

Abstract
This work investigates the role of charge of the phosphorylated aspartate, Asp369, of Na+,K+-ATPase on E1 ↔ E2 conformational changes. Wild type (porcine α1/His10-β1), D369N/D369A/D369E, and T212A mutants were expressed in Pichia pastoris, labeled with fluorescein 5′-isothiocyanate (FITC), and purified. Conformational changes of wild type and mutant proteins were analyzed using fluorescein fluorescence (Karlish, S. J. (1980) J. Bioenerg. Biomembr. 12, 111–136). One central finding is that the D369N/D369A mutants are strongly stabilized in E2 compared with wild type and D369E or T212A mutants. Stabilization of E2(Rb) is detected by a reduced K0.5Rb for the Rb+-induced E1 ↔ E2(2Rb) transition. The mechanism involves a greatly reduced rate of E2(2Rb) → E1Na with no effect on E1 → E2(2Rb). Lowering the pH from 7.5 to 5.5 strongly stabilizes wild type in E2 but affects the D369N mutant only weakly. Thus, this “Bohr” effect of pH on E1 ↔ E2 is due largely to protonation of Asp369. Two novel effects of phosphate and vanadate were observed with the D369N/D369A mutants as follows. (a) E1 → E2·P is induced by phosphate without Mg2+ ions by contrast with wild type, which requires Mg2+. (b) Both phosphate and vanadate induce rapid E1 → E2 transitions compared with slow rates for the wild type. With reference to crystal structures of Ca2+-ATPase and Na+,K+-ATPase, negatively charged Asp369 favors disengagement of the A domain from N and P domains (E1), whereas the neutral D369N/D369A mutants favor association of the A domain (TGES sequence) with P and N domains (E2). Changes in charge interactions of Asp369 may play an important role in triggering E1P(3Na) ↔ E2P and E2(2K) → E1Na transitions in native Na+,K+-ATPase.
Introduction
The kinetic mechanism of P-type cation pumps is now well established. Active cation transport involves covalent phosphorylation by ATP and dephosphorylation of an aspartate residue coupled to cation movements mediated by E1 ↔ E22 conformational changes. The molecular mechanism is, of course, the central question of energy transduction. Molecular structures of the sarcoplasmic reticulum Ca2+-ATPase (SERCA1) in several conformations are available (1–3). Two crystal structures of the Na+,K+-ATPase, consisting of α, β, and FYXD subunits (4), in an E2(2Rb)-MgF42− conformation (equivalent to E2(2Rb)·P) have been published recently (5, 6). The architecture of the α subunit is very similar to the Ca2+-ATPase, consisting of head, stalk, and membrane sectors, with 10 trans-membrane segments and a cytoplasmic sector consisting of N (nucleotide binding), P (phosphorylating), and A (anchor or actuator) domains. Compared with the first published structure of the Na+,K+-ATPase at 3.5 Å (5), the more recent structure at 2.4 Å (6) reveals greater detail of the cation binding domain, resolution of the β subunit ectodomain, and an FXYD protein ectodomain.
In the case of Na+,K+-ATPase, three Na+ ions and two K+ ions are transported in the E1ATP → E1P(3Na) →E2P and the E2P → E2(2K) → E1ATP halves of the cycle, respectively, at the expense of one molecule of ATP. Binding of ATP with low affinity to E2(2K) is the first step in the catalytic cycle and is followed by accelerated conversion of E2(2K)ATP to E1·ATP in which ATP is bound with high affinity (7–9). In the case of Ca2+-ATPase, two Ca2+ ions and two protons are transported in the E1ATP → E1P → E2P E2P → E2 → E1ATP halves of the cycle, respectively.
The mechanism of energy transduction by P-type cation pumps is best addressed by reference to the Ca2+-ATPase, which has been crystallized in most of the relevant conformations, reviewed recently in detail (1–3). Crystal structures of Ca2+-ATPase show mainly rigid body movements of the N, P, and A domains, mediated by the flexible linkers between the N and P and between the A domain and trans-membrane segment (M1–M3), coupled mechanically to movements of M1–M6 trans-membrane segments that allow alternate access of two Ca2+ ions and two protons to the occlusion sites within M4, M5, M6, and M8. M7–M10 segments appear to act as immovable anchors. In the E1ATP state, N and P domains are in close proximity cross-linked by the bound ATP, whereas the A domain is displaced to one side. Following phosphorylation to E1P, the conformational change to E2P involves a large rotation of the A domain bringing it into close proximity with the P domain, and also the N domain, whereas the N domain is displaced from the P domain. In particular, the conserved sequence TGES (A domain) comes close to the phosphorylated aspartate (P domain). Following hydrolysis of E2P and dissociation of phosphate, ATP bound to E2 induces the change back to the E1 state, with the A domain displaced, and the P and N domains come back into close proximity.
This study was prompted by the following paradox. In the case of Ca2+-ATPase, dissociation of ADP from E1P triggers the E1P → E2P transition. This arises because the ADP (in the E1P structure) and the TGES loop (in the E2P structure) occupy the same position relative to the P domain (3). In the case of Na+,K+-ATPase, ADP, like ATP, is known to stabilize the E1 conformation (9, 10). Thus, it is clear that ADP must dissociate from E1P(3Na) to allow the transition to E2P. However, in the absence of ADP or ATP, an E1 conformation is stable either with bound Na+ or without Na+ in a sufficiently high ionic strength buffer (9, 11–13). This is different from Ca2+-ATPase in which E2 is the principal conformation in the absence of Ca2+ ions at neutral pH (14–16). In other words, for Na+,K+-ATPase a spontaneous conformational change of the dephosphoenzyme E1 → E2 or E1(3Na) → E2 does not occur even when ADP is absent, but it occurs only in the context of the phosphoenzyme E1P(3Na) → E2P. Thus, a question arises whether dissociation of ADP from E1P(3Na) is sufficient or whether the covalently bound phosphate/Mg2+ itself plays a role in triggering the E1P(3Na) → E2P conformational change.
One way to address this issue is to investigate a possible role of the phosphorylated aspartate, Asp369. D369N/D369A/D369T/D369E mutants of Na+,K+-ATPase have been expressed previously in different cells and shown to abolish ATP hydrolysis (17, 18). Pedersen et al. (19) expressed wild type and D369N and D369A mutants in Saccharomyces cerevisiae and found that, although they are inactive, the D369N/D369A mutants have a very high affinity for ATP, compared with wild type, but have a similar ADP binding affinity. By analyzing antagonism of ATP binding by K+ ions, it was also suggested that the E1 ↔ E2 equilibrium is altered, favoring E2 conformations. The D369N mutant was later expressed in Pichia pastoris (20) and studied by specific oxidative cleavages mediated by Fe2+, which substitutes for the catalytic Mg2+ (21). The D369N mutation strongly suppressed specific Fe2+-catalyzed cleavages. Compared with the wild type Ca2+-ATPase, mutants of its corresponding phosphorylated aspartate, D351N/D351A/D351T, also bind ATP with a very high affinity (22). A recent crystal structure of D351A with bound ATP is virtually identical to the wild type, showing that the high affinity is due to removal of electrostatic repulsion between the γ-phosphate of ATP and the Asp351 carboxylate, and not to a conformational change (23).
