

Abstract
Mutations in pyridoxine 5′-phosphate oxidase are known to cause neonatal epileptic encephalopathy. This disorder has no cure or effective treatment and is often fatal. Pyridoxine 5′-phosphate oxidase catalyzes the oxidation of pyridoxine 5′-phosphate to pyridoxal 5′-phosphate, the active cofactor form of vitamin B6 required by more than 140 different catalytic activities, including enzymes involved in amino acid metabolism and biosynthesis of neurotransmitters. Our aim is to elucidate the mechanism by which a homozygous missense mutation (R229W) in the oxidase, linked to neonatal epileptic encephalopathy, leads to reduced oxidase activity. The R229W variant is ∼850-fold less efficient than the wild-type enzyme due to an ∼192-fold decrease in pyridoxine 5′-phosphate affinity and an ∼4.5-fold decrease in catalytic activity. There is also an ∼50-fold reduction in the affinity of the R229W variant for the FMN cofactor. A 2.5 Å crystal structure of the R229W variant shows that the substitution of Arg-229 at the FMN binding site has led to a loss of hydrogen-bond and/or salt-bridge interactions between FMN and Arg-229 and Ser-175. Additionally, the mutation has led to an alteration of the configuration of a β-strand-loop-β-strand structure at the active site, resulting in loss of two critical hydrogen-bond interactions involving residues His-227 and Arg-225, which are important for substrate binding and orientation for catalysis. These results provide a molecular basis for the phenotype associated with the R229W mutation, as well as providing a foundation for understanding the pathophysiological consequences of pyridoxine 5′-phosphate oxidase mutations.
Introduction
Neonatal epileptic encephalopathy (NEE)3 is a severe seizure disorder, which is often unresponsive to treatment with conventional antiepileptic drugs (1). Several cases of NEE have been documented throughout the world especially in the Turkish and Asian populations (1). Clinical phenotypes and other general features observed include fetal distress, hypoglycemia, acidosis, anemia, and asphyxia (1, 2). NEE often manifests within hours of birth with intractable seizures. Progressive deterioration can lead to death within weeks (1). Cerebrospinal fluid and urine analyses indicate reduced activities of vitamin B6-dependent (PLP-dependent) enzymes. Surviving children are usually mentally retarded and have an abnormal dependence on vitamin B6 in the form of pyridoxal 5′-phosphate (PLP) (1).
PLP, the active form of vitamin B6, is used to activate a wide variety of PLP-dependent enzymes involved in amino acid metabolism, as well as those involved in several other metabolic pathways (3–7). Eukaryotes cannot synthesize PLP from small metabolites by a de novo pathway and require the primary dietary B6 vitamers, pyridoxal (PL), pyridoxine (PN), or pyridoxamine (PM), for the synthesis of PLP via a so-called salvage pathway. Two enzymes are responsible for this salvage pathway: pyridoxal kinase (PL kinase) and pyridoxine 5′-phosphate oxidase (PNP oxidase). The first enzyme catalyzes the phosphorylation of the 5′-alcohol group of PN, PM, and PL to form pyridoxine 5′-phosphate (PNP), pyridoxamine 5′-phosphate (PMP), and PLP, respectively. Both PNP and PMP are subsequently converted to PLP in an FMN-dependent oxidative process catalyzed by PNP oxidase. Interest in these B6 salvage enzymes is based upon their key roles in brain metabolism. Synthesis of most neurotransmitters, including dopamine, γ-aminobutyric acid, serotonin, histamine, d-serine, and epinephrine, involves PLP-dependent enzymes. Disruption of the salvage pathway due to pathogenic mutations, inhibition of the salvage enzymes, or lack of nutritional intake of vitamin B6 is suspected to lead to neurological disorders, as well as non-neurological pathologies (1, 2, 8–24).
Clinical studies have shown that seizures resulting from NEE significantly decreased with administration of PLP, whereas PN had no effect (1, 2). This suggested that NEE might involve a defective PNP oxidase, which was unable to convert PNP or PMP to PLP. Sequencing of the PNP oxidase coding gene in patients with NEE identified homozygous missense (R229W), splice site (IVS3-1g > a), or stop codon (X262Q) mutations (1). Studies with Chinese hamster ovary cells showed the R229W mutation resulting in a drastic reduction of PNP oxidase activity (1). The splice and stop codon mutations resulted in null PNP oxidase activity. Recently, other mutations, including a nonsense (A174X), as well as two homozygous missense mutations (R95H and R95C) in PNP oxidase, have also been identified in several patients with classical symptoms of NEE (21, 25–27). The precise mechanism governing how these mutations lead to partial or complete loss of function is not yet understood.
PNP oxidase is the smallest member of the flavin-containing oxidase family. There is considerable sequence homology between these enzymes, and detailed sequence analysis has been reported (28, 29). PNP oxidase has been purified and characterized from various sources, including human, sheep, rat, pig, rabbit, Escherichia coli, and a few other prokaryotes (28–38). The crystal structures of human, E. coli, Mycobacterium tuberculosis, and Desulfovibrio vulgaris enzymes are known, all of which show a similar two-fold related dimer with two FMN molecules bound at the dimer interface (28, 29, 33). The putative catalytic mechanism involves a direct hydride transfer from C4′ of PNP (or PMP) to N5 of FMN to form PLP (33). From the crystal structure of PNP oxidase, several active site residues are apparently involved in binding of the substrates, including His-227 and Arg-225 (33).
