

Abstract
Fatty acid-induced triacylglycerol synthesis produces triacylglycerol droplets with a protein coat that includes perilipin 3/TIP47 and perilipin 4/S3-12. This study addresses the following two questions. Where do lipid droplets emerge, and how are their coat proteins recruited? We show that perilipin 3- and perilipin 4-coated lipid droplets emerge along the endoplasmic reticulum (ER). Blocking membrane trafficking with AlF4− during fatty acid-induced triacylglycerol synthesis drove perilipin 3 to the tubular ER. Forskolin, which like AlF4− activates adenylate cyclase, did not redistribute perilipin 3, but when added together with AlF4− perilipin 3 was recruited to lipid droplets rather than the ER. Thus inhibiting trafficking with AlF4− redistributed perilipin 3 differently under conditions of triacylglycerol synthesis (fatty acid addition) versus hydrolysis (forskolin) suggesting a shared acylglycerol-mediated mechanism. We tested whether diacylglycerol (DG), the immediate precursor of triacylglycerol and its first hydrolytic product, affects the distribution of perilipin 3. Stabilizing DG with the DG lipase inhibitor RHC80267 enhanced the perilipin 3 recruited to lipid droplets and raised DG levels in this fraction. Treating cells with a membrane-permeable DG recruited perilipin 3 to the ER. Stabilizing DG, by blocking its hydrolysis with RHC80267 or its acylation with triacsin C, enhanced recruitment of perilipin 3 to the ER. Expressing the ER enzyme DGAT1, which removes DG by converting it to triacylglycerol, attenuated perilipin 3 DG-induced ER recruitment. Membrane-permeable DG also drove perilipin 4 and 5 onto the ER. Together the data suggest that these lipid droplet proteins are recruited to DG-enriched membranes thereby linking lipid coat proteins to the metabolic state of the cell.
Introduction
Fat storage has become an area of great interest because as a population we are experiencing significant increases in adiposity and its associated metabolic complications. There is a direct correlation between levels of adiposity and fat accumulation outside adipocytes, which is associated with a vast array of pathologies. However, the mechanisms underlying cellular fat deposition are not well understood. One example of a metabolically important but poorly understood process is how fat gets into intracellular droplets and how the single membrane leaflet of amphipathic proteins and lipids assembles around these droplets. In previous work, we have demonstrated that when cells are given fatty acids they rapidly synthesize triacylglycerol (TG),2 and a set of proteins moves from the cytosol to coat the nascent TG droplets (1–5). These fat coat proteins are members of the PAT family, and the name is an acronym derived from the first letter of the nonsystematic names of the original three family members, Perilipin/ADRP/TIP47 (6). Later, two other proteins, S3-12 and OXPAT, were added based on similar sequence and lipid binding behaviors (2, 4). We will use the newly described systematic nomenclature for the PAT proteins (7) as follows: perilipin 1 for perilipin; perilipin 2 for adipophilin/ADRP; perilipin 3 for PP17/TIP47; perilipin 4 for S3-12, and perilipin 5 for MLDP/LSDP5/OXPAT. Unlike some lipid droplet proteins, PAT proteins do not have an ER targeting signal and are not trafficked to lipid droplets through the secretory pathway (8–10). PAT proteins have been described as either constitutively lipid-associated proteins (CPATs) or exchangeable lipid-binding proteins (EPATs) (5). Perilipin 1 and perilipin 2 are CPATs, because they are stabilized by lipid binding, and thus are almost always bound to lipid. Perilipin 3, perilipin 4, and perilipin 5 are EPATs, because they are stable when not bound to lipid, and thus can exchange between the cytosol and lipid droplet based on the metabolic state of the cells. For example, when the lipid surface is expanded by fatty acid-driven TG synthesis, perilipin 3, perilipin 4, and perilipin 5 move from cytosol to the lipid droplet membrane leaflet (5).
The PAT proteins appear to play an important role in regulating intracellular lipid storage, and CPATs in particular appear to protect lipid stores from unregulated hydrolysis. Studies in cultured cells and whole animals show that CPAT levels control the size of static lipid pools. In addition, a growing set of observations implicates one CPAT, perilipin 1, as the hub of a lipolytic complex in adipocytes (11, 12). The EPATs, perilipin 3, perilipin 4, and perilipin 5, coat nascent neutral lipid (1–4) and influence the ability of the cell to accumulate TG. For example, overexpression of perilipin 5 has been shown to increase TG storage capacity (4). However, many important questions remain. These include: from which intracellular structure do the EPAT-coated droplets emerge, and what recruits EPATs to coat the nascent lipid?
Coat proteins might share similar functions whether they associate with a lipid-cored droplet or an aqueous-cored vesicle. Aqueous-cored vesicle protein coats have been shown to function in concentrating cargo in the aqueous-cored vesicle and in mediating cargo-rich vesicle budding (13). Both functions are also required to traffic neutral lipid. A simple model of lipid droplet assembly has been presented in which neutral lipid synthesized in the ER forms a lipid lens that buds into the cytosol (14). This model is based on many observations as follows: the ER is rich in acylation enzymes, particularly neutral lipid synthetic enzymes; most membrane-bound structures emerge from the ER; there is protein traffic between the ER and lipid droplets; and lipoproteins (extracellular neutral lipid-filled particles) emerge from the ER. However, the coat that concentrates the neutral lipid and facilitates budding has not been identified. We previously hypothesized that EPATs are the coat that concentrates neutral lipid into patches in the ER and then buds lipid droplets into the cytosol (5). Overall, the ER coat proteins have been shown to influence the size and number of lipid droplets and trafficking of proteins to lipid droplets (15–17).