Fluorescence probes are very useful tools for studying E1 ↔ E2 conformational changes. They allow detailed characterization of specificities and affinities of ligands that stabilize E1 or E2 forms and especially measurement of rates of individual E1 → E2 and E2 → E1 transitions (8, 11, 13). Fluorescence probes used extensively to study native Na+,K+-ATPase include intrinsic tryptophan fluorescence (11), noncovalently bound formycin nucleotides (8) and eosin (24), as well as covalently bound labels such as FITC3 (13), iodoacetamidofluorescein (25, 26), and the electrochromic shift dye RH421 (27, 28). FITC covalently labels the α subunit of renal Na+,K+-ATPase, predominantly at Lys501 within the ATP-binding site (the N domain) (13, 29). The fluorescein chromophore is sensitive to the pH of its local environment. Because of the rather large fluorescence changes (25–35%) associated with E1 ↔ E2 conformational changes observed for the FITC-labeled renal Na+,K+-ATPase, it was possible to carry out detailed equilibrium and stopped-flow fluorescence studies (13, 30–33). FITC-labeled sarcoplasmic reticulum Ca2+-ATPase (16, 34, 35) and gastric H+,K+-ATPase (36) have also been used extensively to monitor E1 ↔ E2 conformational changes.
The fluorescence methods described above have not been applied to recombinant Na+,K+-ATPase. Recent publications from this laboratory describe expression of Na+,K+-ATPase in P. pastoris and purification of the recombinant protein in a detergent-soluble functional state in quantities up to 1 mg (20, 37, 38). The high yield of purified protein now makes it possible to carry out detailed biochemical and biophysical work. Development of fluorescein-labeled recombinant Na+,K+-ATPase described here has allowed us to examine directly effects of charge neutralization on Asp369 on E1 ↔ E2 equilibria and to investigate its mechanism.
EXPERIMENTAL PROCEDURES
Materials
Escherichia coli XL-1 blue strain was used for propagation and preparation of various plasmid constructs. Yeast Lytic Enzyme from ICN Biomedicals, Inc. (catalogue number 152270), was used for transformation of yeast. P. pastoris protease-deficient strain SMD1165 (his4, prb1) was used for transformation. DDM (catalogue number D310) and C12E8 (25% w/w, catalogue number O330) were purchased from Anatrace. Synthetic SOPS (sodium salt)) was obtained from Avanti Polar Lipids and stored as chloroform solutions. BD TalonTM metal affinity resin (catalogue number 635503) was obtained from Clontech. Cholesterol, ouabain (O3125), and FITC (catalogue number F7250) were from Sigma. [32P]ATP and [3H]ouabain were obtained from Amersham Biosciences. All other materials were of analytical grade.
Media
YPD is 1% bacto-yeast extract, 2% bacto-peptone, 2% dextrose. To solidify the medium 2% Bacto-agar was added. YNB is yeast nitrogen base without amino acids (Difco). BMG is 1.34% YNB, 0.04% biotin, 0.1 m potassium phosphate buffer, pH 6.0, glycerol 0.2–1%. BMM is 1.34% yeast nitrogen base without amino acids, 0.04% biotin, 0.1 m potassium phosphate buffer, pH 6.0, 0.5% methanol.
Construction of Yeast Plasmids for the Expression of α and β Subunits
The pHIL-D2(α1/His10-β1) construct contained cDNAs encoding wild type or mutant porcine α1 (GenBankTM accession number X03938) and porcine β1 (GenBankTM accession number X04635) with a 10× His tag at the N′ terminus of the β subunit is described in Ref. 37. D369N/D369A/D369E, T212A, and E214A/E214Q mutants were prepared by the methods described previously (39).
Yeast Transformation and Selection; Screening for Expression; Yeast Growth and Induction of Protein Synthesis
10 μg of linear DNA, obtained by digestion of the pHIL-D2 (wild type or mutant α1/His10-β1) with NotI, were used to transform spheroplasts of P. pastoris SMD1165, and His+Muts transformants were selected (20). Muts clones were grown in 5 ml of BMG cultures, and expression was induced with methanol for 5 days. Small scale membrane preparations were made. Samples were run on SDS-PAGE, and relative expression in the different Muts clones was determined by Western blots, using the antibody anti-KETYY antibody (at a dilution of 1:3000), which recognizes the C terminus of the α subunit. Ouabain binding was also carried out to determine the highest expressing clones that were subsequently used routinely.
For large scale growth and expression of the recombinant Na+,K+-ATPase (20, 37), single colonies propagated in YPD medium were inoculated into 5 ml of BMG liquid medium, which was incubated for 48 h with vigorous shaking at 30 °C. The culture was further diluted 100-fold to a final volume of 300 ml and grown in the BMG medium (glycerol 1%) for a further 24 h to an A600 of 2–6. Cells were then poured into 3 liters of BMG medium (with 0.2% glycerol) in Bellco Spinner FlasksTM with air supplement (0.5 liter/min) and magnetic stirring at 250 rpm, and temperature was maintained at 30 °C. After 24 h, cell growth had stopped (A600 = 2–4). Expression of the Na+,K+-ATPase was induced by adding 0.5% methanol daily for 5 days. Routinely, the wild type was induced at 25 °C and mutants at 20 °C.
The maximal A600 achieved under these conditions was ∼10–15. On the 6th day cells were collected and stored at −20 °C.
Membrane Preparations and FITC Labeling of Membranes
Cells were broken with glass beads, and membranes were prepared as described previously (20). Membranes were stored at −80 °C in a solution containing 10 mm MOPS/Tris, pH 7.4, and 25% glycerol, with a mixture of protease inhibitors (1 mm phenylmethylsulfonyl fluoride, 10 μg/ml pepstatin, chemostatin, and leupeptin). Roughly 1 g of membrane protein was obtained per 3 liters of culture. For FITC labeling (see Ref. 13, 31), membranes were suspended at 2 mg/ml in a buffer containing 50 mm NaCl, 1 mm EDTA, 20 mm Tris, pH 9.2, with protease inhibitors. Fluorescein 5′-isothiocyanate (usually 1–2 μm FITC dissolved in DMSO) was added, and membranes were incubated for 1 h at 20 °C in the dark. The suspension was then diluted 3-fold with an ice-cold solution of 100 mm MOPS, pH 6.45, mixed for 10 min, and centrifuged at 100,000 × g for 80 min. The pelleted labeled membranes were resuspended at 1 mg/ml in 10 mm MOPS/Tris, pH 7.4, and 25% glycerol. Fluorescein bound to α subunit, separated by SDS-PAGE, was detected with a Typhoon fluorescence imager with excitation supplied by a blue laser and emission at 520 nm set by a monochromator.
Solubilization and Purification of FITC-labeled Recombinant Na+,K+-ATPase
The methods have been described in detail (37, 38, 40). Briefly, unlabeled or FITC-labeled membranes (1–2 mg/ml) were homogenized well for 15 min on ice with DDM (DDM/protein, 2:1 w/w) in a medium containing 250 mm NaCl, 20 mm Tris-HCl, pH 7.4, 5 mm imidazole, 0.5 mm phenylmethylsulfonyl fluoride, and 10% glycerol. Unsolubilized material was removed by ultracentrifugation. The soluble material was incubated with shaking overnight at 4 °C with BD TalonTM beads (Co2+-chelate), at a ratio of 1 ml of beads per supernatant from 100 mg of membrane protein, together with 50–100 μm EDTA. Beads were washed twice with 5 volumes of buffer containing 125 mm NaCl, 10 mm Tricine, pH 7.4, 5% glycerol, 10 mm imidazole, 0.1 mg/ml C12E8, 0.05 mg/ml SOPS, 0.01 mg/ml cholesterol. The Na+,K+-ATPase (0.2–3 mg/ml) was eluted by mixing with 1 volume of a solution containing 150 mm imidazole, 20 mm Tricine, pH 7.4, 0.1 mg/ml C12E8, 0.05 mg/ml SOPS, 0.01 mg/ml cholesterol, 25% glycerol for 45 min at 0 °C and stored on ice.
