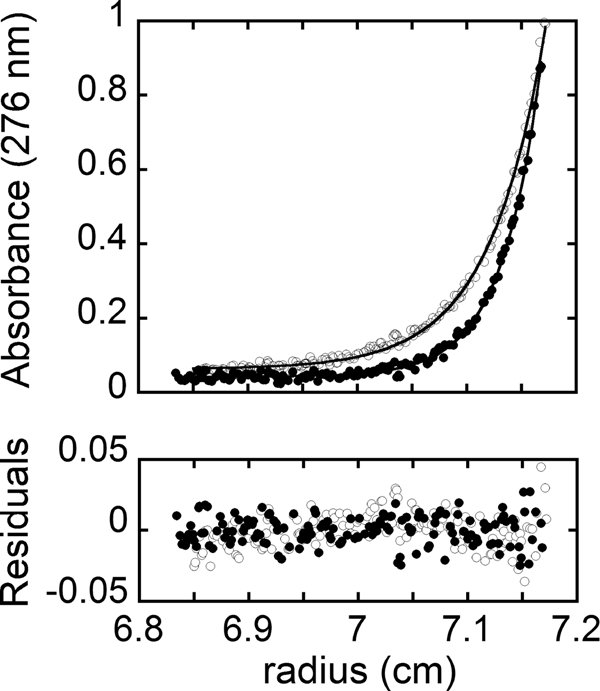
Concentration distribution for 3.8 μm 58-HCS in reaction buffer at 20 °C. Absorbance versus radial position obtained at rotor speeds of 18,000 rpm (○) and 22,000 rpm (●). The solid lines represent the best fit of the data acquired at 3.8 and 1.9 μm 58-HCS and the two rotor speeds to a single species model. The residuals of the fit are provided in the bottom panel.
The E. coli biotin protein ligase undergoes a monomer-dimer transition that is dependent on bio-5′-AMP binding (20). To investigate the effect of the intermediate on the self-association of the two HCS enzymes, sedimentation equilibrium measurements were performed in the presence of excess bio-5′-AMP. The experimentally obtained molecular masses for the intermediate bound species were 79 ± 8 kDa for 58-HCS and 83 ± 7 kDa for FL-HCS, consistent with a monomeric species. Thus, both the unliganded and intermediate-bound forms of FL and 58-HCS are monomeric.
Kinetic Analysis of the Two-step HCS-catalyzed Reaction
In the HCS-catalyzed reaction, biotin is covalently linked to a specific lysine residue on the biotin carboxyl carrier protein domain of five distinct carboxylases. The physiologically relevant substrates contain multiple copies of this domain. To develop a simple assay for measuring the catalytic activity of the purified HCS enzymes, the p67 domain of propionyl-CoA carboxylase was employed as the model acceptor substrate. This protein corresponds to the carboxyl-terminal 67 residues of the α subunit of human propionyl-CoA carboxylase, which contains sufficient information for recognition by the human biotin protein ligase (22).
The two kinetic assays used in this work rely on measurements of incorporation of radiolabeled biotin into p67. In the first assay, [14C]biotin incorporation into p67 was monitored. The [14C]biotin-labeled p67 was separated from the unincorporated biotin by electrophoresis, and radioactivity in the bands corresponding to p67 was quantitated by phosphorimaging (Fig. 4A). The initial rate at several ATP concentrations and constant biotin and p67 concentrations exhibit the expected increase in the rate of [14C]biotin incorporation with increasing ATP concentration (Fig. 4B).
FIGURE 4.