In this study we use OP9 preadipocytes, which express both perilipin 3 and perilipin 4 (18). These preadipocytes have small lipid droplets as well as large flat processes with defined ER, which makes them suitable for imaging the relationship between these organelles. Using various manipulations to disrupt trafficking and lipid storage, we provide several lines of evidence that EPATs traffic onto the cytosolic leaflet of the ER and that lipid droplets emerge from the ER coated with the EPAT perilipin 3. In addition, our data also suggest that during TG synthesis EPATs are recruited to DG-enriched ER membrane leaflets and under lipolytic conditions they are recruited to DG-enriched lipid droplet membrane leaflets.
MATERIALS AND METHODS
Reagents
1-Oleoyl-2-acetyl-sn-glycerol (OAG) was from Sigma (catalog number O6754) or Cayman Chemical (Ann Arbor, MI, catalog number 62600). Acyl-CoA synthase inhibitor triacsin C (19) was from Sigma (catalog number T4540); diacylglycerol lipase inhibitor RHC80267 (20) was from Tocris (Ellisville, MO, catalog number 1842), and bovine serum albumin from Equitech-Bio (Kerrville, TX, catalog number BAH66).
Oleate Albumin Complexes
Oleic acid was solubilized with sodium hydroxide and bound to bovine serum albumin at a ratio of 5.5 oleate/bovine serum albumin.
Antibodies
The rabbit antibody and antiserum against the amino terminus of perilipin 4 and perilipin 3 were described previously (2, 3). An antibody to the carboxyl terminus of perilipin 3 (anti-perilipin 3C) was generated using a similar approach to the previously described antibody to the amino terminus of perilipin 3 (3) except the immunizing peptide was SGPFAPGITEKTPEGK. This antibody is characterized in supplemental Fig. 1. The M5 mouse monoclonal antibody against the FLAG epitope was purchased from Sigma (catalog number F4042); the guinea pig antiserum against perilipin 2 was from Fitzgerald Industries (Concord, MA, catalog number RDI-PROGP40); the rabbit antibody against calnexin was from StressGen (Ann Arbor, MI catalog number SPA-860), and the guinea pig antiserum against perilipin 5 was from American Research Products (Belmont, MA, catalog number 03-GP31).
Construction of DNAs to Generate Viral Particles
myc-perilipin 5 was constructed as described previously (4). DGAT1 was tagged with the FLAG epitope on the amino terminus. The forward primer was ggcgtcgacgaattcgccaccatgggcgattataaagatgacgatgacggcgaccgcggaggcgcgggaagctctc and the reverse primer was ggggcggccgctcatacccccactggggcatcgtac (21). FLAG DGAT1 was then ligated into the Δ U3 retroviral vector (22). The GFP-KDEL construct was made by excising the lysozyme-GFP-KDEL from the pXs-lysozyme-GFP-KDEL (23) construct with BamHI and NotI. Then this fragment was ligated into the Δ U3 retroviral vector (22).
Cell Culture
OP9 mouse stromal cells, which have the potential to rapidly differentiate into adipocytes, were cultured as described previously (18). However, these cells were used as preadipocytes in all experiments.
Generation of myc-Perilipin 5, FLAG-DGAT1, and GFP-KDEL OP9 Cell Lines
To generate stably transfected lines, pseudotyped retroviral particles were produced as described previously (22). Then viral particles were used to make stable OP9 cell lines.
Pharmacological Manipulations
AlF4− was generated by adding 30 mm AlCl3 at 1 μl/ml to media while vortexing and then adding 10 μl/ml 1 m NaF while vortexing (24). Forskolin, RHC80267, membrane-permeable DGs, and staurosporine were added to cell incubations in small aliquots from 1000× stocks in DMSO. Appropriate DMSO controls were included.
Fractionation of OP9 Cells
30% confluent 150-mm plates of OP9 cells were maintained in media with 100 μm oleate complexed to albumin (four per condition). The cells were washed, scraped, sedimented, resuspended in 800 μl of lysis buffer (10 mm HEPES and 1 mm EDTA titrated to pH 7.4 with NaOH), and allowed to swell for 10 min. Swollen cells were disrupted by four passes through a 27-gauge needle. To allow the layering of buffer on top of the suspended cell fragments, the resulting homogenate was brought to 10% (w/w) sucrose by the addition of 200 μl of 40% (w/w) sucrose. This sucrose-weighted homogenate was centrifuged for 10 min at 1000 × g at 4 °C in a 1700-μl microcentrifuge tube. Then 800 μl of the supernatant was transferred to a clean microcentrifuge tube, and 700 μl of lysis buffer was layered on top of the sucrose-weighted supernatant. The tubes were centrifuged for 2 h at 21,000 × g at 4 °C. The tubes were then frozen at −80 °C, and the frozen tubes were cut at 300 μl from the top. The floating fraction was collected by transferring the top ∼300-μl piece of ice to a pre-weighed tube, and then the tube was reweighed and brought to 800 mg (800 μl) over the tube weight with lysis buffer. The bottom of the tube was allowed to thaw, and 500 μl was collected from near the pellet to get an undiluted sample of the soluble fraction. The pellets were then resuspended to 800 μl.
Isolation of ER Enriched Membranes
Cells were disrupted as described under “Fractionation of OP9 Cells” except that one plate of OP9 cells grown in standard media was used per data point, and cells were resuspended in 1 ml of lysis buffer. The supernatant (800 μl) was then layered on 500 μl of 10% (w/w) sucrose and centrifuged for 1 h at 21,000 × g. The tubes were frozen, and the bottom portions containing the membrane pellets were cut off and thawed, and the sucrose-containing buffer was removed. Then the membrane pellet was resuspended in 30 μl of PAGE sample buffer.
Immunofluorescence Microscopy
Cells were fixed and stained as described previously (2). Slides were imaged on a Nikon Eclipse TE2000-U microscope. Images were captured using a Photometrics Coolsnap cf camera driven by MetaMorph version 6.2r6 software (Molecular Devices, Downingtown, PA).