Protein was determined by scanning Coomassie-stained gels for the α subunit content of the purified recombinant enzyme and compared with known amounts of pig kidney Na+,K+-ATPase. Ouabain binding to yeast membranes was assayed using [3H]ouabain essentially as described (19). Na+,K+-ATPase activities and covalent phosphorylation were done as described (38, 40, 41).
Equilibrium and Stopped-flow Fluorescence Measurements
Equilibrium fluorescence changes were measured in a Varian fluorimeter at room temperature (20–23 °C). 10–15 μg of FITC-labeled purified recombinant enzyme were incubated for 30 min at room temperature and then added to a stirred cuvette containing 2 ml of the following solution: 150 mm choline chloride, 10 mm Hepes (Tris), pH 7.5 (and other pH values between 7 and 8), or 10 mm Mes (Tris) (pH range 5.5–6.5). The same buffer with no enzyme was used to bring the fluorescence level to zero before addition of the enzyme. Excitation was at 495 nm, and emission was at 520 nm, with both slits adjusted to 5 nm. After stabilization of the fluorescence, ligands such as RbCl, KCl or NaCl, Pi(Tris), and vanadate (Tris) were added until no further fluorescence change occurred. For equilibrium titrations, graded concentrations of ligands such as RbCl were added, and the fluorescence change was recorded. For each addition, a correction was made for the dilution effects of the added volume.
Stopped-flow fluorescence measurements at 23 °C were made using an Applied Photophysics stopped-flow fluorimeter. Excitation via a monochromator was at 495 nm, and emission was measured via a 515-nm cutoff filter. The two syringes were loaded with 1.2 ml of solution. Each push utilized 70 μl per syringe. 3–4 pushes were used to fill the flow tubes, thus allowing up to 10 replicates for repetitive measurement in each condition. Dead time was ∼1.5 ms. Purified enzyme was incubated for 30 min at room temperature before mixing with the relevant solution. Traces were smoothed with the use of an “oversample” function of the machine. 4–9 such traces were then averaged to produce a single trace and analyzed as described below.
Solutions
Total ionic strength of all solutions in syringe 1 and 2 was maintained at 175 mm, consisting of 10 mm Hepes, pH 7.5, and choline chloride, RbCl, KCl, NaCl, or other ligands such Pi (Tris) or vanadate (Tris) at a total ionic strength of 165 mm. Solutions not containing monovalent cations or other ligands consisted of 10 mm Hepes, pH 7.5, and 165 mm choline chloride. The final concentrations of all ligands not present in both syringes was half that given below. The design of the experiments follows that developed previously (8, 11, 13).
E2(2Rb) → E1Na
Syringe 1 contained FITC-labeled wild type or 20–30 μg of D369N/D369A mutants and 20 mm RbCl or KCl (wild type) or 4 mm mutants. Syringe 2 contained 80 mm NaCl.
E1 → E2(2Rb)
Syringe 1 contained FITC-labeled wild type or 20–50 μg of D369N/D369A mutants. Syringe 2 contained 40, 60, and 165 mm RbCl (wild type and D369N/D369A mutants) or 20–165 mm KCl (wild type).
E1 → E2·P and E1 → E2·Vanadate
Syringe 1 contained FITC-labeled wild type or 15–30 μg of D369N mutants and 4 mm MgCl2, and 15–30 μg of D369A without MgCl2. Syringe 2 contained 50 mm Pi (Tris), pH 7.5, or 2 mm vanadate (Tris) and 4 mm MgCl2.
Derivation of Kinetic Parameters
All fits were done using the KaleidaGraph program (Synergy Software).
1) Rb+ titrations in equilibrium measurements were fitted to the following form of the Hill Equation 1,
where n = Hill coefficient, and K0.5 = concentration of Rb+ ions required for half-maximal fluorescent signal. For each curve ΔFmax was found by fitting the raw data, and the ratio of ΔF/ΔFmax was then recalculated. Values of ΔF/ΔFmax for replicate experiments were combined, and the best fit average parameters nH ± S.E. and K0.5 ± S.E. were recalculated. This normalization enables comparison of the curves in different conditions or for wild type and mutant proteins.
2) Time courses in the stopped-flow fluorescence experiments were fitted either to single or double exponential function as shown in Equations 2 and 3,
or
The “end point” is the constant fluorescence intensity after the reaction has reached equilibrium; k1 and k2 are rates of first and second exponentials. a1 and a2 are amplitudes of first and second exponentials.
RESULTS
Expression of Wild Type and Mutants of Na+,K+-ATPase in P. pastoris
Expression of pig α1β1 subunits and T212A, D369A, D369N, and D369E mutants was done as described under “Experimental Procedures” and as described previously (20). The wild type was expressed optimally at 25 °C, and mutants were expressed at 20 °C. The expression levels of the proteins in the yeast membranes were estimated by immunoblots of the α subunit and also ouabain binding. The range of ouabain binding was as follows: wild type, 20–40 pmol/mg protein; D369A, 20–30 pmol/mg protein; D369N, 15–35 pmol/mg protein; D369E, 3–7 pmol/mg protein; and T212A, 20–30 pmol/mg protein. Attempts to express E214A and E214Q mutants were not successful.
Labeling and Purification of Fluorescein-labeled Na+,K+-ATPase; Optimization of Fluorescence Signals
We have taken two possible approaches to labeling the recombinant enzyme with FITC to detect fluorescence signals associated with E1 ↔ E2 conformational changes (see Fig. 3 for examples of the latter). As one approach, we purified the protein and then attempted to label it with FITC at pH 9.0. However, K+- and Na+-induced fluorescein fluorescence changes were not observed, probably because the high pH during labeling inactivates the protein. The protein is much more stable in the yeast membrane than in the detergent-soluble state (40) and should survive the incubation at pH 9.0. Therefore, we labeled the Na+,K+-ATPase in the yeast membranes with FITC before purifying the fluorescein-labeled enzyme. This strategy turned out to be successful. The expressed Na+,K+-ATPase constitutes no more than 0.5–1% of the yeast membrane proteins, and therefore FITC could label many proteins at pH 9.0. However, at low concentrations of FITC, one could expect to label Na+,K+-ATPase more selectively at Lys501 than at other lysines and minimize labeling of other proteins. Unrelated fluorescein-labeled proteins should be removed during the purification. With these assumptions, extensive optimization of labeling, purification of labeled Na+,K+-ATPase, and K+- and Na+-dependent fluorescence changes were undertaken. FITC labeling prevents ATP binding and inactivates covalent phosphorylation by ATP (13). As seen in Table 1, incubation of the yeast membranes with 10 μm FITC for 1 h at pH 9.0 inactivated phosphorylation by 84.6%, whereas 50 μm inactivated fully. Therefore, 10 μm FITC was chosen initially, although subsequent optimization experiments showed that lower concentrations of FITC were preferable. A more sensitive criterion for selective labeling by FITC at Lys501 is that it should be suppressed by ATP (13). Fig. 1 shows an experiment in which membranes expressing wild type and the mutant proteins were incubated with 1 μm FITC at pH 9.0 for 1 h in the absence and presence of 1–1000 μm ATP. The fluorescence of the bound fluorescein was detected at 520 nm. Indeed, the labeling of the α subunit was progressively suppressed by increasing concentrations of ATP. This is indicative of selective labeling. For comparison with labeling of the α subunit, labeling of an unrelated protein (Fig. 1, asterisk) was not affected by the presence of ATP. Several other unrelated bands were also labeled unselectively and are not shown. As a semi-quantitative measure of the effect of ATP concentrations, the fluorescence intensity associated with the α subunit was scanned for each concentration of ATP, and the values relative to the control are recorded below each lane in Fig. 1. One obvious finding was that the concentration of ATP required to suppress labeling of the D369A mutant was much lower than that required to suppress labeling of the wild type, D369E, and T212A mutants (compare the values at 1 μm). Because the ATP binding affinity of the D369A mutant is known to be greatly increased compared with that of the wild type (19), this observation provides a further indication for selective labeling of Lys501. It is also noticeable that 25–50% of the labeling in the different clones was not suppressed by ATP, even at 1000 μm. This indicates that some of labeling of the α subunit is unselective, even in optimal conditions.