Kinetic assays used to determine Michaelis-Menten constants for HCS-catalyzed biotin transfer with respect to p67, biotin, and ATP. A, incorporation of [14C]biotin into p67 was used to determine the kinetic constants with respect to p67 and ATP. The biotinylated p67 and free biotin are separated on a 16% acrylamide gel. B, biotin incorporation versus time at 150 nm 58-HCS, 50 μm p67, 10 μm [14C]biotin, and a range of ATP concentrations as follows: (■, 5 μm; ▴, 30 μm; ▾, 90 μm; ♦, 250 μm; ●, 500 μm; □, 750 μm; and ▵, 1000 μm ATP. C, incorporation of [3H]biotin into p67 used to determine the kinetic constants with respect to biotin. Biotinylated p67 is separated from unincorporated biotin by filtration through a nitrocellulose membrane. D, initial rates of biotin incorporated measured at 5 nm FL-HCS, 150 μm p67, 16.67 nm [3H]biotin and variable unlabeled biotin concentrations as follows: ▾, 20 μm; ■, 10 μm; ♦, 7 μm; ●, 3 μm; and ▴, 1 μm biotin. The inset depicts the traces obtained for the following: ▿, 400 nm; Δ, 150 nm; and 50 nm total biotin. B and D, the lines were obtained from linear regression of the measured biotin incorporation versus time.
The second assay for HCS catalytic activity monitors [3H]biotin incorporation into p67. The [3H]biotin-labeled p67 was separated from the unincorporated biotin by filtration through nitrocellulose and quantitated by liquid scintillation counting (Fig. 4C). The relatively low Km value for biotin, the detection limit of the 14C radiation using phosphorimaging, and a new lack of commercial availability of the [14C]biotin motivated development of this second assay. Results of measurements of the initial rates of HCS-catalyzed [3H]biotin incorporation at constant ATP and p67 concentrations and varying total biotin concentrations are shown in Fig. 4D. The time courses used for initial rate measurements at low biotin concentrations were carried out for a shorter time than those acquired at the higher biotin concentration. This is due to the higher specific activity of the substrate at the low cold biotin concentration, which allowed acquisition of data over shorter time periods.
To investigate if the amino-terminal 57-amino acid residues impact the basic catalytic properties of the human biotin protein ligase, the kinetic parameters associated with the process were measured for both FL-HCS and 58-HCS. Measurements of the initial rates of biotin transfer as a function of p67 concentration are shown in Fig. 5, A and B. The results indicate Km values of 19 and 21 μm, respectively, for FL-HCS and 58-HCS (Table 1) and values of kcat that are similar at 0.47 and 0.35 s−1, respectively. The magnitudes of kcat/Km are, within error, identical.
FIGURE 5.

Steady state kinetic analysis of biotin incorporation into p67 catalyzed by 58-HCS and FL-HCS. Samples of steady state analysis of the reaction catalyzed by 58-HCS (A) and FL-HCS (B) as a function of p67 concentration are shown. Data were obtained as a function of ATP concentration for 58-HCS (C) and FL-HCS (D). E and F display results of steady state measurements for 58-HCS and FL-HCS as a function of biotin concentration. In these two panels each data point represents the average of the initial rate measured at each biotin concentration in three independent experiments. The error bars represent the standard deviation of each three points. In each panel the solid line represents the best fit of the data to the Michaelis-Menten equation.
TABLE 1.
Kinetic parameters for the overall HCS-catalyzed reaction
The kinetic parameters were determined as described under “Experimental Procedures.”
| HCS form | Substrate | Km | kcat | kcat/Km |
|---|---|---|---|---|
| μm | s−1 | ×104m−1s−1 | ||
| 58 | p67 | 19 ± 4a | 0.35 ± 0.02a | 1.8 ± 0.4 |
| ATP | 41 ± 10a | 0.33 ± 0.02ab | 0.8 ± 0.2 | |
| (0.24 ± 0.01)ac | 0.6 ± 0.1 | |||
| Biotin | 0.7 ± 0.3d | 0.11 ± 0.01d | 16 ± 7 | |
| FL | p67 | 21 ± 2a | 0.47 ± 0.01a | 2.2 ± 0.2 |
| ATP | 47 ± 8a | 0.45 ± 0.02ab | 1.0 ± 0.2 | |
| (0.32 ± 0.01)ac | 0.7 ± 0.1 | |||
| Biotin | 0.8 ± 0.2d | 0.17 ± 0.01d | 21 ± 5 |
a The errors correspond to the standard deviation of two independent experiments.
b The calculated value is based on the experimentally obtained kcat at 50 μm p67.
c Experimentally obtained kcat was at 50 μm p67.
d The error represents the standard error obtained from globally fitting data from three independent experiments.