We describe the crystal structure of the defective human PNP oxidase R229W at 2.5 Å resolution. The mutation has resulted in structural changes in the active site geometry with loss of hydrogen-bond and/or salt-bridge interactions from Arg-229 and Ser-175 to FMN. In addition, the mutation has led to loss of two essential hydrogen-bond interactions from His-227 and Arg-225 to the substrate PNP. These findings are consistent with steady-state kinetics, as well as FMN affinity measurements performed with the wild-type enzyme and the two R229 variants we studied (R229W and R229Q). Taken together, these results provide molecular insight into how the R229W missense mutation affects PNP oxidase catalytic activity.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis, Expression, and Purification
The R229W and R229Q mutants were made on a cDNA coding for human PNP oxidase, which was cloned into the expression vector pET28a(+) as described previously (29). Site-directed mutagenesis was performed using the QuikChange kit from Stratagene (La Jolla, CA). Primers were synthesized by Integrated DNA Technologies: R229W forward, 5′-CCGCCTGCATGACTGGATAGTCTTTCGGCG-3′, reverse, 5′-CGCCGAAAGACTATCCAGTCATGCAGGCG-3′; R229Q forward, 5′-CCGCCTGCATGACCAGATAGTCTTTCGGCG-3′, reverse, 5′-CGCCGAAAGACTATCTGGTCATGCAGGCG-3′, with the underlined nucleotides corresponding to the codon changes. The mutations were confirmed by sequence analysis of the constructs. Plasmids were purified using a QIAprep miniprep kit from Qiagen. Competent E. coli Rosetta(λDE3)pLysS cells (Novagen) were transformed with recombinant pET28(a)+ carrying the R229W or R229Q human PNP oxidase insert. Transformants were grown in LB medium containing kanamycin (40 μg/ml) and chloramphenicol (34 μg/ml) to an absorbance of 0.7 and then induced with 50–100 μm isopropyl-β-d-thiogalactopyranoside for 28 h at 18 °C. Cells were harvested by centrifugation and rapidly ruptured by high pressure homogenization in an AVESTIN cell disrupter. After centrifugation, the expressed mutant proteins were purified by metal chelation chromatography as described previously (29).
Kinetic Studies
Routine enzymatic assays of PNP oxidase were performed in 50 mm Tris-BES buffer, pH 7.6, with 10 μm FMN and 1 mm dithiothreitol. Assays were done in a water-jacketed 10-cm path length cuvette at 37 °C. The reaction was initiated with the addition of PNP, and the progress of the reaction was followed at 414 nm, where the characteristic aldimine product PLP-Tris absorbs maximally with a molar absorbance coefficient of 5,900 cm−1 m−1. Kinetic constant measurements for the mutant enzymes were performed in a reaction mixture of 12 ml containing a fixed concentration of the R229 mutant (typically, 10 μm) and varying PNP concentrations between 0.1 and 2.5 mm. Initial velocities were determined over the first 60 s of the reaction. The values of Km and kcat were determined from double reciprocal plots of the initial velocity and substrate concentration. A similar kinetic study was conducted with the wild-type enzyme.
Measurement of the Dissociation Constant (Kd) of FMN Binding
The Kd values for FMN binding to the wild-type and the R229W and R229Q mutant enzymes were analyzed, taking advantage of FMN fluorescence quenching, observed upon binding of the cofactor to apoPNP oxidase. ApoPNP oxidase (from 5 nm to 3 μm) was added to FMN samples (45 nm) at 25 °C in 50 mm sodium phosphate buffer, pH 7.2, containing 1 mm dithiothreitol. Preliminary experiments demonstrated that the binding equilibrium was established within 5 min from mixing with all enzyme forms. Fluorescence emission spectra (470–570 nm; 4-nm emission slit) were recorded with a Horiba Jobin-Ivon FluoroMax-3 spectrofluorometer, with excitation wavelength set at 450 nm (1-nm excitation slit), using a 1-cm path length quartz cell. Data were analyzed according to Equation 1, in which Frel is the measured relative fluorescence at 525 nm, F0 is fluorescence in the absence of apoPNP oxidase, Finf is fluorescence at infinite apoPNP oxidase concentration, [APO] is the total apoenzyme concentration, [FMN] the total cofactor concentration, and Kd is the dissociation constant of the equilibrium APO + FMN ↔ HOLO
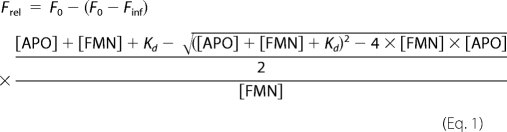 |
The fraction in Equation 1 corresponds to the fraction of enzyme-bound FMN at equilibrium ([HOLOeq]/[FMN]). [HOLOeq] was derived from the equation for the dissociation constant of the binding equilibrium,
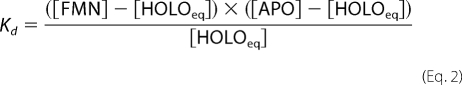 |
as one of the two solutions of the quadratic equation
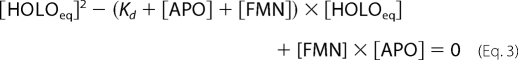 |
Determination of Enzyme Stability
Protein samples (apo and holo forms) of wild-type and R229W enzymes (2.3 μm) in 50 mm sodium Hepes buffer, pH 7.2, containing 0.2 μm dithiothreitol and 0.1 μm EDTA, were heated from 30 to 95 °C with a heating rate of 1 °C/min controlled by a Jasco programmable Peltier element. The dichroic activity at 220 nm was monitored continuously every 0.5 °C. All thermal scans were corrected for solvent contribution at the different temperatures. The observed transitions were irreversible, and the spectra measured at the end of the cooling phase differed from those of the protein at 30 °C. Melting temperature (Tm) values were calculated by taking the first derivative of the ellipticity at 220 nm with respect to temperature. All denaturation experiments were performed in triplicate.