FIGURE 3.

K+- and Na+-induced E1 ↔ E2conformational changes of wild type and mutant Na+,K+-ATPase. Typical fluorescein fluorescence changes of wild type (WT) and mutant proteins labeled with FITC in optimal conditions. The concentrations of Rb+ and Na+ were chosen to give maximal responses for each protein. a.u., arbitrary units.
TABLE 1.
Inhibition by FITC of phosphorylation of recombinant Na+,K+-ATPase
Wild-type membranes suspended in a medium containing 50 mm NaCl, 1 mm EDTA, 20 mm Tris, pH 9.2, were incubated with FITC at 0.10 or 50 μm for 1 h at room temperature. The membranes were then centrifuged to remove the FITC and resuspended in the normal medium. Phosphorylation by ATP was then measured.
| Ouabain binding | Phosphoenzyme |
||
|---|---|---|---|
| Nonlabeled | +10 μm FITC | +50 μm FITC | |
| pmol/mg | pmol of ATP/mg of membranes | ||
| 25.7 | 18.5 | 2.85 (84.6% inhibition) | 0 (100% inhibition) |
FIGURE 1.

Selective labeling of recombinant Na+,K+-ATPase by FITC and protection by ATP. FITC-labeled membranes show fluorescein label in the α subunit, with ATP 0–1000 μm. The asterisk indicates a contaminant protein. The fluorescence associated with the α subunit was measured with the Typhoon fluorescence imager. The values at each concentrations of ATP are recorded below the appropriate lane, as a percent of control without ATP. WT, wild type.
After labeling the protein with FITC, the next step was solubilization by β-DDM and the purification of the wild type and mutant Na+,K+-ATPase as described under “Experimental Procedures.” The proteins were eluted in a medium containing 150 mm imidazole, 0.1 mg/ml C12E8, 0.05 mg/ml SOPS, and 0.01 mg/ml cholesterol, as described, but the elution buffer was nominally devoid of both Na+ and K+ ions, because the fluorescence experiments involved measurement of responses after addition of Na+ and K+. Fig. 2 presents an example of purification of wild type and two mutant proteins (D369N and T212A) after labeling membranes in the optimal conditions discussed below. The proteins are all about 80–90% pure, and the α subunit was clearly labeled as detected by the fluorescein fluorescence (the β subunit does not bind FITC and is not fluorescent). D369A was purified to the same extent as the proteins shown in Fig. 2, whereas D369E was less pure due to the lower level of expression (data not shown). The final step was the detection and optimization of fluorescein fluorescence changes. Fig. 3 shows a standard equilibrium fluorescence experiment with excitation at 495 nm and emission at 520 nm. The detergent-soluble fluorescein-labeled Na+,K+-ATPase was added to a medium containing 166 mm choline chloride, which stabilizes an E1 conformation. Upon addition of sufficient Rb+ or K+ ions, the protein is converted to an E2(2Rb)/(2K) conformation, and the fluorescence drops. Upon subsequent addition of sufficient Na+, the protein is re-converted to an E1Na conformation, and the fluorescence change reverts to the original level. Fig. 3 shows that this behavior is seen for the wild type and all the mutants, indicating that E1 ↔ E2 transitions can be detected in each case. One difference already evident in these initial experiments is that reversion of the signal in D369N is incomplete at 50 mm NaCl. The experiment in Fig. 3 shows the signals for typical optimized preparations and denotes the amplitude of the signal changes, for example 13.7% in the case of the wild type protein. However, in initial experiments a fluorescence change of only 7% was achieved with the wild type protein. Optimization of the labeling conditions included variation of pH during labeling, FITC concentration, and time and temperature of the labeling reaction. Eventually, optimal signal amplitudes were obtained after labeling the membranes at room temperature with 1 μm FITC for 1 h at pH 9.0 (the conditions in Fig. 1).
FIGURE 2.
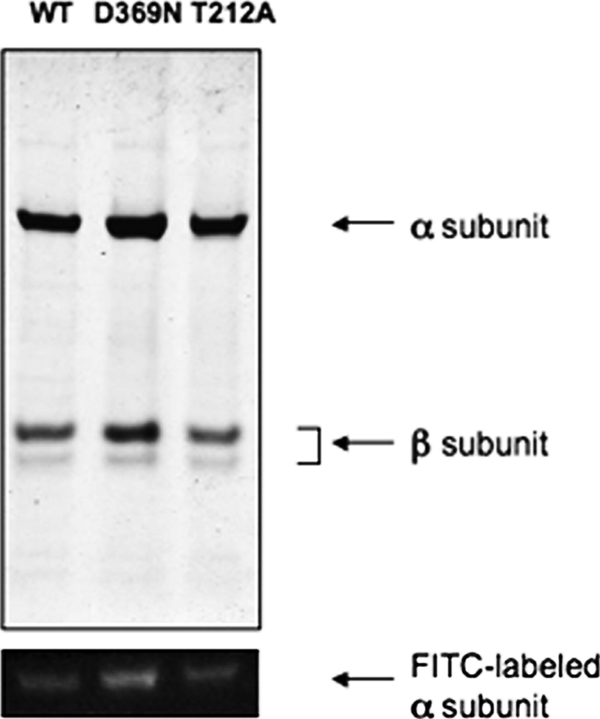
Purification of fluorescein-labeled wild type (WT) and mutant Na+,K+-ATPase. Coomassie-stained gel of purified proteins. The fluorescein fluorescence in the α subunit of the purified complexes was visualized with a UV lamp.