A similar analysis as a function of ATP concentration (Fig. 5, B and D) provides Km values for ATP of 41 and 47 μm for 58-HCS and FL-HCS, respectively. The magnitude of kcat associated with ATP obtained from direct fitting of the data is lower than that obtained from measurements of the p67 concentration dependence of the reaction. This is attributable to the necessity, due to the appearance of a doublet for the labeled p67 at high concentrations, of using an acceptor protein concentration of 50 μm in the ATP concentration dependence measurements. Normalization of the resolved kcat values obtained from analysis of the ATP concentration dependence of the reactions to those obtained from the measurements of the p67 concentration dependence reveals that they are identical (Table 1).
The FL-HCS and 58-HCS enzymes were subjected to Michaelis-Menten analysis of the biotin concentration dependence of the reaction using [3H]biotin. The results of measurements of the initial rates versus biotin concentration indicate that both FL- and 58-HCS-catalyzed reactions are well described by a Michaelis-Menten model (Fig. 5, E and F). The initial rate at each biotin concentration represents the average of at least three independent measurements. Results of nonlinear least squares analysis of the data indicate that the two enzymes are similar with respect to their rate versus biotin concentration profiles. The kcat values resolved from the analysis are ∼3-fold lower than those obtained from analysis of data obtained using SDS-PAGE to separate products from reactants. Because p67 and ATP concentrations used in the measurements were saturating, the differences in the values of kcat reflect differences intrinsic to the two assays.
Pre-steady State Analysis of Bio-5′-AMP Synthesis
The overall reaction catalyzed by HCS can be classified as a ping-pong mechanism, which allows measurement of the two individual half-reactions. Upon bio-5′-AMP synthesis, decreases in intrinsic protein fluorescence of ∼14% for FL-HCS and 23% for 58- HCS are observed (Fig. 6A). A stopped flow experiment was developed to investigate the first half-reaction, in which biotin-bound HCS was rapidly mixed with ATP. Measurement of the time dependence of the fluorescence decrease revealed that it was well described by a double exponential model (Fig. 6A, inset). A loss in amplitude with increasing ATP concentration was observed, which was attributed to the faster phase becoming increasingly fast and outside of the dead time of the stopped flow instrument. The apparent rates for the two phases were plotted as a function of ATP concentration (Fig. 6B). The observed linear dependence of the faster phase on ATP concentration and the saturable slower phase are consistent with Reaction 3,
 |
in which the slope of the linear dependence of the apparent rate of the fast phase on nucleotide concentration provides an estimate of the rate of association of ATP with the HCS·biotin complex, k1 (Table 2). Nonlinear least squares analysis of the concentration dependence of the second phase using the Michaelis-Menten model yielded kcat and Km values for the reaction. The parameters obtained for 58-HCS and FL-HCS from these two analyses are similar in magnitude (Table 2).
FIGURE 6.

Pre-steady state analysis of bio-5′-AMP synthesis. A, fluorescence emission spectra of 1.5 μm HCS and 15 μm biotin before (solid line) and after (dashed line) addition of 100 μm ATP. Inset, representative stopped flow fluorescence trace obtained upon mixing 2 μm HCS and 30 μm biotin with 25 μm ATP. B, dependence of the apparent rate of bio-5′-AMP synthesis on ATP concentration. The line is the fit to the Michaelis-Menten equation. Inset, dependence of the apparent rate of ATP association with HCS-biotin on ATP concentration.
TABLE 2.
Kinetic parameters for ATP dependence of bio-5′-AMP synthesis
The kinetic parameters were determined by mixing a complex of HCS-biotin with ATP as described under “Experimental Procedures.”
| HCS form | k1 | Km | kcat | kcat/Km |
|---|---|---|---|---|
| ×103m−1s−1 | μm | s−1 | ×103m−1s−1 | |
| 58 | 4.9 ± 0.7a | 50 ± 30a | 0.12 ± 0.02a | 2 ± 1 |
| (0.39 ± 0.06)b | 8 ± 5 | |||
| FL | 6 ± 1c | 50 ± 20c | 0.11 ± 0.02c | 2 ± 1 |
| (0.36 ± 0.06)b | 7 ± 3 |
a The errors correspond to the standard deviation for two independent experiments.
b The calculated kcat values were obtained at 37 °C assuming that the rate doubles every 10 °C.
c The errors correspond to the standard deviation for three independent experiments.