Thin Layer Chromatography
200 μl of each cell fraction was extracted as described previously (25). The extracted material was resuspended in hexane/ether (1:1) and spotted on Whatman Partisil LK6D Silica Gel TLC plates. Lipids were resolved in hexane/ether/acetic acid (70:30:1.2), and lipids were stained with molecular iodine.
Immunoblotting
SDS-PAGE, transfer to membranes, and probing of membranes were done as described previously (3). Membranes were imaged using the LI-COR Odyssey system and infrared fluorescing antibodies (LI-COR Biosciences, Lincoln, NE).
RESULTS
EPAT-coated Droplets Emerge along the ER
Various observations have led to the longstanding but still unproven notion that lipid droplets emerge from the ER. The availability of specific antibodies to proteins that coat emerging lipid droplets allowed us to test this notion. To determine the site of lipid droplet emergence, we used OP9 cells that express two EPATs, perilipin 3 and perilipin 4. These proteins coat nascent lipid droplets and thus mark the site of lipid droplet emergence. To definitively label the ER, we engineered OP9 cells to retain a GFP-labeled protein in the ER lumen. The ER in these cells forms a clearly visible tubular reticulum at the cell periphery as shown in Fig. 1. To test the hypothesis that lipid droplets emerge from the ER, we drove TG synthesis with fatty acid and then co-imaged the GFP-labeled ER and the EPATs perilipin 3 or perilipin 4. We have previously shown that in OP9 cells and most other cells under standard culture conditions, perilipin 3 is found diffusely throughout the cytosol (1, 3). We found (Fig. 1) that under conditions of fatty acid-driven TG synthesis, the EPATs perilipin 3 (Fig. 1A) and perilipin 4 (Fig. 1B) form puncta falling along the tubular ER. In fact, when TG precursors (fatty acids) are added, the EPAT-coated structures are seen alongside ER tubules. The puncta do not overlap with the ER lumen, suggesting they are localized to the cytosolic face of the tubules. These data suggest that during lipid storage EPAT-coated nascent lipid droplets emerge from the ER. We cannot resolve the relationship of ER with lipid droplets in the perinuclear region because of the thickness and dense packing of the ER and lipid droplets in this region. Thus it is not clear if lipid droplet emergence is restricted to a certain region of the ER.
FIGURE 1.

Perilipin 3 and 4 puncta fall along the tubular ER. OP9 cells transfected with KDEL-GFP to visualize the ER were seeded on coverslips. Then 1.8 mm albumin-bound oleate (5:1) was added to the media. After 1.5 h the cells were fixed and stained for endogenous lipid droplet proteins. A shows a cell stained with anti-perilipin 3C (1.6 μg/ml), and B shows cells with anti-N-perilipin 4 (1.1 μg/ml). Antibody staining is shown as red, and GFP fluorescence is shown as green. Insets are expanded 270%. Bar, 10 μm in all micrographs in this paper.
Perilipin 3 Can Be Driven onto the Tubular ER
We hypothesized that EPATs form the coat that traffics intracellular lipid in a manner similar to the coats that traffic soluble cargo in aqueous-cored vesicles. The use of the G protein inhibitor AlF4− has been useful in dissecting the trafficking of aqueous-cored vesicle transport. AlF4− treatment often blocks the uncoating of vesicles, resulting in the accumulation of coat protein on the membrane from which that vesicle buds (26). These AlF4−-sensitive vesicles include proteins that mediate trafficking between the ER and lipid droplets (15–17, 26). We hypothesized that AlF4− treatment would reveal the membrane of lipid droplet emergence. We treated cells with AlF4− while driving fat storage with fatty acid supply (Fig. 2C). As shown in Fig. 2A in untreated OP9 cells, perilipin 3 is mostly diffuse, and perilipin 3 staining of lipid droplets is rare. Fatty acids alone, as previously shown (compare Fig. 2, A and B), drive perilipin 3 onto lipid droplets (1, 3). Treatment with AlF4− in combination with fatty acid caused perilipin 3 in some cells to coat a peripheral reticulum that is reminiscent of smooth ER (Fig. 2C), a staining pattern that was not observed in any of the control cells. AlF4− alone did not appreciably perturb perilipin 3 staining as shown in Fig. 2, D and J. These data suggest that some cytosolic perilipin 3 translocated to a region near the peripheral tubular ER following oleic acid addition in the presence of AlF4−, suggesting that lipid droplets emerge from ER structures. Thus, AlF4− likely revealed an otherwise inconspicuous compartment of perilipin 3 traffic.
FIGURE 2.

Pharmacologic manipulation of perilipin 3 trafficking. OP9 cells were seeded on coverslips. A–F were labeled with anti-N-perilipin 3 (600 ng/ml). A shows untreated cells. All other panels show cells fixed after 3 h of the following treatments: B, 1.8 mm albumin-bound oleate; C, AlF4− and 1.8 mm albumin-bound oleate; D and G, AlF4−; E and H, AlF4− and 10 μm forskolin (Forsk); and F and I, AlF4−, 10 μm forskolin, and 10 μm RHC80267 (RHC). G–I were labeled with serum against perilipin 2 (diluted 1:2000). J shows the percent cells with lipid droplet staining as scored by a blinded observer after 3 h of the indicated combination of AlF4−, 10 μm forskolin, 10 μm RHC80267, and fatty acid (1.8 mm albumin-bound oleate). Each bar is the average of eight fields (number of coverslips in parentheses above the bar). Analyses of variance followed by Bonferroni's t tests show that the treatments fall into four groups (I–IV) that differ significantly (p < 0.01). Error bar, S.E.