The effects of Rb+ (K+) and Na+ in the high ionic strength buffer are quite similar to those observed previously using the native membrane-bound renal Na+,K+-ATPase except for a lower amplitude, typically 12–15% for recombinant wild type, compared with 25–35% for renal Na+,K+-ATPase (13). Another similar property is that the emission maximum for the fluorescein-labeled recombinant Na+,K+-ATPase was 520 nm and did not change upon addition of Rb+ or K+ ions (not shown). Thus, the fluorescence change represents a change in fluorescence intensity of the bound fluorescein and not a spectral shift, as also found previously for renal Na+,K+-ATPase. These features provide strong evidence that the basic E1 ↔ E2 mechanism is preserved in the purified detergent-soluble recombinant protein and can be studied using fluorescein fluorescence changes.
Analysis of E1 ↔ E2(2Rb) Conformational Transitions of Wild Type and D369N/D369A Mutants
This section compares equilibrium fluorescence titrations of the E1 ↔ E2(2Rb) transitions and rates of the individual transitions E2(2Rb) → E1Na and E1 → E2(2Rb) measured in a stopped-flow fluorimeter. Rb+ ions are congeners of K+ ions and are used routinely in place of K+ ions because the rates of transitions are slower, but some experiments have also been done with K+ ions.
Equilibrium titrations of the dependence on Rb+ concentration of the fluorescence change accompanying the E1 ↔ E2(2Rb) transition were analyzed by recording the response to graded additions of RbCl. Fig. 4 shows typical Rb+ titrations for wild type, D369A, D369N, D369E, and T212A mutants. It is obvious that the curves for wild type, D369E, and T212A mutants differ significantly from those for the D369N and D369A mutants. The titration curves were sigmoid and were fitted to a Hill function leading to a value of the K0.5Rb and Hill coefficient n, summarized in Table 2 (and also K+ titration data for the wild type). The clear finding is that the K0.5Rb for the D369A and D369N mutants is severalfold lower (6.22-fold for D369N and 3.3-fold for D369A) than for the wild type, D369E, and T212A, which are quite similar. The Hill coefficients, in the range 1.6–2.3, are not significantly different between wild type and mutants. The Hill equation is used here empirically to obtain a value for K0.5Rb and n. Although, in principle, no mechanistic significance can be attributed to nH, a value close to 2 is, of course, suggestive of occlusion of two Rb+ ions, as is known to be the case (42).
FIGURE 4.

Equilibrium titrations of the E1 ↔ E2(2Rb) conformational change. Representative titrations of the E1 ↔ E2(Rb) change for wild type (WT) and mutants. The data have been normalized, as described under the “Experimental Procedures,” to permit comparison between the different proteins. Solid lines represent curves fitted to the data and provide the parameters in Table 2 for Rb+.
TABLE 2.
Equilibrium titrations of the E1 ↔ E2(2Rb) conformational change
Parameters were obtained by fitting the normalized data in Fig. 4 to the Hill equation. The K+ titrations for the wild-type are derived from a separate experiment.
| K0.5Rb(K) | nH | |
|---|---|---|
| mm ± S.E. | ||
| Wild type, Rb+ | 5.60 ± 0.09 | 1.8 ± 0.04 |
| Wild type, K+ | 3.90 ± 0.04 | 1.7 ± 0.08 |
| D369N, Rb+ | 0.90 ± 0.02 | 2.1 ± 0.1 |
| D369A, Rb+ | 1.70 ± 0.07 | 1.98 ± 0.14 |
| D369E, Rb+ | 6.33 ± 1.0 | 1.63 ± 0.17 |
| T212A, Rb+ | 6.6 ± 0.08 | 2.3 ± 0.05 |
The following simple kinetic Scheme 1 and the accompanying Equation 4 depict occlusion of two Rb+ ions coupled to the conformational change and illustrate an important point on the analysis of the mutants.
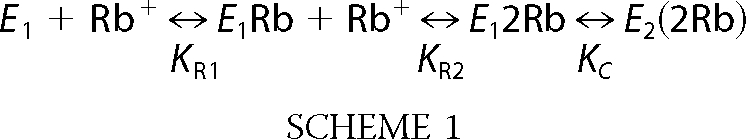 |
KR1 and KR2 are the dissociation constant for the two Rb+ ions and KC is a conformational equilibrium constant. In an equilibrium fluorescence titration between E1 and E2(2Rb) the following relationship will hold (Equation 4),
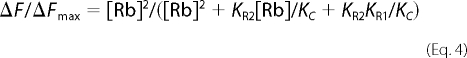 |
It can be shown that K0.5Rb is a function of all three constants KR1, KR2, and KC, and without independent information it is not possible to estimate the individual constants. Thus, the Hill equation is more useful as an empirical measure of the “apparent affinity” of the Rb+ ions. However, the point of showing the equation is that the difference in K0.5Rb between wild type and D369N and D369A mutations could reflect differences in either or both the intrinsic dissociation constants, KR1 and KR2, and the conformational equilibrium constant, KC. Thus, it is necessary to obtain independent evidence to distinguish between the possibilities.
Measurement of the rates of the individual transitions E2(2Rb) → E1Na and E1 → E2(2Rb) by stopped-flow fluorescence allows analysis of the mechanism of the change in K0.5Rb seen in equilibrium titrations. It is assumed that the cation binding and dissociation steps are rapid by comparison with the conformational change in either direction. Thus, the rate constants of the observed fluorescence changes are limited by the rates of the conformational changes. Fig. 5 shows an experiment that measures the rate of E2(2Rb) → E1Na. The enzyme was pre-stabilized in the E2(2Rb) conformation and then mixed rapidly with Na+ ions, which stabilize the E1Na conformation detected by the progressive rise in fluorescence. The rate for D369N and D369A is much slower than for the wild type. The traces were fitted to single exponential functions and gave the values of the rate constants in Table 3. The data show that the rate of E2(2Rb) → E1Na for D369A is 13-fold and that for D369N is 20-fold slower than for the wild type. It was also a repeated finding that a larger effect on the conformational change was seen for D369N compared with D369A (see Tables 2 and 3). The rate of E2(2K) → E1Na was also measured for wild type and is faster than that for E2(2Rb) → E1Na (Table 3).
FIGURE 5.

Stopped-flow traces of E2(2Rb) → E1Na for wild type and D369N/D369A mutants. Each trace for the wild type (WT) and D369N/D369A mutant proteins represents the average of 5–10 replicates. The curves have been fitted to single exponential functions. Rate constants are shown in Table 3. Wild type, black; D369N, gray; D369A, light gray.
TABLE 3.
Rates of E2(2Rb) → E1Na and E1 → E2(2Rb) conformational changes
Fitted rate constants for the traces in Figs. 5 and 6 are given. Rate constants of E2(2Rb) → E1Na for wild type and Asp369 represent averages from several independent experiments. The data for E2(2K) → E1Na are from a separate experiment and that for E1 → E2(2K) are derived from Fig. 7.