Single Turnover Analysis of the Biotin Transfer Reaction
The second half-reaction, transfer of biotin from bio-5′-AMP to p67, was also measured using stopped flow fluorescence. An increase in intrinsic protein fluorescence, which reports on depletion of the enzyme-intermediate species, occurs upon addition of p67 to the pre-formed HCS·bio-5′-AMP complex (Fig. 7A). In the measurements, HCS was first allowed to synthesize bio-5′-AMP from ATP and biotin. To ensure single turnover conditions, the biotin concentration used was half that of the HCS concentration. Moreover, because at micromolar protein concentrations HCS is at stoichiometric conditions with respect to bio-5′-AMP binding, virtually all of the intermediate is associated with HCS. The HCS·intermediate complex was rapidly mixed with varying concentrations of p67 under pseudo first-order conditions, and the resulting time-dependent increase in fluorescence was well described by a single exponential model (Fig. 7A, inset). The amplitudes of the transients displayed no dependence on p67 concentration, indicating that the reaction goes to completion at all concentrations. Measurements performed over a large range of p67 concentrations up to 100 μm yield apparent rates that show no indication of leveling off (Fig. 7B). As a result, the fluorescent signal was interpreted as reporting on the initial p67 binding event that is governed by k1 in Reaction 4,
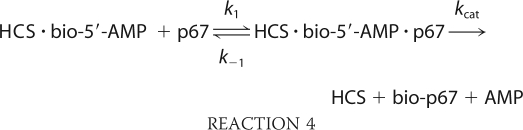 |
The slope of the line relating the dependence of the apparent rate on p67 concentration provides a bimolecular rate constant for association of p67 with FL-HCS that is ∼2-fold greater than for 58-HCS (Table 3).
FIGURE 7.

Single turnover measurements of biotin transfer from bio-5′-AMP to p67. A, fluorescence emission spectra of 1 μm 58-HCS, 0.5 μm biotin, and 100 μm ATP before (solid line) and after (dashed line) addition of 10 μm p67. Inset, representative stopped flow fluorescence trace obtained upon mixing 1 μm 58-HCS, 0.5 μm biotin, and 100 μm ATP with 20 μm p67. B, dependence of the apparent association rate of FL-HCS-bio-5′-AMP (●) or 58-HCS-bio-5′-AMP (■) with p67 on acceptor protein concentration. Error bars represent the mean ± S.E. of at least five measurements performed at each [p67].
TABLE 3.
Bimolecular association rate constants for HCS·bio-5′-AMP with p67
The kinetic parameters were determined as described under “Experimental Procedures.”
a The errors correspond to the spread of the values obtained from two independent experiments.
DISCUSSION
Elucidation of the biochemical details of human holocarboxylase synthetase function has implications for understanding the mechanism of the metabolic disorder, multiple carboxylase deficiency, as well as how this metabolic enzyme functions in transcription regulation. The significance of the existence of multiple forms of the enzyme for its distinct roles also merits further study. The results presented in this work indicate that although the two HCS isoforms catalyze the first of the two half-reactions in post-translational biotin addition with similar rates, they differ in the second half-reaction. Thus, the 57 amino-terminal residues appear to be involved in biotin acceptor protein recognition.
FL-HCS and 58-HCS Are Monomeric
Results of sedimentation equilibrium measurements indicate that in the micromolar concentration range both enzymes are monomeric regardless of ligation state. This result contrasts with that obtained for the E. coli enzyme, which homodimerizes in response to the adenylate binding (20). However, homodimerization of the E. coli enzyme is associated with the site-specific DNA binding function of the enzyme, a function that the human enzyme does not possess. Studies of a number of simple monofunctional bacterial biotin protein ligases that do not possess a DNA binding domain indicate that they can exist as monomers or constitutive dimers. The structures of the dimeric enzymes, which utilize a surface distinct from that used by the E. coli enzyme to homodimerize, reinforce the idea that linkage of dimerization of the E. coli enzyme to bio-5′-AMP binding is idiosyncratic to its additional role in site-specific DNA binding.