Crystallization, Data Collection, and Structure Refinement
Crystals of R229W were obtained by the hanging drop vapor diffusion method with 5-μl drops consisting of a 1:1 ratio of protein:reservoir solution equilibrated against 700 μl of reservoir solution at 15 °C. Protein concentrations varied from 10 to 28 mg/ml in 50 mm sodium phosphate, pH 7.0, containing 1 mm dithiothreitol. The optimal reservoir solution was 0.5 m ammonium dihydrogen phosphate in 0.1 m sodium citrate buffer, pH 5.7. Suitable crystals of R229W for x-ray analysis appeared in about 2 weeks. The cell constants and space group are isomorphous with wild-type crystals of human PNP oxidase. The crystals belong to the trigonal space group P3121 with unit cell parameters: a = b = 82.3, c = 58.8 Å. X-ray data sets were collected at 100 K on an R-Axis IV++ image plate detector using CuKα X-rays (λ = 1.54 Å) from a Rigaku MicroMaxTM-007 x-ray source equipped with Rigaku/MSC Varimax confocal optics operating at 40 kV and 20 mA. The crystals were transferred with a cryoloop into 0.7 m ammonium dihydrogen phosphate solution in 70 mm sodium citrate buffer, pH 5.7, containing additional 26% ethylene glycol and subsequently flash-cooled in a cold nitrogen gas stream from an X-stream cryosystem (Rigaku/MSC). Crystals diffracted to 2.5 Å resolution. The data were processed and scaled with the Rigaku/MSC d*TREK software program.
An initial model of the wild-type human PNP oxidase structure (PDB accession number 1NRG), mutating Arg-229 to Ala and omitting bound FMN, PLP, phosphate, and water molecules, was placed in the unit cell of the R229W crystal with rigid body refinement as implemented in CNS version 1.0. This model was refined via alternating cycles of manual fitting into SIGMAA-weighted 2Fo − Fc maps in COOT (39) and computational refinement in CNS (40). Repeated refinement and model building yielded a model that included residues 48–238, 242–261, one each of FMN and PLP molecules, three phosphate anions, and 50 water molecules, with final R and Rfree values of 0.226 and 0.287, respectively, for all reflections at the resolution range of 22.6–2.5 Å (Table 1).
TABLE 1.
Crystallographic data and refinement statistics
| Data collectiona | |
| Space group | P3121 |
| Cell dimensions (Å) | 82.3, 82.3, 58.8 |
| Resolution (Å) | 35.63-2.5 (2.59-2.5) |
| No. of measurements | 79,382 (8447) |
| Unique reflections | 8,215 (800) |
| I/sigma I | 22.5 (6.5) |
| Completeness (%) | 99.8 (100) |
| Rmerge (%)b | 5.6 (33.0) |
| Refinement | |
| Resolution limit (Å) | 22.02–2.5 (2.61–2.5) |
| R-factor (%) | 22.6 (32.4) |
| Rfree (%)c | 28.7 (39.9) |
| No. of reflections | 8,195 (1000) |
| Completeness (%) | 99.9 (100) |
| r.m.s.d. standard geometry | |
| Bond lengths (Å)/angles (°) | 0.008/1.3 |
| Dihedral angles | |
| Most favored/allowed regions | 88.8/11.2 |
| Average B-factors | |
| All atoms/Protein/water | 61.3/61.2/60.3 |
| FMN/PLP/phosphate | 43.2/69.9/98.6 |
a Numbers in parentheses refer to the outermost resolution bin.
b Rmerge = ΣhklΣi|Ihkli − 〈Ihkli〉|/ΣhklΣi〈Ihkli〉.
c Rfree calculated with 5% of excluded reflection from the refinement.
RESULTS AND DISCUSSION
PNP Oxidase R229W and R229Q Mutations Significantly Affect the Catalytic Efficiency of the Enzyme
A previous study with Chinese hamster ovary cells showed that the R229W mutation resulted in a 70% reduction in PNP oxidase catalytic activity as compared with the wild-type enzyme (1). To precisely understand how this mutation affects the enzymatic activity, the PNP oxidase R229W mutant was constructed, expressed, and purified. The R229Q variant was also made for comparative studies. Unlike Trp, Gln has a similar size to Arg, and thus steric interference may not be a factor for any observable changes in the oxidase activity. Size-exclusion chromatography showed the mutant and wild-type enzymes to have similar elution profiles. CD spectra also showed no significant secondary structure differences between the enzymes. These data suggest that the mutations did not affect the oligomerization state or the overall structural integrity of the enzyme (data not shown).