| Conformational change | Wild type | D369N | D369A |
|---|---|---|---|
| k s−1 ± S.E. | k s−1 ± S.E. | k s−1 ± S.E. | |
| E2(2Rb) → E1Na | 0.544 ± 0.087(n = 5) | 0.026 ± 0.005(n = 2) | 0.039 ± 0.001 s−1 |
| E1 → E2(2Rb) | |||
| 20 mm Rb+ | 5.1 ± 0.02 s−1 | 3.7 ± 0.01 s−1 | |
| 30 mm Rb+ | 13.5 ± 0.5 s−1 | 8.8 ± 0.15 s−1 | 8.4 ± 0.1 s−1 |
| 83 mm Rb+, Exp. 1 | 33.8 ± 0.6 s−1 | 28.2 ± 0.5 s−1 | 37 ± 0.7 s−1 |
| 83 mm Rb+, Exp. 2 | 28.9 ± 0.15 s−1 | 27.4 ± 0.2 s−1 | |
| E2(2K) → E1Na | 3.3 ± 0.03 s−1 | ||
| E1 → E2(2K) | 125 ± 15 s−1 | ||
| Extrapolated maximum value | |||
Fig. 6 shows experiments to measure the rates of the opposite conformational transition E1 → E2(2Rb) at several Rb+ concentrations (20, 30, and 83 mm) for wild type and D369N or D369A mutants. In this type of experiment, the proteins are suspended in an Na+-free high ionic strength medium and mixed with different concentrations of Rb+ ions in the stopped-flow fluorimeter. The conversion of the E1 to the E2(2Rb) conformation is associated with a decrease in fluorescence. Evidently, the rates of this transition for wild type and mutants are quite similar. The rate constants obtained from fits to an exponential function are plotted against Rb+ concentrations in Fig. 7 A, which shows clearly that there is little or no difference between the wild type and either mutant (exact values ± S.E. are given in Table 3). According to the kinetic scheme above, for a Rb+(K+)-induced conformational transition the observed rate constant shows a saturating dependence on Rb+(K+) concentration. The data in Fig. 7A show the rates of the transition increased as the Rb+ concentration was raised, in the case of all the wild type and mutant recombinant proteins, although saturation was not reached at the highest Rb+ concentration used (but see Fig. 7B for saturation by K+ ions). A technical limitation of this experiment was that the accessible range of Rb+ concentrations was limited by the total ionic strength, and the number of Rb+ concentrations that could be analyzed, particularly for the mutants, was limited due to the necessity to use large amounts of protein. However, because the rate of E1 → E2(2Rb) was unaffected at any of several Rb+ concentrations, it is unlikely that the D369N/D369A mutations affect either the maximal rate of E1 → E2(2Rb) or the intrinsic affinity for Rb+ ions. Thus, the simple conclusion is that the mechanism involves a large reduction in the rate of E2(2Rb) → E1Na with little or no effect on the rate of E1 → E2(2Rb) or Rb+ affinity.
FIGURE 6.

Stopped-flow traces of E1 → E2(2Rb) for wild type and D369N/D369A mutants. Each trace for the wild type and D369N/D369A mutant proteins represents the average of 5–10 replicates. Final Rb+ concentrations are indicated. The curves have been fitted to single exponential functions. Rate constants are shown in Table 3. Wild type (WT), black; D369N, gray; D369A, light gray. Note that the solution in syringe 1 consists of 20–30 μg of FITC-labeled enzymes, choline chloride, plus RbCl (wild type 20 mm, mutants 4 mm) to a total concentration of 165 mm plus 10 mm Hepes, pH 7.5, and that the solution in syringe 2 consists of 165 mm NaCl plus 10 mm Hepes, pH 7.5.
FIGURE 7.

Rate constants of E1 → E2(2×) at different concentrations of the monovalent cation. A, rate constants of E1 → E2(2Rb) at 20, 30, and 83 mm RbCl for wild type (WT) and D369N/D369A mutants. The graph depicts the fitted rate constants from the data in Table 3. The values of the rate constants for D369N and D369A are virtually identical and cannot be seen as separate points. Note that the solution in syringe 1 consists of 20–50 μg of FITC-labeled enzymes, 165 mm choline chloride, plus 10 mm Hepes, pH 7.5, and the solution in syringe 2 consists of choline chloride + RbCl to a total concentration of 165 mm plus 10 mm Hepes, pH 7.5. B, comparison of rate constants of E1 → E2(2×) at different Rb+ or K+ concentrations for wild type. The data for K+ represents fitted rate constants from an experiments similar to that in Fig. 6 for a range of K+ concentrations from 10 to 83 mm. The solid line for K+ represents the hyperbolic fit with the parameters kE2(K) → E1 125 ± 15 s−1 and the KK 39 ± 10.5 mm. The data for Rb+ are taken from Table 3.
Fig. 7B provides an interesting mechanistic insight into the lack of effect of the mutations on E1 → E2(Rb). For the wild type protein, we compared the rates of E1 → E2(2K) and E1 → E2(2Rb) at increasing concentrations of K+ and Rb+ ions, respectively. The raw data for E1 → E2(2K) were fitted to exponential decay curves, and the rate constants are plotted as a function of K+ concentration. The data for the Rb+ are the same as in Fig. 7A. The figure shows that the rate for E1 → E2(2K) is about 3-fold higher than for E1 → E2(2Rb) and also shows saturation behavior in the accessible range of K+ concentrations. The data for E1 → E2(2K) have been fitted to a hyperbola with kE1 → E2(K) 125 ± 15 s−1, and the KK was 39 ± 10.5 mm. As we discuss below, the difference with K+ and Rb+ ions shows that the rate of E1 → E2(2×) is limited by events in the cation- binding sites rather than in the active site.
Effects of pH on E1 ↔ E2(2Rb) Conformational Transitions of Wild Type and D369N Mutant
It was reported many years ago that pH affects the conformational equilibrium E1 ↔ E2(2Rb), by a “Bohr”-like effect (12). In particular it was observed that acidic pH favors E2(2Rb) and alkaline pH favors E1. The observations in Figs. 8 and 9 and Table 4 provide a mechanistic explanation of this Bohr-like effect. The Rb+ titrations in Fig. 8 (and the fitted parameters in Table 4) show that a reduction of the pH from 7.5 to 5.5 has a large effect on the K0.5Rb of the wild type, the value being reduced by 10-fold from 3.45 ± 0.1 to 0.35 ± 0.03 mm. By contrast, the K0.5Rb for the D369N mutant is reduced only from 0.91 ± 0.02 at pH 7.5 to 0.37 ± 0.01 mm at pH 5.5, a factor of 2.45-fold. An increase in apparent affinity of the wild type for Rb+ caused by raising the proton concentration by 100-fold must be due to an effect of the pH on the conformation of the protein. (Note for comparison that raising the pH even further to 8.0 actually causes a fall in K0.5Rb, which might be explained by a reduction of direct competition between Rb+ ions and protons between pH 7.5 and 8.0.) Consistent with the notion that lowering the pH below 7.5 stabilizes the E2(2Rb) form, the stopped-flow measurements in Fig. 9 show that the rate of E2(2Rb) → E1Na for the wild type is much slower at pH 6.0 compared with 7.5 (pH 7.5 as follows: kE2(Rb) → E1 = 0.42 ± 0.002; pH 6.0 and kE2(Rb) → E1 = 0.073 ± 0.001 s−1), quite similar to the effect of the D369A and D369N mutants at pH 7.5. In summary the effect of lowering pH from 7.5 to 5.5 on the conformational transition of the wild type is similar to the effect of neutralizing the charge on Asp369 in the D369N mutation. Furthermore, the effect of pH is largely abolished in the D369N mutation itself.
FIGURE 8.

Equilibrium titrations of E1 ↔ E2(Rb) at varying pH. A, wild type; B, D369N. The data have been normalized as described under “Experimental Procedures.” Solid lines represent curves fitted to these data and provide the parameters in Table 4. At pH values lower than 7.0, a linear drift was subtracted from the fluorescence signal changes.
FIGURE 9.