Sedimentation analysis of the two HCS isoforms indicates that the HCS amino terminus plays no role in mediating homo-oligomerization of the protein. Several eukaryotic biotin protein ligases have been characterized at the sequence level. However, very few have been purified. Among these are the Saccharomyces cerevisiae and one of the two Arabidopsis thaliana enzymes. The chloroplastic A. thaliana enzyme has been overexpressed in and purified from E. coli and found to be monomeric in its native form (23). No characterization of the oligomeric state of the yeast enzyme was reported. Like the human enzyme, these eukaryotic enzymes are characterized at the primary structure level by a carboxyl terminus that is homologous to the monofunctional bacterial enzymes and the central and carboxyl-terminal domains of the bifunctional bacterial enzymes. Furthermore, they all possess amino-terminal segments of varying lengths. One potential role for these amino-terminal segments is in self-association of the protein. However, the characterization of the purified human and plant enzymes indicates that this is not the case.
Steady State Analysis of the Overall HCS-catalyzed Reaction
The FL- and 58-HCS isoforms exhibit similar kinetic properties in the overall biotin transfer reaction. However, the kcat values for FL-HCS are consistently larger than those measured for the truncated protein. For example, the kcat values of 0.35 ± 0.02 and 0.47 ± 0.01 s−1 for 58-HCS versus FL-HCS, respectively, were resolved from measurements of the p67 concentration dependence of the reaction. Similarly modest differences in kcat are observed in measurements of the dependence of the rate on ATP and biotin concentrations.
The values of the kinetic constants for two of the three substrates compare reasonably well with those reported previously for the human enzyme. However, the caveat to any comparison is that all previous measurements were performed in cellular extracts. Previously reported values of the Km for biotin range from 15 nm to 1 μm and thus are in the same range as the values of 700–800 nm reported in this work (24–27). The Km values for ATP measured for FL-HCS and 58-HCS are ∼40–50 μm, although previously reported values range from 190 to 250 μm (24, 25). Finally, the Michaelis constants for p67 reported in this work cannot be compared with any other values because none exist in the literature.
Analysis of HCS Two Half-reactions
Results of the pre-steady state measurements of the first half-reaction indicate that the two forms synthesize bio-5′-AMP with similar rates and Km for ATP. Furthermore, the Michaelis constants for ATP for the half-reaction are the same as the values obtained for the overall reaction. Because the fluorescence signal at this temperature was more reliable, measurements of the half-reactions were performed at 20 °C. Assuming that the rate doubles every 10 °C, the values of kcat for the first half-reaction at 37 °C are predicted to be 0.39 ± 0.06 s−1 for 58-HCS and 0.36 ± 0.06 s−1 for FL-HCS. These values agree with those obtained from measurements of the overall reaction and therefore indicate that bio-5′-AMP synthesis is the rate-limiting step in the two-step reaction.
Stopped-flow fluorescence measurements of second half-reaction, which report only on association of p67 with the enzyme·intermediate complex, indicate that FL-HCS associates with the acceptor protein at a rate twice that measured for 58-HCS. Furthermore, the absence of a leveling off of the rate as p67 concentration is increased is consistent with the conclusion that bio-5′-AMP synthesis is the rate-limiting step in the two-step reaction. Simulations of the kinetic time courses in the context of the model used for the analysis (see Reaction 4) indicate a lower limit for the rate of biotin transfer and product release of 1 s−1, a value significantly greater than the kcat value of 0.1 s−1 determined for the first half-reaction.
The distinct rates of association of FL- and 58-HCS with p67 are qualitatively consistent with the results of previous studies in which a series of truncation proteins of 58-HCS were characterized with respect to their abilities to catalyze biotin linkage to the biotin acceptor domain of the human propionyl-CoA carboxylase, p67, and the E. coli biotin carboxylase carrier protein fragment, BCCP87, of acetyl-CoA carboxylase (28). Those studies indicate that the amino-terminal region of the enzyme functions in dictating the relative specificity of the enzyme for the alternative acceptor protein substrates. However, in those studies, the physiologically relevant HCS forms, FL and 58, were not compared, and no time dependence was investigated.