Enzymatic activity measurements of the wild-type and the R229 mutant enzymes were carried out by following the formation of the aldimine complex of PLP with Tris as published previously (29). Km and kcat values of wild-type and mutant forms are listed in Table 2. Assays were performed in the presence of an excess concentration of FMN with respect to the protein to ensure that each enzyme was saturated with this cofactor. The results show that the kcat of R229W and R229Q mutants is 4.5- and 3.6-fold lower, respectively, than the kcat of the wild-type enzyme. The Km for PNP decreased by 192- and 129-fold, respectively, with respect to the wild-type enzyme. Kinetic studies clearly show that the mutations lead to a significant loss of catalytic efficiency (kcat/Km) of the enzymes, which is about 865- and 470-fold less for the R229W and R229Q enzymes, respectively, as compared with the wild-type enzyme.
TABLE 2.
Kinetic constants for wild-type and mutant human PNP oxidase
Enzymatic activity of wild-type, R229W, and R229Q enzymes was measured by following the formation of aldimine complex of PLP with Tris at 414 nm. Km and kcat values are average of at least 3 measurements.
| Enzyme | Km for PNP | kcat |
|---|---|---|
| μm | s−1 | |
| Wild type | 2.4 ± 0.2 | 0.18 ± 0.03 |
| R229W | 461 ± 27 | 0.04 ± 0.01 |
| R229Q | 310 ± 20 | 0.05 ± 0.01 |
PNP Oxidase R229W and R229Q Mutations Reduce FMN Affinity
The crystal structure of wild-type PNP oxidase shows the substrate (PNP) or product (PLP) situated at the re face of FMN with close hydrophobic contacts between the two molecules (29, 36). Arg-229 contributes to a highly conserved region of the active site of the PNP oxidase family. This residue makes an ion-pair interaction with the FMN phosphate. The question we ask is whether the loss of this interaction, as well as the change in the electrostatics of the FMN phosphate binding site due to the mutation, would lead to attenuation of FMN binding. To measure the dissociation constant of the FMN binding equilibrium, we performed fluorescence titration experiments with the apo form of wild-type, R229W, and R229Q enzymes. The FMN was excited at 450 nm, and the change in FMN fluorescence, due to binding to apoPNP oxidase, was monitored between 470 and 570 nm (Fig. 1). The dissociation constants of FMN obtained for the wild-type enzyme and the R229 mutants are shown in Table 3. Expectedly, the R229W mutant binds the cofactor with a dissociation constant that is ∼50-fold higher as compared with that of the wild-type enzyme, whereas the R229Q mutant showed ∼3-fold higher Kd as compared with the wild-type enzyme.
FIGURE 1.

Fluorescence emission spectra of FMN (excitation wavelength of 450 nm) upon binding to apo forms of PNP oxidase. Continuous line, wild type; dashed line, R229Q; dotted line, R229W.
TABLE 3.
Dissociation constants (Kd) for FMN binding to human PNP oxidase
Dissociation constant for FMN with apo forms of wild-type, R229W, and R229Q enzymes were obtained by monitoring the decrease in the relative fluorescence emission intensity of FMN at 525 nm (excitation λ = 450 nm) due to fluorescence quenching of the flavin upon binding to the enzyme. Kd is an average of at least three measurements.
| Enzyme | Kd |
|---|---|
| nm | |
| Wild type | 13.8 ± 1.8 |
| R229W | 672 ± 65 |
| R229Q | 36.6 ± 7.6 |
The R229W Mutation Does Not Affect Protein Stability
The thermal stability of wild-type and R229W PNP oxidases was investigated by monitoring the change of ellipticity at 220 nm in the far-UV CD spectrum of the enzymes, when the temperature was increased from 30 to 95 °C. The observed sigmoid transitions were irreversible. The parameter chosen to compare the transition curves was the apparent melting temperature (Tm), defined as the mid-point of the sigmoid denaturation process and calculated by plotting the first derivative of the molar ellipticity values as a function of temperature. The Tm of holo wild-type and R229W PNP oxidases turned out to be similar to each other (79.5 and 82 °C, respectively). Consistently, the activity of the mutant enzymes, like that of the wild type, stayed constant over several weeks after purification when the protein was kept at −20 °C. In addition, the integrity of all proteins remained intact when checked by SDS-PAGE. The apoenzymes gave Tm values of 82 and 86 °C, respectively.
Crystallographic Studies
We undertook structural studies by crystallizing the R229W mutant form in complex with FMN to elucidate the structural basis of the reduced enzymatic activity. The R229W complex crystallized in a trigonal cell. We have not been able to obtain x-ray quality crystals of the R229Q mutant form. The R229W structure was refined to 2.5 Å resolution using the published isomorphous wild-type structure (PDB code 1NRG). The overall protein fold and active site architecture of the wild-type human PNP oxidase have been discussed elsewhere (29). Comparison of mutant and wild-type structures shows identical overall folds (Fig. 2A) with a root mean square deviation of ∼0.6 Å. Both structures (in trigonal cells) are missing almost the first 47 N-terminal amino acid residues due to disorder. We also observe missing N-terminal residues in a previously published wild-type E. coli PNP oxidase structure in a trigonal cell (28, 36). We note that the missing N-terminal residues are resolved in a monoclinic cell of another E. coli structure as a result of stabilization by the symmetry-related molecules (33).