Stopped-flow traces of E2(2Rb) → E1Na for wild type at pH 7. 5 and 6. Conditions were as under “Experimental Procedures,” except that solutions at pH 6.0 contained 10 mm Mes. Each trace represents the average of 5–10 replicates. The rate was measured at pH 6.0 rather than at 5.5 to avoid a fluorescence drift that interferes with the measurement.
TABLE 4.
Equilibrium titrations of the E1 ↔ E2(2Rb) conformational change at different pH values
Parameters were obtained by fitting the normalized data in Fig. 8 to the Hill equation.
| pH | Wild type K0.5Rb ± S.E. | D369N K0.5Rb ± S.E. |
|---|---|---|
| mm | mm | |
| 5.5 | 0.35 ± 0.03 | 0.37 ± 0.01 |
| 6.5 | 0.71 ± 0.02 | |
| 7.0 | 1.06 ± 0.04 | |
| 7.5 | 3.45 ± 0.1 | 0.91 ± 0.02 |
| 8.0 | 1.5 ± 0.03 |
E1 ↔ E2 Transitions of Wild Type and D369N/D369A Mutant Induced by Phosphate and Vanadate
As shown previously, addition of inorganic phosphate/Mg+ or vanadate/Mg+ to fluorescein-labeled renal Na+,K+-ATPase in an E1 conformation, without Na+ and K+ ions, stabilizes an E2 conformation, detected by fluorescence quenching, and subsequent addition of Na+ ions reverses the fluorescence change (13, 30, 32). The equilibrium data in Fig. 10 and stopped-flow fluorescence data in Fig. 11 and Table 5 compare fluorescence responses to phosphate and vanadate of the wild type and mutant proteins and reveal two new phenomena. For the wild type, the fluorescence quench induced by phosphate or vanadate occurs only in the presence of Mg2+ ions (Fig. 10), similar to the response seen for renal Na+,K+-ATPase (13). The new finding is that for both the D369A and D369N mutants, the fluorescence quench response to phosphate is observed without added Mg2+ ions, i.e. Mg2+ ion is not required for the phosphate-dependent stabilization of E2 (Fig. 10). Vanadate differs from phosphate in that the response of D369N and D369A is like the wild type protein and requires the presence of Mg2+ ions (Fig. 10). Addition of Na+ ions at high concentrations (100 mm), after vanadate/Mg2+, partially reversed the fluorescence change of the wild type, as found previously for renal Na+,K+-ATPase (30). Interestingly, the vanadate/Mg2+-induced signal change for D369A was also partially reversed by 100 mm NaCl but that for D369N was not reversed by NaCl, showing again that the D369N is more strongly poised toward E2. The concentrations of phosphate and vanadate required to induce these signal changes were in the several millimolar and tens of micromolar range, respectively, but attempts to determine accurate concentration dependences were not satisfactory due to a slow time course or small signal size and difficulty in determining the end points after graded addition of phosphate or vanadate.
FIGURE 10.

Phosphate- and vanadate-induced E1 ↔ E2 conformational changes of wild type (WT) and D369N/D369A mutants.
FIGURE 11.

Stopped-flow traces of rates of phosphate- and vanadate-induced E1 → E2 conformational changes for wild type and D369N/D369A mutants. A, phosphate: wild type (WT) and D369N, 25 mm phosphate (Tris), 4 mm MgCl2, D369A, 25 mm phosphate (Tris). B, vanadate: wild type and mutants 1 mm vanadate (Tris), 4 mm MgCl2. Traces represent averages of 4–8 replicates and are fitted to a double exponential function; see Table 5.
TABLE 5.
Rates of E1 → E2·P and E1 → E2·vanadate conformational transitions
Rate constants obtained by double exponential fits of the data in Fig. 11.
| Wild type | D369N | D369A | |
|---|---|---|---|
| s−1 ± S.E. | s−1 ± S.E. | s−1 ± S.E. | |
| E1 → E2·P | k1, 0.026 ± 0.001 | k1, 0.67 ± 0.09 | k1, 0.89 ± 0.05 |
| k2, 0.004 ± 0.0001 | k2, 0.036 ± 0.002 | k2, 0.038 ± 0.001 | |
| E1 → E2·vanadate | k1, 0.086 ± 0.001 | k1, 12.18 ± 0.37 | k1, 0.97 ± 0.18 |
| k2, 0.011 ± 0.0001 | k2, 0.1 ± 0.006 | k2, 0.17 ± 0.017 |
We have also looked at the rate of fluorescence quenching in the stopped-flow fluorimeter when the proteins were mixed with phosphate/Mg2+ (wild type, D369N) or phosphate alone (D369A) (E1 → E2·P) or vanadate/Mg2+ (E1 → E2·vanadate) at high concentrations that induce the full response (Fig. 11). These experiments show a second clear difference between wild type and mutants. As seen in Fig. 11, the rate of quenching by either phosphate/Mg2+ or vanadate/Mg2+ is very slow for the wild type protein, but a much faster component was observed for both D369N and D369A with either phosphate or vanadate. The rate constants of all the curves were fitted to double exponential functions, giving the values of k1 and k2 recorded in Table 5. In the case of the wild type, k2 is very slow and may represent an artifact due to photo-bleaching of the bound fluorescein. For quenching induced by phosphate, k1 for the D369N and D369A mutants is 25–34-fold faster than the k1 of the wild type. It is noticeable that k for these mutants has about the same rate as the k1 of the wild type. For quenching induced by vanadate/Mg2+, the k1 was about 11-fold (D369A) or 141-fold (D369N) faster than the k1 of the wild type, and again, the k2 for the mutants has about the same rate as the k1 of the wild type. Possible explanations of these observations are given below.
DISCUSSION
This study introduces the use of the purified fluorescein-labeled recombinant Na+,K+-ATPase and its mutants for detailed study of E1 ↔ E2 conformational changes. This represents a significant advance in functional analysis compared with experiments on Na+,K+-ATPase expressed at rather low levels in mammalian cells or Xenopus oocytes or even after expression in yeast S. cerevisiae or P. pastoris membranes without purification, which permit studies such as ligand binding (ATP, ouabain), Rb+ occlusion, or proteolytic and Fe2+-catalyzed oxidative cleavages (20, 43). The selective effects of ligands (K+, Na+, Pi/Mg2+, vanadate/Mg2+, effects of pH) on the fluorescein fluorescence responses of the purified detergent-soluble recombinant Na+,K+-ATPase are quite similar to those found previously for native membrane-bound renal Na+,K+-ATPase, although the amplitudes of the responses are smaller (12–15% for wild type compared with 25–35% for renal Na+,K+-ATPase). The specific effects of the ligands are diagnostic so that the basic enzymatic mechanism is preserved. In a complementary approach, fluorescence signals of the electrochromic shift dye RH421 have also been detected recently for the detergent-soluble purified recombinant Na+,K+-ATPase,4 similar to RH421 fluorescence changes for native renal Na+,K+-ATPase (27). RH421 signals reflect changes in the local electrical field accompanying cation binding and dissociation and are also indicative of an intact catalytic mechanism. Tryptophan fluorescence changes of glycerol-stabilized purified detergent-soluble Ca2+-ATPase have also been described recently, as another example of the utility of the fluorescence approach (44).