Forskolin in Presence of AlF4− and RHC80267 Increases Perilipin 3 in Lipid Droplets
In addition to blocking vesicular trafficking, AlF4− activates adenylate cyclase and increases cAMP levels (27). Increasing cellular cAMP levels activates TG hydrolysis (28) and in 3T3-L1 adipocytes drives the EPAT perilipin 4 to lipid droplets (29). To test if the AlF4− effect was due to activation of adenylate cyclase, we stimulated the enzyme with forskolin. We found that forskolin alone had no effect on perilipin 3 (Fig. 2J). However in the presence of AlF4−, forskolin drives some perilipin 3 staining onto perilipin 2-coated lipid droplets (Fig. 2, E, H, and J). Fig. 2, E, F, and J, shows that the effect of forskolin/AlF4− is markedly and significantly enhanced by simultaneous addition of the lipase inhibitor RHC80267. This compound is most potent against DG lipases (20) suggesting that stabilizing the DG resulting from forskolin-induced lipolysis promotes perilipin 3 recruitment to lipid droplets.
In summary AlF4− treatment, under conditions associated with either TG synthesis (oleate) or breakdown (forskolin), caused perilipin 3 to coat either the cytosolic surface of the ER or that of the lipid droplet, respectively. The adenylate cyclase activator forskolin, when added alone, does not redistribute perilipin 3 suggesting that increasing cAMP levels is not sufficient to redistribute perilipin 3 in OP9 preadipocytes. The observation that inhibiting DG hydrolysis during TG mobilization enhances perilipin 3 recruitment to lipid droplets suggested a role for DG in the recruitment process.
DG Levels Regulate Perilipin 3 Distribution
Both fat synthesis and lipolysis share acylglycerol intermediates, most notably DG. Indeed DG is both the immediate precursor to TG and its first hydrolytic product. We therefore examined the possibility that perilipin 3 might be recruited to DG-rich membranes. We tested whether local enrichment in DG might promote perilipin 3 coating of lipid droplets or ER.
Biochemical Characterization of Perilipin 3 Recruitment and DG Levels in Lipid Droplets
To directly examine the possibility that perilipin 3 associated with DG-rich membrane leaflets, we treated cells with the AlF4−, forskolin, and DG lipase inhibitor mixture and then fractionated the cells (Fig. 3). As shown in Fig. 3 this treatment generated easily detectable DG from an undetectable level in the lipid droplet fraction. Under the same conditions perilipin 3 was recruited from the cytosol to the lipid droplet fraction. In contrast, the relative contents of membrane and lipid droplet markers did not change; specifically, the ER marker calnexin, perilipin 2, and TG were not altered by the treatment. The demonstration that DG levels are increased on lipid droplets, to which perilipin 3 is recruited, further implicates DG in perilipin 3 recruitment.
FIGURE 3.
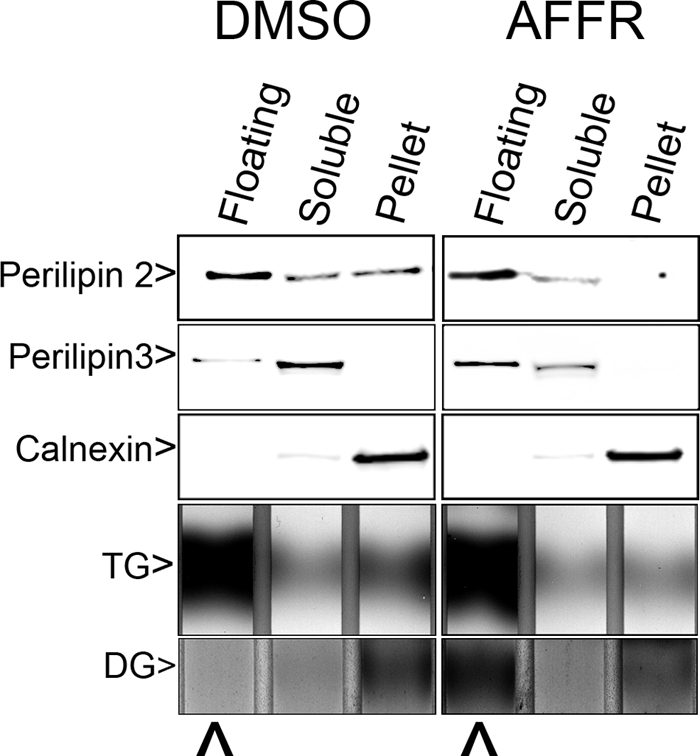
AlF4−-forskolin-RHC80267 treatments affect subcellular distribution of DG and TIP47. OP9 cells were treated with 2 μl/ml DMSO or AlF4−, 10 μm forskolin, 20 μm RHC80267 (AFFR) for 3 h. Then the cells were fractionated as described under “Materials and Methods.” Proteins were extracted and immunoblotted, and the extracted lipids were resolved by TLC. Floating, Soluble, and Pellet indicate samples are from the gradient top, bottom, or pellet, respectively. DG labels the region where 1,2-diolein migrates on the TLC lane. Immunoblots were loaded with proteins solubilized from 15 μl of each fraction, and TLC plates were loaded with lipid extracted from 200 μl of fraction. The immunoblots were probed with anti-perilipin 2 at 1 μg/ml, anti-N-perilipin 3 (400 ng/ml), or calnexin antiserum (diluted 1000-fold) as indicated. ⋀ points to 1,2-DG in the lipid floating fractions from each gradient.
Membrane-permeable DGs Drive Perilipin 3 onto the ER
To further test the hypothesis that perilipin 3 is recruited to DG-rich membrane leaflets, we treated cells with two membrane-permeable DGs, OAG and sn-1,2-dioctanoylglycerol (Fig. 4). OAG caused perilipin 3 to coat the ER as did sn-1,2-dioctanoylglycerol (data not shown). Adding OAG to the cells redistributes perilipin 3 dramatically (Fig. 4C) in a manner different from that observed with oleate addition (Fig. 4B). Furthermore when OAG is stabilized with triacsin C (inhibits acylation slowing OAG conversion to TG) or with RHC80267 (inhibits DG lipases slowing OAG conversion to monoacylglycerol), both the amount (data not shown) and frequency of perilipin 3 coating the ER increased. In addition the increase in perilipin 3 ER coating in the presence of both DG-stabilizing compounds is additive (Fig. 4H).