FIGURE 2.

Structure of PNP oxidase enzyme. All figures were drawn using PyMOL (45). A, least-squares superposition of human PNP oxidase R229W dimeric structure in complex with FMN and PLP (red) with that of human PNP oxidase wild-type dimeric structure in complex with FMN and PLP (blue). B, a difference electron density map (with coefficients Fo − Fc shown at the 2.2 σ level) of the R229W model calculated before FMN, PLP, and the Trp-229 side chain were added to the model. C, an electron-density map (with coefficients 2Fo − Fc shown at the 0.8 σ level) of the active site of the refined R229W model. All maps are superimposed with the final refined models. D, stereo view comparison of the FMN phosphate (Phosph) binding site (residue 229 environment) of the wild-type structure (blue) and R229W structure (red). Residue 229 is labeled as X229. E, stereo view comparison of the active site of the wild-type structure (blue) and R229W structure (red). Note the significant movements of the R229W mutant residues His-227 and Arg-225 away from the wild-type position.
The R229W structure also shows clear density for the Trp-229 residue and FMN (Fig. 2, B and C), with the position and orientation of the cofactor identical in both wild-type and mutant structures (Fig. 2, D and E). In the wild-type structure, FMN binds at the dimer interface and is involved in both direct and water-mediated intersubunit interactions with 14 different amino acids; 12 of them, including Arg-229, are strictly conserved across all species. These interactions stabilize FMN binding, and they probably ensure correct FMN orientation for optimal substrate oxidation. At least half of the protein interactions, including those established by Arg-229, are made to the FMN phosphate moiety. The side chain of Arg-229 is located in a tightly packed pocket surrounded by the residues Arg-141, Glu-217, Ser-175, Trp-219, Gln-139, Gln-174, Pro-261, Val-231, Ala-260, and Leu-259, with the first six residues making direct or water-mediated hydrogen-bond interactions with FMN. The substituted Trp-229 is too big to be accommodated at this FMN binding pocket, necessitating movement of some of the above surrounding residues to relieve the steric interaction (Fig. 2D). In addition to the loss of the hydrogen-bond/salt-bridge interaction from Arg-229, the structural perturbation has also led to the loss of the wild-type hydrogen-bond interaction between Ser-175 and FMN. Glu-217, which has moved significantly from its wild-type position, also seems to have lost its water-mediated hydrogen-bond interaction with FMN; however, there is a phosphate ion close by that could explain the missing water molecule (Fig. 2D). The loss of these two direct hydrogen bonds, and possibly the water-mediated interactions, could explain the reduced affinity of the R229W variant for FMN. Consistently, we noticed that the R229W enzyme lost FMN quite easily during purification. Nonetheless, in the R229W mutant, the 11 remaining protein residue interactions with FMN are able to ensure FMN binding and also maintain its catalytic orientation. It is plausible that the substitution of Arg with Gln in R229Q would not result in a significant steric perturbation of the residues around residue 229 as observed in the R229W structure, allowing Ser-175 to make a hydrogen-bond interaction with FMN. Most likely, Gln-229 in the R229Q mutant could still make a hydrogen-bond interaction with the phosphate, albeit significantly weaker than Arg-229. These analyses are consistent with the observed higher affinity of the R229Q mutant for FMN, as compared with the R229W mutant, but with slightly lower affinity with respect to the wild-type enzyme.
The PNP Oxidase R229W Mutation Alters the Conformation of Key Residues Essential for Substrate Binding and Catalysis
Although no substrate or PLP was added during the crystallization experiment, we observed a difference electron density at the active site, highly suggestive of a weak binding of PLP or possibly PNP during protein expression in E. coli cells. A PLP molecule was modeled into the density and refined. The phosphate moiety of PLP shows strong density and is relatively well ordered, whereas the rest of the PLP molecule is weak (Fig. 2, B and C). Similar weak density was also observed when the mutant enzyme was co-crystallized with PLP (2.7 Å). Both structures have been refined, showing similar PLP density and binding orientation. The overall structures are also indistinguishable. We chose to present the 2.5 Å structure here due to its better resolution. The interpretation of this density as PLP is also consistent with similar density observed for wild-type PNP oxidase when crystallized in the presence of PLP or PNP (29). However, unlike the mutant structure, refinement of the wild-type structure resulted in a more well defined pyridine density.
In the wild-type structure, there are very close hydrophobic contacts between the substrate pyridine ring and the FMN isoalloxazine ring, as well as hydrogen-bond interactions between the strictly conserved residues His-227 and Arg-225 and the substrate. The side chain of the former residue forms a hydrogen-bond interaction with the O3′ of PNP. In a study using the E. coli enzyme, removing this hydrogen bond in the corresponding His to Ala mutant resulted in a 233-fold decrease in substrate affinity (33). The Arg-225 side chain also forms a salt-bridge interaction with the phosphate group of PNP. The corresponding E. coli PNP oxidase Arg to Glu variant also resulted in an 8,000-fold increase and a 16-fold decrease in Km and kcat, respectively (33). As suggested previously, the interactions between FMN and the two residues His-227 and Arg-225 ensure both substrate binding and proper orientation that places the PNP pyridine ring parallel to the FMN isoalloxazine ring. The distance of ∼3.3 Å between the C4′ of PNP and the N5 of FMN apparently allows for efficient hydride transfer (33). Arg-225, which is located at about ∼3.4 Å from the C4′ carbon of PNP, has also been shown to play an important role in controlling the stereospecific transfer of the pro-R hydrogen of the substrate to FMN (33).