Implications of the Distinct Rates of Association of 58-HCS and FL-HCS with p67
The most significant difference observed between FL-HCS and 58-HCS lies in the parameters describing their association with p67, with FL-HCS associating with the biotin acceptor substrate at a rate ∼2-fold faster than 58-HCS. Although this difference is small, it may have consequences for the metabolic activities of carboxylases. The low biotin concentration in human tissue (nanomolar range) limits the supply of the HCS·bio-5′-AMP complex (29, 30). Once the complex is formed, biotin utilization by the carboxylases depends, in part, on the efficiency with which it is transferred from the intermediate to the acceptor protein substrates. There are five biotin-dependent carboxylases present in mammalian cells that are involved in a variety of distinct metabolic processes. A hierarchy of recognition of these carboxylases by HCS may exist, depending on how vital the reaction is to the survival of the cell. In addition, there is currently no information about the relative abundance of FL- and 58-HCS, each with its distinct rate of association with acceptor protein targets. Tissue-specific variation in the availability of the two isoforms could potentially provide a means of regulating activities of biotin-dependent carboxylases.
Other possible roles exist for distinct forms of the HCS. The function of HCS in transcription regulation is consistent with a nuclear role for the enzyme. Indeed, immunofluorescence studies indicate that the enzyme is localized to both the cytosol and the nucleus (31). The alternative HCS forms could be functionally distinct with regard to nuclear localization and, consequently, their relative significance for metabolism and transcription regulation.
In conclusion, the work presented here has established that two naturally occurring isoforms of HCS behave similarly in most respects, but they have distinct activities in biotin acceptor substrate binding, thus indicating a role for the amino-terminal amino acids in carboxylase recognition.
Acknowledgment
We thank Dr. Roy Gravel for providing the plasmid containing the 58-HCS coding sequence and the overexpression plasmid for p67.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-GM46511 and S10-RR15899.
- HCS
- holocarboxylase synthetase
- SUMO protein
- small ubiquitin-like modifier protein
- FL-HCS
- full-length holocarboxylase synthetase
- 58-HCS
- protein product of translation initiation at methionine 58 of the full-length protein
- p67
- carboxyl-terminal fragment of the propionyl-CoA-carboxylase α subunit
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- bio-5′-AMP
- biotinyl 5′-AMP
- MALDI-TOF
- matrix-assisted laser desorption ionization time-of-flight.
REFERENCES
- 1.Lane M. D., Rominger K. L., Young D. L., Lynen F. (1964) J. Biol. Chem. 239, 2865–2871 [PubMed] [Google Scholar]
- 2.Sherwood W. G., Saunders M., Robinson B. H., Brewster T., Gravel R. A. (1982) J. Pediatr. 101, 546–550 [DOI] [PubMed] [Google Scholar]
- 3.Sweetman L., Burri B. J., Nyhan W. L. (1985) Ann. N.Y. Acad. Sci. 447, 288–296 [DOI] [PubMed] [Google Scholar]
- 4.Dakshinamurti K., Cheah-Tan C. (1968) Arch. Biochem. Biophys. 127, 17–21 [DOI] [PubMed] [Google Scholar]
- 5.Chauhan J., Dakshinamurti K. (1991) J. Biol. Chem. 266, 10035–10038 [PubMed] [Google Scholar]