FIGURE 4.

Membrane-permeable DG drives perilipin 3 onto the ER. OP9 cells (A–C) or OP9-FLAG-DGAT1 cells (D–G) were seeded on coverslips. Cells were either fixed without treatment (A) or fixed 30 min after the following treatments (B–G): B, 1.8 mm albumin-bound oleate; C–E, 0.5 mm OAG; and F and G, 0.5 mm OAG and 10 μm triacsin C. Insets are expanded 150%. H shows the percent of cells with reticular staining (ER) as scored by a blinded observer after 30 min of treatment with the indicated combination of 0.5 mm OAG, 10 μm triacsin C, and 10 μm RHC80267. Each bar is the average of 12 fields from four coverslips. Analyses of variance followed by Bonferroni t tests show that OAG + triacsin C and OAG + RHC80267 are not different, and OAG and OAG + triacsin C are not different. All other pairs of conditions are different (p < 0.01). I shows OP9 FLAG-DGAT1 cells treated with 1 mm OAG and 20 μm RHC80267 for 30 min. From a single coverslip, two separate populations of cells were scored. First, cells were randomly selected and scored as to having low, medium, or high expression of FLAG-DGAT1. Second, cells showing reticular (ER) perilipin 3 staining were scored using the same criteria. The bars show the percent of cells scored as having low, medium, and high levels of FLAG-DGAT1. The open bars show the FLAG-DGAT1 level of randomly selected cells (n = 79). The solid bars show cells with reticular perilipin 3 staining (n = 40). The χ2 analysis shows that these distributions are different (p < 10−6).
In summary OAG drives perilipin 3 to the ER, and fatty acid drives it onto lipid droplets. These data are consistent with perilipin 3 having affinity for DG-rich membrane leaflets.
Decreasing ER DG Content by Expressing DGAT1 Attenuates ER Coating of Perilipin 3
We next tested the effect of removing DG by enhancing its conversion to TG. We engineered OP9 cells with increased capacity to clear DG from the ER by ectopically expressing the ER enzyme DGAT1, which acylates DG to make TG. We used an amino-terminal FLAG epitope to verify that DGAT1 localized to the ER (supplemental Fig. 2). Because OAG is a DG, it is presumably cleared by DGAT1. Thus, if perilipin 3 is recruited to OAG-rich membrane leaflets and OP9 FLAG-DGAT1 cells are able to clear the OAG faster, then these cells will be resistant to OAG-driven perilipin 3 ER coating. We found that OP9 FLAG-DGAT1 cells are in fact resistant to the effect of OAG. This difference is illustrated in Fig. 4, D and E, and quantified in the bar graph shown in Fig. 4I. The cell in Fig. 4E, top left, expresses FLAG-DGAT1 (Fig. 4E) and shows perilipin 3 coating round patent-cored structures, typical of lipid droplets. The cell in the middle of Fig. 4E lacks FLAG-DGAT1 and shows strong ER staining (Fig. 4D). These results further support the interpretation that perilipin 3 is recruited to DG-rich membranes.
DGAT1 enzymatic activity is presumably needed for DGAT1 to inhibit the perilipin 3 coating of the ER. To determine whether this is indeed the case, we treated FLAG-DGAT1 OP9 cells prior to OAG exposure with triacsin C, which inhibits DGAT1 by blocking formation of the DGAT1 substrate acyl-CoA. We found that under these conditions perilipin 3 coated the ER of OP9 FLAG-DGAT1-expressing cells (Fig. 4F), which then showed a staining similar to cells not expressing FLAG-DGAT1 (Fig. 4G). This result shows that DGAT1 activity is necessary for FLAG-DGAT1 expression to block perilipin 3 coating of the ER.
Cell Fractionation and Biochemical Validation
The above findings were validated further by conducting cell fractionation to examine the effect of OAG on perilipin 3 distribution. OP9 cells were treated with OAG, and a pellet enriched in ER was isolated. Content of perilipin 3 was related to that of the ER membrane marker calnexin. As shown in Fig. 5 OAG treatment caused an 8.5-fold increase of the ER perilipin 3 to calnexin ratio (p > 0.05).
FIGURE 5.

Membrane-permeable DG drives perilipin 3 onto an ER-enriched membrane pellet. A shows immunoblots of ER-enriched membrane pellets from OP9 cells. Untreated were not treated before fractionation. The following indicate additions to the culture media: oleate, 1.8 mm albumin-bound oleate; OAG, 0.5 mm OAG; OAG + Stauro., 0.5 mm OAG and 100 nm staurosporine. The top immunoblot was probed with anti-N-perilipin 3 (400 ng/ml), and the bottom immunoblot was probed with calnexin antiserum (1000-fold dilution). B is a summary of the data from four experiments such as that shown in A. To normalize between experiments the highest perilipin 3/calnexin measurement in each of the four independent experiments was corrected to 1, and this correction factor was applied to all measurements in that experiment. The asterisk indicates that for the No OAG condition, the perilipin 3/calnexin ratio is different from either OAG alone or OAG with staurosporine (St.) (analysis of variance, p < 0.05). The difference between OAG and OAG with staurosporine was not statistically significant.