The structural perturbation due to the R229W mutation is not only confined to the FMN phosphate binding site (Fig. 2, D and E) but is transmitted to the substrate binding site (Fig. 2E). Most notable is a rotation of the β6-strand-turn-β7-strand (residues 220–227) from the active site, removing the two critical hydrogen-bond interactions between the substrate and the residues His-227 and Arg-225. The loss of these hydrogen-bond interactions seems to have altered the substrate binding orientation as the PLP and FMN ring moieties are no longer parallel to each other (Fig. 2E). Additionally, the C4′ of the substrate and the N5 of FMN no longer face each other but are separated by about 4.5 Å as compared with the ∼3.3 Å observed in the wild-type enzyme. Finally, the Arg-225 guanidinium group is no longer close to the PLP C4′ group. In effect, the inability of Arg-225 and His-227 to make contact with the substrate has led to the destabilization of the substrate, preventing its proper orientation relative to FMN for efficient hydride transfer. Consistently, the kinetic studies (Table 2) show the R229W variant to display a 192-fold increase of Km and a 4.5-fold decrease of kcat as compared with wild-type enzyme. This translates to about an 850-fold less efficient mutant enzyme. Interestingly, in R229W, other protein interactions with the phosphate moiety of PLP are conserved, including those from Tyr-157, Arg-161, Ser-165, and Lys-10, that have allowed the phosphate moiety to almost retain its wild-type position. The weak pyridine ring density of PLP suggests a mobility that could result in the pyridine ring assuming an orientation closer to that of the wild type, probably accounting for the observed residual activity in the R229W mutant enzyme. In the absence of a crystal structure for the R229Q variant, we can only speculate that this mutation may also be causing some structural changes at the substrate binding site, leading to a reduction of catalytic efficiency, albeit smaller than that observed for the R229W variant.
Conclusion: Implications for Other Neurological Disorders Associated with PLP Deficiency
A homozygous missense mutation (R229W) in PNP oxidase is one of several pathogenic mutations linked to neonatal epileptic encephalopathy disorder (1, 2). The results presented here suggest that the R229W mutation influences the enzyme affinity for the cofactor due to the absence of two hydrogen-bond interactions from Arg-229 and Ser-175 to FMN. More importantly, the mutation prevents the proper binding mode of PNP for efficient catalysis due to the absence of two essential hydrogen-bond interactions from His-227 and Arg-225 to PNP. These observations clearly explain why the use of PN was not effective in treating NEE and suggests that the current treatment of PLP or PL is appropriate because the salvage enzyme PL kinase can metabolize the latter vitamer into PLP. Note that ingested PLP undergoes dephosphorylation to PL in the body before it is reconverted back to PLP. It is possible, although yet to be proven, that the use of riboflavin (vitamin B2) and PN, in conjunction with PL, may offer some improvement over the current treatment protocol of PL or PLP alone due to the apparent loss of FMN from the R229W variant. FMN is a phosphate derivative of riboflavin. It is synthesized from riboflavin and ATP, catalyzed by riboflavin kinase. This study also clearly shows that the requirement of the cells for PLP cannot be solely met by PL kinase, underscoring the importance of both salvage enzymes to provide sufficient PLP to satisfy the need of the cells to activate PLP requiring apoenzymes.
As noted earlier, there are several other neurological pathologies, not to mention non-neurological ones, that are suspected to be due to mutations in either PNP oxidase or PL kinase genes. Such mutations may result in defective enzymatic activity, low protein expression levels, and/or impaired regulation mechanisms causing PLP deficiency. Human PNP oxidase and PL kinase genes are located on chromosomes 17q21.2 and 21q22.3, respectively. As observed for NEE, individuals with chromosome 21 trisomy (Down syndrome) have also been reported to display a significant alteration in B6 metabolism (17). The autoimmune polyglandular disease type I has also been linked to chromosome 21q22.3 (19). A recent study has reported a strong association between several polymorphisms in the PNP oxidase gene and schizophrenia in the Japanese population (18). These patients have unusually high concentrations of homocysteine, which is known to be a risk factor for schizophrenia (41). Both cystathionine β-synthase and cystathionine γ-lyase, which function in the transsulfuration pathway that converts homocysteine to cysteine, are PLP-dependent enzymes. Therapy with vitamin B6 reduces homocysteine levels and appears to improve the symptoms experienced by chronic schizophrenic patients (18, 42). Another study found that children with the autism spectrum show consistently higher concentrations of primary B6 vitamers and significantly lower levels of PLP as compared with normal subjects (15). This study concluded that the autistic patients probably have defective PL kinase that was unable to metabolize the primary vitamers into PLP. Several other neurological disorders, including seizures, attention deficit hyperactive disorder, Alzheimer disease, Parkinson disease, learning disability, and anxiety disorders, have also been associated with PLP deficiency (20–24).