- 6.Solórzano-Vargas R. S., Pacheco-Alvarez D., León-Del-Río A. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 5325–5330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zempleni J. (2005) Annu. Rev. Nutr. 25, 175–196 [DOI] [PubMed] [Google Scholar]
- 8.Suzuki Y., Aoki Y., Ishida Y., Chiba Y., Iwamatsu A., Kishino T., Niikawa N., Matsubara Y., Narisawa K. (1994) Nat. Genet. 8, 122–128 [DOI] [PubMed] [Google Scholar]
- 9.León-Del-Rio A., Leclerc D., Akerman B., Wakamatsu N., Gravel R. A. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 4626–4630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang X., Aoki Y., Li X., Sakamoto O., Hiratsuka M., Kure S., Taheri S., Christensen E., Inui K., Kubota M., Ohira M., Ohki M., Kudoh J., Kawasaki K., Shibuya K., Shintani A., Asakawa S., Minoshima S., Shimizu N., Narisawa K., Matsubara Y., Suzuki Y. (2001) Hum. Genet. 109, 526–534 [DOI] [PubMed] [Google Scholar]
- 11.Hiratsuka M., Sakamoto O., Li X., Suzuki Y., Aoki Y., Narisawa K. (1998) Biochim. Biophys. Acta 1385, 165–171 [DOI] [PubMed] [Google Scholar]
- 12.Abbott J., Beckett D. (1993) Biochemistry 32, 9649–9656 [DOI] [PubMed] [Google Scholar]
- 13.Gill S. C., von Hippel P. H. (1989) Anal. Biochem. 182, 319–326 [DOI] [PubMed] [Google Scholar]
- 14.Marblestone J. G., Edavettal S. C., Lim Y., Lim P., Zuo X., Butt T. R. (2006) Protein Sci. 15, 182–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mossessova E., Lima C. D. (2000) Mol. Cell 5, 865–876 [DOI] [PubMed] [Google Scholar]
- 16.Healy S., Heightman T. D., Hohmann L., Schriemer D., Gravel R. A. (2009) Protein Sci. 18, 314–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nenortas E., Beckett D. (1996) J. Biol. Chem. 271, 7559–7567 [DOI] [PubMed] [Google Scholar]
- 18.Roark D. E. (1976) Biophys. Chem. 5, 185–196 [DOI] [PubMed] [Google Scholar]
- 19.Johnson M. L., Correia J. J., Yphantis D. A., Halvorson H. R. (1981) Biophys. J. 36, 575–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eisenstein E., Beckett D. (1999) Biochemistry 38, 13077–13084 [DOI] [PubMed] [Google Scholar]
- 21.Schägger H., von Jagow G. (1987) Anal. Biochem. 166, 368–379 [DOI] [PubMed] [Google Scholar]
- 22.Leon-Del-Rio A., Gravel R. A. (1994) J. Biol. Chem. 269, 22964–22968 [PubMed] [Google Scholar]
- 23.Tissot G., Pepin R., Job D., Douce R., Alban C. (1998) Eur. J. Biochem. 258, 586–596 [DOI] [PubMed] [Google Scholar]
- 24.Burri B. J., Sweetman L., Nyhan W. L. (1985) Am. J. Hum. Genet. 37, 326–337 [PMC free article] [PubMed] [Google Scholar]
- 25.Burri B. J., Sweetman L., Nyhan W. L. (1981) J. Clin. Invest. 68, 1491–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakamoto O., Suzuki Y., Li X., Aoki Y., Hiratsuka M., Suormala T., Baumgartner E. R., Gibson K. M., Narisawa K. (1999) Pediatr. Res. 46, 671–676 [DOI] [PubMed] [Google Scholar]
- 27.Aoki Y., Suzuki Y., Li X., Sakamoto O., Chikaoka H., Takita S., Narisawa K. (1997) Pediatr. Res. 42, 849–854 [DOI] [PubMed] [Google Scholar]
- 28.Campeau E., Gravel R. A. (2001) J. Biol. Chem. 276, 12310–12316 [DOI] [PubMed] [Google Scholar]
- 29.Baker H., Frank O., Matovitch V. B., Pasher I., Aaronson S., Hutner S. H., Sobotka H. (1962) Anal. Biochem. 3, 31–39 [DOI] [PubMed] [Google Scholar]
- 30.Mock D. M., deLorimer A. A., Liebman W. M., Sweetman L., Baker H. (1981) N. Engl. J. Med. 304, 820–823 [DOI] [PubMed] [Google Scholar]
- 31.Narang M. A., Dumas R., Ayer L. M., Gravel R. A. (2004) Hum. Mol. Genet. 13, 15–23 [DOI] [PubMed] [Google Scholar]