DG Effect Is Not Mediated by Protein Kinase C (PKC) Activation
DG is a signaling molecule, and OAG is often used to activate PKC through DG binding (30). Thus it is possible that DG mediates perilipin 3 binding to the ER through the PKC signaling pathway. To test this possibility we blocked PKC signaling with the PKC inhibitor staurosporine (31) and treated OP9 cells with OAG. Staurosporine did not decrease the perilipin 3 ratio in the membrane pellet (Fig. 5). In addition when OAG redistribution of perilipin 3 was assessed morphologically, staurosporine treatment did not have a discernable effect (data not shown).
EPATs Perilipin 4 and Perilipin 5 Can Be Recruited to the ER by Increasing DG Levels
Perilipin 3 is described as an EPAT, because of its sequence and lipid binding behavior. There are two other proteins, perilipin 4 and perilipin 5, that share sequence similarity and lipid binding behavior with perilipin 3. To test whether OAG can drive the ER recruitment of perilipin 4, we treated OP9 cells with OAG and stained for perilipin 4 (Fig. 6A). Levels of perilipin 4 in subconfluent OP9 cells are low (18), so we were less able to detect it. However, we still found that in some cells perilipin 4 clearly coats the ER. To determine whether OAG drives the EPAT perilipin 5 to the ER, we engineered OP9 cells to ectopically express myc-perilipin 5. We treated these cells with OAG and found that myc-perilipin 5 coats the ER in response to OAG treatment. Furthermore perilipin 3 and myc-perilipin 5 distributions largely overlap (Fig. 6B and Fig. 5C). These observations suggest that association with DG-rich membranes is a characteristic shared by EPATs.
FIGURE 6.

Membrane-permeable DG drives all three EPATs onto the ER. OP9 cells (A) or OP9-myc-perilipin 5 (B and C) cells were seeded on coverslips. After 30 min of 0.5 mm OAG treatment, cells were fixed and labeled with anti-N-perilipin 4 at 1.6 μg/ml (A), anti-N-perilipin 3 at 600 ng/ml (B), or perilipin 5 sera diluted 1000-fold (C). Insets are expanded 200% (A) and 300% (B and C).
DISCUSSION
To define the relationships between lipid droplets and the ER, we engineered OP9 cells with GFP-labeled ER. These cells revealed that fatty acid-driven TG synthesis induces the emergence of perilipin 3- and perilipin 4-coated droplets at the branch points of tubular ER. We also show that when TG synthesis is driven in the presence of the membrane traffic inhibitor AlF4−, perilipin 3 coats the tubular ER, likely revealing an otherwise unidentifiable compartment of perilipin 3 trafficking. Through the use of various manipulations to alter DG content of ER or lipid droplet membranes, we show that a local increase in DG content is instrumental in recruiting the EPATs to either ER or lipid droplet cytosolic membrane leaflets. Treatments that generate DG or that promote DG accumulation by preventing its hydrolysis or its acylation to form TG caused perilipin 3 to coat the membranes where DG content is increased (Figs. 2–6). Specifically, membrane-permeable DGs cause perilipin 3 to coat the ER, and this coating is increased when either DG conversion to TG (triacsin C) or DG catabolism (RHC80267) was blocked with inhibitors. Consistent with a key role for DG level, the effects these inhibitors are additive when used in combination (Fig. 2J and Fig. 4H). Furthermore overexpression of the ER enzyme DGAT1, which reduces the DG pool by converting it to TG, attenuates DG-induced perilipin 3 coating of ER (Fig. 4, D, E, and I). This DGAT1-driven attenuation requires DGAT1 enzymatic activity (Fig. 4, F and G).
The DG-dependent perilipin 3 recruitment is not unique to the ER; perilipin 3 associates with lipid droplets when DG is generated though lipolysis, and membrane trafficking is blocked with AlF4−. Furthermore perilipin 3 coating of lipid droplets increases when DG hydrolysis is prevented with RHC80267 (Fig. 2J and Fig. 4H). The most parsimonious explanation for these data is that perilipin 3 associates with DG-rich membrane leaflets, a property that also appears to apply to a number of signaling proteins (32, 33).
The ER has been proposed as the site of lipid droplet emergence based on several observations. It is the site from which lipoproteins originate, and it is rich in acylation and neutral lipid synthetic enzymes (34–37). Ultrastructural studies also show lipid droplets in tight association with the ER (38, 39). In addition overexpression of apoB forms a lipid-filled structure with the PAT protein perilipin 2 (40) on the cytosolic side and apoB on the ER luminal side. However all the above evidence is indirect, and this model is still not universally accepted (41). Our results using cells in which the ER was visualized provide evidence that perilipin 3- and perilipin 4-coated structures emerge next to ER tubules. In addition we demonstrate that perilipin 3 can be driven onto peripheral ER tubules by various manipulations that alter DG levels. Thus observations in this work more directly support the notion that lipid droplets emerge from the ER.
The influence of DG levels on subcellular distributions of signaling proteins (32, 33) is proposed to involve an increase in the affinity of specific proteins for membranes and/or downstream events caused by membrane recruitment of proteins with DG affinity (42). Our data do not reveal the mechanism by which EPATs are recruited to DG-rich membrane leaflets. The two acyl groups of DG and the tiny polar head group, however, disrupt the hydrophilic face of the membrane leaflet. Thus EPATs, which are composed largely of amphipathic helices, may embed these in the membrane leaflets once DG destabilizes the hydrophilic face of the membrane leaflet. An equally plausible explanation is that EPAT recruitment is indirect. Sensitivity of EPAT trafficking to AlF4− implicates G proteins, and the small G protein ARF1 (ADP-ribosylation factor 1) has been shown to play a role in trafficking to lipid droplets (15–17, 43, 44). In addition ARFs have been shown to bind membranes via amphipathic helices (45), and it has been suggested that ARF-GAP1 membrane recruitment is DG-dependent (46). Finally G proteins and direct binding of EPATs may mediate the assembly of the protein coat around intracellular fat.