This has inspired considerable interest in treating the symptoms of some of these disorders with vitamin B6. However, studies on the effect of B6 supplementation show conflicting results. It is quite clear that therapy with PN or PM in patients with a defective PNP oxidase will not be successful because this enzyme is needed to oxidize PNP and PMP into PLP after the kinase has changed them into PNP and PMP. Also, therapy with PL, PN, or PM in patients with a dysfunctional PL kinase will not work because these vitamers would have to be phosphorylated by the kinase. We note that PLP has also been used with some success in the treatment of a range of epilepsies from infantile spasms to status epilepticus (43, 44). Although there is no direct evidence linking some of these neurological disorders to mutations in either the kinase or the oxidase, these genes are clearly candidates for mutational analysis in affected patients. If such an error is identified, studies could be performed to investigate the role of the mutation in the enzymatic activity and the associated phenotypes. This perhaps would point to the correct pharmacologic intervention.
The work was supported by a grant from the AD Williams Fund (to M. K. S.), the Jeffress Research Grant Award (to M. K. S.) and grants from PRIN2007 and Istituto Pasteur – Fondazione Cenci Bolognetti (to M. L. D. S. and R. C.).
The atomic coordinates and structure factors (code 3HY8) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- NEE
- neonatal epileptic encephalopathy
- PLP
- pyridoxal 5′-phosphate
- PL
- pyridoxal
- PN
- pyridoxine
- PM
- pyridoxamine
- PNP
- pyridoxine 5′-phosphate
- PMP
- pyridoxamine 5′-phosphate
- PNP oxidase
- pyridoxine 5′-phosphate oxidase
- PL kinase
- pyridoxal kinase
- BES
- 2-[bis(2-hydroxyethyl)amino]ethanesulfonic acid.
REFERENCES
- 1.Mills P. B., Surtees R. A., Champion M. P., Beesley C. E., Dalton N., Scambler P. J., Heales S. J., Briddon A., Scheimberg I., Hoffmann G. F., Zschocke J., Clayton P. T. (2005) Hum. Mol. Genet. 14, 1077–1086 [DOI] [PubMed] [Google Scholar]
- 2.Clayton P. T., Surtees R. A., DeVile C., Hyland K., Heales S. J. (2003) Lancet 361, 1614–1614 [DOI] [PubMed] [Google Scholar]
- 3.John R. A. (1995) Biochim. Biophys. Acta 1248, 81–96 [DOI] [PubMed] [Google Scholar]
- 4.Leklem J. E. (1991) in Handbook of Vitamins (Machlin L. ed) pp. 341–378, Marcel Dekker, Inc., New York [Google Scholar]
- 5.McCormick D. B. (1996) in Encyclopedia of Molecular Biology and Molecular Medicine (Meyers R. A. ed) pp. 396–406, Wiley-VCH, Weinheim, Germany [Google Scholar]
- 6.Jansonius J. N. (1998) Curr. Opin. Struct. Biol. 8, 759–769 [DOI] [PubMed] [Google Scholar]
- 7.Eliot A. C., Kirsch J. F. (2004) Annu. Rev. Biochem. 73, 383–415 [DOI] [PubMed] [Google Scholar]
- 8.Steichen O., Martinez-Almoyna L., De Broucker T. (2006) Rev. Mal. Respir. 23, 157–160 [DOI] [PubMed] [Google Scholar]
- 9.Kuwahara H., Noguchi Y., Inaba A., Mizusawa H. (2008) Rinsho Shinkeigaku 48, 125–129 [DOI] [PubMed] [Google Scholar]
- 10.Lainé-Cessac P., Cailleux A., Allain P. (1997) Biochem. Pharmacol 54, 863–870 [DOI] [PubMed] [Google Scholar]
- 11.Ubbink J. B., Delport R., Bissbort S., Vermaak W. J., Becker P. J. (1990) J. Nutr. 120, 1352–1359 [DOI] [PubMed] [Google Scholar]
- 12.Delport R., Ubbink J. B., Vermaak W. J., Becker P. J. (1993) Res. Commun. Chem. Pathol. Pharmacol. 79, 325–333 [PubMed] [Google Scholar]
- 13.Seto T., Inada H., Kobayashi N., Tada H., Furukawa K., Hayashi K., Hattori H., Matsuoka O., Isshiki G. (2000) No to. Hattatsu. 32, 295–300 [PubMed] [Google Scholar]
- 14.Clayton P. T. (2006) J. Inherit. Metab. Dis. 29, 317–326 [DOI] [PubMed] [Google Scholar]
- 15.Adams J. B., George F., Audhya T. (2006) J. Altern. Complement. Med. 12, 59–63 [DOI] [PubMed] [Google Scholar]
- 16.Adams J. B., Holloway C. (2004) J. Altern. Complement. Med. 10, 1033–1039 [DOI] [PubMed] [Google Scholar]