Our data documented that DG recruits EPATs to lipid droplets, but the functional implications of this recruitment remain to be determined. After lipid droplet emergence from the ER several observations suggest that nascent lipid droplets are specialized TG synthesis compartments. The droplets grow in size from under 1 μm to several microns in diameter by a mechanism other than fusion (3), suggesting that acylation occurs on nascent droplets. DGAT2 moves from the ER to lipid droplets when cells are treated with fatty acid (36, 47),3 and acyl-CoA synthase has been found in the lipid droplet fractions (29, 48–50). Isolated lipid droplets from fatty acid-treated DGAT2-transfected cells contain a large fraction of the cellular TG synthesis capacity (33). Finally, the bulk of labeled fatty acid pulses is transiently found incorporated into DG in lipid droplets (47). Thus all of the components necessary to convert DG to TG are found on lipid droplets. The recruited EPATs might contribute to the protection or appropriate trafficking of the nascent lipid. DG recruitment of EPATs to the ER may control the branch point between lipid storage and membrane expansion, thus linking droplet formation to energy availability and cell growth.
Supplementary Material
Acknowledgments
We thank Dr. Brian Finck for helpful discussions, Dr. Pamela Skinner for careful editing, and Dr. Leroy Wolins for designing and performing the statistical analysis. N. E. W. thanks Dr. Perry Bickel for past support without which this work would not have been possible.
This work was supported, in whole or in part, by National Institutes of Health Grant R01DK 33301 (to N. A. A.). This work was supported by American Diabetes Association Grant 7-06-JF-69 (to N. E. W.) and by Clinical Nutrition Research Unit Grant DK56351 through a pilot and feasibility grant (to N. E. W.) for use of the Adipocyte Biology Core facility.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
N. E. Wolins, unpublished observations.
- TG
- triacylglycerol
- DG
- diacylglycerol
- PAT
- perilipin, adipophilin, TIP47
- EPAT
- exchangeable PAT lipid droplet proteins
- CPAT
- constitutive PAT lipid droplet proteins
- DGAT
- diacylglycerol acyltransferase
- OAG
- 1-oleoyl-2-acetyl-sn-glycerol
- GFP
- green fluorescent protein
- BAT
- brown adipose tissue
- WAT
- white adipose tissue
- ER
- endoplasmic reticulum
- PKC
- protein kinase C.
REFERENCES
- 1.Wolins N. E., Rubin B., Brasaemle D. L. (2001) J. Biol. Chem. 276, 5101–5108 [DOI] [PubMed] [Google Scholar]
- 2.Wolins N. E., Skinner J. R., Schoenfish M. J., Tzekov A., Bensch K. G., Bickel P. E. (2003) J. Biol. Chem. 278, 37713–37721 [DOI] [PubMed] [Google Scholar]
- 3.Wolins N. E., Quaynor B. K., Skinner J. R., Schoenfish M. J., Tzekov A., Bickel P. E. (2005) J. Biol. Chem. 280, 19146–19155 [DOI] [PubMed] [Google Scholar]
- 4.Wolins N. E., Quaynor B. K., Skinner J. R., Tzekov A., Croce M. A., Gropler M. C., Varma V., Yao-Borengasser A., Rasouli N., Kern P. A., Finck B. N., Bickel P. E. (2006) Diabetes 55, 3418–3428 [DOI] [PubMed] [Google Scholar]
- 5.Wolins N. E., Brasaemle D. L., Bickel P. E. (2006) FEBS Lett. 580, 5484–5491 [DOI] [PubMed] [Google Scholar]
- 6.Miura S., Gan J. W., Brzostowski J., Parisi M. J., Schultz C. J., Londos C., Oliver B., Kimmel A. R. (2002) J. Biol. Chem. 277, 32253–32257 [DOI] [PubMed] [Google Scholar]
- 7.Kimmel A. R., Brasaemle D. L., McAndrews-Hill M., Sztalryd C., Londos C. (2009) J. Lipid Res., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brasaemle D. L., Barber T., Kimmel A. R., Londos C. (1997) J. Biol. Chem. 272, 9378–9387 [DOI] [PubMed] [Google Scholar]
- 9.Abell B. M., Hahn M., Holbrook L. A., Moloney M. M. (2004) Plant J. 37, 461–470 [DOI] [PubMed] [Google Scholar]
- 10.Turró S., Ingelmo-Torres M., Estanyol J. M., Tebar F., Fernández M. A., Albor C. V., Gaus K., Grewal T., Enrich C., Pol A. (2006) Traffic 7, 1254–1269 [DOI] [PubMed] [Google Scholar]
- 11.Tansey J. T., Sztalryd C., Hlavin E. M., Kimmel A. R., Londos C. (2004) IUBMB Life 56, 379–385 [DOI] [PubMed] [Google Scholar]
- 12.Brasaemle D. L. (2007) J. Lipid Res. 48, 2547–2559 [DOI] [PubMed] [Google Scholar]
- 13.Bonifacino J. S., Lippincott-Schwartz J. (2003) Nat. Rev. Mol. Cell Biol. 4, 409–414 [DOI] [PubMed] [Google Scholar]
- 14.Murphy D. J., Vance J. (1999) Trends Biochem. Sci. 24, 109–115 [DOI] [PubMed] [Google Scholar]