- 17.Coburn S. P., Mahuren J. D., Schaltenbrand W. E. (1991) J. Ment. Defic. Res. 35, 543–547 [DOI] [PubMed] [Google Scholar]
- 18.Song H., Ueno S., Numata S., Iga J., Shibuya-Tayoshi S., Nakataki M., Tayoshi S., Yamauchi K., Sumitani S., Tomotake T., Tada T., Tanahashi T., Itakura M., Ohmori T. (2007) Schizophr. Res. 97, 264–270 [DOI] [PubMed] [Google Scholar]
- 19.Aaltonen J., Björses P., Sandkuijl L., Perheentupa J., Peltonen L. (1994) Nat. Genet. 8, 83–87 [DOI] [PubMed] [Google Scholar]
- 20.Sandyk R., Pardeshi R. (1990) Int. J. Neurosci. 52, 225–232 [DOI] [PubMed] [Google Scholar]
- 21.Khayat M., Korman S. H., Frankel P., Weintraub Z., Hershckowitz S., Sheffer V. F., Ben Elisha M., Wevers R. A., Falik-Zaccai T. C. (2008) Mol. Genet. Metab. 94, 431–434 [DOI] [PubMed] [Google Scholar]
- 22.Mousain-Bosc M., Roche M., Polge A., Pradal-Prat D., Rapin J., Bali J. P. (2006) Magnes. Res. 19, 53–62 [PubMed] [Google Scholar]
- 23.Rajesh R., Girija A. S. (2003) Indian Pediatr. 40, 633–638 [PubMed] [Google Scholar]
- 24.Nogovitsina O. R., Levitina E. V. (2006) Eksp. Klin. Farmakol. 69, 74–77 [PubMed] [Google Scholar]
- 25.Ruiz A., García-Villoria J., Ormazabal A., Zschocke J., Fiol M., Navarro-Sastre A., Artuch R., Vilaseca M. A., Ribes A. (2008) Mol. Genet. Metab. 93, 216–218 [DOI] [PubMed] [Google Scholar]
- 26.Bagci S., Zschocke J., Hoffmann G. F., Bast T., Klepper J., Müller A., Heep A., Bartmann P., Franz A. R. (2008) Arch. Dis. Child. Fetal Neonatal Ed. 93, F151–152 [DOI] [PubMed] [Google Scholar]
- 27.Hoffmann G. F., Schmitt B., Windfuhr M., Wagner N., Strehl H., Bagci S., Franz A. R., Mills P. B., Clayton P. T., Baumgartner M. R., Steinmann B., Bast T., Wolf N. I., Zschocke J. (2007) J. Inherit. Metab. Dis. 30, 96–99 [DOI] [PubMed] [Google Scholar]
- 28.Safo M. K., Mathews I., Musayev F. N., di Salvo M. L., Thiel D. J., Abraham D. J., Schirch V. (2000) Structure 8, 751–762 [DOI] [PubMed] [Google Scholar]
- 29.Musayev F. N., Di Salvo M. L., Ko T. P., Schirch V., Safo M. K. (2003) Protein Sci. 12, 1455–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horiike K., Tsuge H., McCormick D. B. (1979) J. Biol. Chem. 254, 6638–6643 [PubMed] [Google Scholar]
- 31.Yang E. S., Schirch V. (2000) Arch. Biochem. Biophys. 377, 109–114 [DOI] [PubMed] [Google Scholar]
- 32.Kazarinoff M. N., McCormick D. B. (1973) Biochem. Biophys. Res. Commun. 52, 440–446 [DOI] [PubMed] [Google Scholar]
- 33.di Salvo M. L., Ko T. P., Musayev F. N., Raboni S., Schirch V., Safo M. K. (2002) J. Mol. Biol. 315, 385–397 [DOI] [PubMed] [Google Scholar]
- 34.Choi S. Y., Churchich J. E., Zaiden E., Kwok F. (1987) J. Biol. Chem. 262, 12013–12017 [PubMed] [Google Scholar]
- 35.Kwon O., Kwok F., Churchich J. E. (1991) J. Biol. Chem. 266, 22136–22140 [PubMed] [Google Scholar]
- 36.Safo M. K., Musayev F. N., di Salvo M. L., Schirch V. (2001) J. Mol. Biol. 310, 817–826 [DOI] [PubMed] [Google Scholar]
- 37.Safo M. K., Musayev F. N., Schirch V. (2005) Acta Crystallogr. D Biol. Crystallogr. 61, 599–604 [DOI] [PubMed] [Google Scholar]
- 38.Pédelacq J. D., Rho B. S., Kim C. Y., Waldo G. S., Lekin T. P., Segelke B. W., Rupp B., Hung L. W., Kim S. I., Terwilliger T. C. (2006) Proteins 62, 563–569 [DOI] [PubMed] [Google Scholar]
- 39.Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 40.Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 41.Reif A., Schneider M. F., Kamolz S., Pfuhlmann B. (2003) J. Neural Transm. 110, 1401–1411 [DOI] [PubMed] [Google Scholar]
- 42.Miodownik C., Lerner V., Vishne T., Sela B. A., Levine J. (2007) Clin. Neuropharmacol. 30, 13–17 [DOI] [PubMed] [Google Scholar]
- 43.Takuma Y. (1998) Epilepsia 39, Suppl. 5, 42–45 [DOI] [PubMed] [Google Scholar]
- 44.Nakagawa E., Tanaka T., Ohno M., Yamano T., Shimada M. (1997) Neurology 48, 1468–1469 [DOI] [PubMed] [Google Scholar]
- 45.DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA [Google Scholar]