- 15.Beller M., Sztalryd C., Southall N., Bell M., Jäckle H., Auld D. S., Oliver B. (2008) PLoS Biol. 6, e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo Y., Walther T. C., Rao M., Stuurman N., Goshima G., Terayama K., Wong J. S., Vale R. D., Walter P., Farese R. V. (2008) Nature 453, 657–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soni K. G., Mardones G. A., Sougrat R., Smirnova E., Jackson C. L., Bonifacino J. S. (2009) J. Cell Sci. 122, 1834–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolins N. E., Quaynor B. K., Skinner J. R., Tzekov A., Park C., Choi K., Bickel P. E. (2006) J. Lipid Res. 47, 450–460 [DOI] [PubMed] [Google Scholar]
- 19.Lewin T. M., Kim J. H., Granger D. A., Vance J. E., Coleman R. A. (2001) J. Biol. Chem. 276, 24674–24679 [DOI] [PubMed] [Google Scholar]
- 20.Sutherland C. A., Amin D. (1982) J. Biol. Chem. 257, 14006–14010 [PubMed] [Google Scholar]
- 21.Cheng D., Meegalla R. L., He B., Cromley D. A., Billheimer J. T., Young P. R. (2001) Biochem. J. 359, 707–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ory D. S., Neugeboren B. A., Mulligan R. C. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 11400–11406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Snapp E. L., Iida T., Frescas D., Lippincott-Schwartz J., Lilly M. A. (2004) Mol. Biol. Cell 15, 4512–4521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolins N., Bosshart H., Küster H., Bonifacino J. S. (1997) J. Cell Biol. 139, 1735–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwartz D. M., Wolins N. E. (2007) J. Lipid Res. 48, 2514–2520 [DOI] [PubMed] [Google Scholar]
- 26.Finazzi D., Cassel D., Donaldson J. G., Klausner R. D. (1994) J. Biol. Chem. 269, 13325–13330 [PubMed] [Google Scholar]
- 27.Sternweis P. C., Gilman A. G. (1982) Proc. Natl. Acad. Sci. U.S.A. 79, 4888–4891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carey G. B. (1998) Adv. Exp. Med. Biol. 441, 157–170 [DOI] [PubMed] [Google Scholar]
- 29.Brasaemle D. L., Dolios G., Shapiro L., Wang R. (2004) J. Biol. Chem. 279, 46835–46842 [DOI] [PubMed] [Google Scholar]
- 30.Wang R., Zhang L., Yin D., Mufson R. A., Shi Y. (1998) J. Immunol. 161, 2201–2207 [PubMed] [Google Scholar]
- 31.Tamaoki T., Nomoto H., Takahashi I., Kato Y., Morimoto M., Tomita F. (1986) Biochem. Biophys. Res. Commun. 135, 397–402 [DOI] [PubMed] [Google Scholar]
- 32.Kazanietz M. G. (2002) Mol. Pharmacol. 61, 759–767 [DOI] [PubMed] [Google Scholar]
- 33.Colón-González F., Kazanietz M. G. (2006) Biochim. Biophys. Acta 1761, 827–837 [DOI] [PubMed] [Google Scholar]
- 34.Lin S., Cheng D., Liu M. S., Chen J., Chang T. Y. (1999) J. Biol. Chem. 274, 23276–23285 [DOI] [PubMed] [Google Scholar]
- 35.Joyce C. W., Shelness G. S., Davis M. A., Lee R. G., Skinner K., Anderson R. A., Rudel L. L. (2000) Mol. Biol. Cell 11, 3675–3687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stone S. J., Levin M. C., Zhou P., Han J., Walther T. C., Farese R. V., Jr. (2009) J. Biol. Chem. 284, 5352–5361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shockey J. M., Gidda S. K., Chapital D. C., Kuan J. C., Dhanoa P. K., Bland J. M., Rothstein S. J., Mullen R. T., Dyer J. M. (2006) Plant Cell 18, 2294–2313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blanchette-Mackie E. J., Dwyer N. K., Barber T., Coxey R. A., Takeda T., Rondinone C. M., Theodorakis J. L., Greenberg A. S., Londos C. (1995) J. Lipid Res. 36, 1211–1226 [PubMed] [Google Scholar]
- 39.Cushman S. W. (1970) J. Cell Biol. 46, 326–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohsaki Y., Cheng J., Suzuki M., Fujita A., Fujimoto T. (2008) J. Cell Sci. 121, 2415–2422 [DOI] [PubMed] [Google Scholar]
- 41.Robenek H., Buers I., Hofnagel O., Robenek M. J., Troyer D., Severs N. J. (2008) Biochim. Biophys. Acta, in press [DOI] [PubMed] [Google Scholar]
- 42.Carrasco S., Mérida I. (2007) Trends Biochem. Sci. 32, 27–36 [DOI] [PubMed] [Google Scholar]
- 43.Nakamura N., Banno Y., Tamiya-Koizumi K. (2005) Biochem. Biophys. Res. Commun. 335, 117–123 [DOI] [PubMed] [Google Scholar]
- 44.Nakamura N., Akashi T., Taneda T., Kogo H., Kikuchi A., Fujimoto T. (2004) Biochem. Biophys. Res. Commun. 322, 957–965 [DOI] [PubMed] [Google Scholar]
- 45.Lundmark R., Doherty G. J., Vallis Y., Peter B. J., McMahon H. T. (2008) Biochem. J. 414, 189–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nie Z., Randazzo P. A. (2006) J. Cell Sci. 119, 1203–1211 [DOI] [PubMed] [Google Scholar]
- 47.Kuerschner L., Moessinger C., Thiele C. (2008) Traffic 9, 338–352 [DOI] [PubMed] [Google Scholar]
- 48.Liu P., Ying Y., Zhao Y., Mundy D. I., Zhu M., Anderson R. G. (2004) J. Biol. Chem. 279, 3787–3792 [DOI] [PubMed] [Google Scholar]
- 49.Beller M., Riedel D., Jänsch L., Dieterich G., Wehland J., Jäckle H., Kühnlein R. P. (2006) Mol. Cell. Proteomics 5, 1082–1094 [DOI] [PubMed] [Google Scholar]
- 50.Sato S., Fukasawa M., Yamakawa Y., Natsume T., Suzuki T., Shoji I., Aizaki H., Miyamura T., Nishijima M. (2006) J. Biochem. 139, 921–930 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.