Comparison of E1 ↔ E2 Conformational Transitions of Wild Type and D369N/D369A/D369E Mutants
Replacement of the negatively charged aspartate by asparagine and alanine in the D369N/D369A mutants reveals a clear-cut stabilization of the E2 conformation (Figs. 4–7 and Table 2). One important observation is that substitution of the aspartate with the negatively charged glutamate, D369E, led to behavior much like the wild type, as judged by the equilibrium fluorescence titrations (Fig. 4). Another mutant, T212A, which does not change the charge within the active site, behaved similarly. Thus, the observed changes in conformational transitions of the D369N/D369A mutants can be attributed to neutralization of the negative charge of the aspartate. The present observations are consistent with the proposal (19) that D369N/D369A mutants stabilize E2, but the direct measurements show that the effect is much larger than inferred from effects of K+ ions on ATP binding. More importantly, the fluorescence measurements allow elucidation of the detailed mechanism. Stabilization of the E2 conformation was detected by a lower K0.5Rb in equilibrium titrations, 3.3-fold for D369A and 6.2-fold for D369N (Fig. 4 and Table 2). The origin of this change in apparent affinity for Rb+ ions is a large reduction in the rate of E2(2Rb) → E1Na, 13-fold for D369A and 20-fold for D369N, with little or no effect on the rate of E1 → E2(2Rb) or intrinsic Rb+ binding affinity (Figs. 5–7 and Table 3). Although the absolute values of the conformational equilibrium constant KC with Rb+ ions could not be calculated due to lack of saturation of E1 → E2(2Rb), one can calculate a lower limit value as kE1 → E2(2Rb) 83 mm, kE2(2Rb) → E1Na. The values calculated from the rate constants in Table 3 are for wild type, 57.62 ± 1.1; D369N, 1069 ± 19.2; D369A, 948 ± 30.2, i.e. 16.5–18.5-fold in favor of E2(2Rb) for the mutants. For the wild type, the value of KC with K+ ions could be calculated as kmaxE1 → E2(2K):kE2(2K) → E1Na and equals KC = 37.9 ± 4.6 (see Table 3).5
For the wild type, lowering the pH from 7.5 to 5.5 had a similar effect on the K0.5Rb in equilibrium titrations and rate of E2(2Rb) → E1Na as the D369N/D369A mutants (Fig. 8 and Table 4). Furthermore, because replacing the protonatable carboxyl by the neutral carboxamide group in D369N abrogated about 75% of the effect of pH on K0.5Rb compared with the wild type, the inference is that the effect of reducing pH on the wild type is due largely to protonation of the carboxyl of Asp369 and neutralization of its charge. Thus, in essence, the membrane Bohr effect described in Ref. 12 has the same origin as the neutralization of the charge in the D369N/D369A mutations.
Structural Basis for Effect of D369N/D369A Mutations on E1 ↔ E2(2Rb) Conformational Changes
Fig. 12 depicts residues in the active site of Na+,K+-ATPase in the recent structures of the E2·P(2Rb)-like conformation with bound MgF42−, an analogue of bound phosphate, and Mg2+ ion or depicted also without the bound MgF42− and Mg2+ (Fig. 12, A and B) (5, 6). The conserved TGES sequence of the A domain (Fig. 12, red) comes close to the P domain (green), and in particular Glu214 (A domain) is poised above the MgF42−. As seen in the 2ZXE structure with bound MgF42− and Mg2+ removed (Fig. 12B, double-headed arrow), Glu214 is the closest charged residue of either the A or N domain to the active site Asp369. Because an important characteristic of the E2 conformation is the interaction of A-P and A-N domains, a simple hypothesis is that removal of electrostatic repulsion between the negative charges of Asp369 and Glu214 stabilizes the E2 conformation.
FIGURE 12.

Active site of Na+,K+-ATPase in E2·P conformation and Ca2+-ATPase in the E1ATP conformation. A, active site of Na+,K+-ATPase in E2·MgF42−. Mg2+ conformation (PDB code 2XZE). B, as in A without the bound MgF42−·Mg2+. Arrows indicate Asp369 and Glu214 and Arg544–Glu216 and Arg685–Glu231 pairs. C, active site of Ca2+-ATPase in E1·AMPPCP (PDB code 1VFP) showing proximity of Arg560 and Arg678 to the bound AMPPCP. The figure was drawn with PyMOL. P domain, green; A domain, red; N domain, blue.
Using Coulomb's law, we can calculate approximately the change in electrostatic repulsion between Asp369 and Glu214, when the charge on Asp369 is removed, and predict the effect on the conformational equilibrium. This follows a similar calculation, which determined the effects of neutralization of the charge on Asp351 of Ca2+-ATPase on ATP affinity (23). The nearest approach distance between Asp369 and Glu214 (PDB code 2ZXE) is 6.2 Å, and in the homologous structure of Ca2+-ATPase (PDB code 1WPG), the distance between Asp351 and Glu183 is 6.5 Å. In the E2 conformation of Ca2+-ATPase (PDB code 1IWO) without bound MgF42−/Mg2+, the P and A domains are more separated, and the closest distance between Asp351 and Glu183 is 10.8 Å. For the calculation, we will assume that the distance between Asp369 and Glu214 in an E2(2Rb) conformation of Na+,K+-ATPase without MgF42−/Mg2+ lies in the range 6.5–10 Å. The electrostatic potential between Asp369 and Glu214 is calculated from Coulomb's law as in Ref. 23. The range of values is 111 to 72 mV.6 Assuming that the effect of the D369N/D369A mutations on KC is due only to removal of the electrostatic repulsion with Glu214, the predicted effect is calculated from the Nernst equation,6 and the range of values is 85- to 18-fold at 6.5 or 10 Å distance, respectively. The experimental range of the 16.5–18.5-fold change in poise of KC toward E2, calculated from the kinetic data, falls in this predicted range with a distance between Asp369 and Glu214 close to 10 Å. Of course, these calculations depend on assumptions of the dielectric constant and the distance within the active site, but they certainly seem to be reasonable.
Fig. 12B depicts another important point, namely that outside of the active site the A domain makes very few contacts with the P and N domains. However, two salt bridges, Arg544–Glu216 (N-A) and Arg685–Glu231 (P-A) can be clearly detected. The interactions of the residues in these salt bridges in different conformations provide a simple but convincing explanation of the well established effect of ATP to accelerate the rate of E2(2K) → E1Na in wild type Na+,K+-ATPase and, as a consequence, the effects of D369N/D369A on the rate of E2(2K) → E1Na. Comparison of the Na+,K+-ATPase structure in the E2(2Rb)·P conformation with the Ca2+-ATPase structure with a bound ATP analogue in an E1-AMPPCP conformation (Fig. 12C) (45) shows that the homologous arginines of Ca2+-ATPase, Arg678 (Arg685), and Arg560 (Arg544), directly bind the AMPPCP. Mutations of Arg544 in the Na+,K+-ATPase have also shown that this residue is crucial for high affinity binding of ATP and ADP (46), and Arg678 in Ca2+-ATPase was shown to be cross-linked to the N domain by glutaraldehyde, and this is disturbed by ATP binding (47).Thus it is clear that in E2(2K) binding of ATP to Arg544 and Arg685 will compete with Glu216 and Glu231 of the A domain for these residues. In other words, ATP and the A domain compete for the interaction with the P and N domains, and attraction of ATP for Arg544 and Arg685 and repulsion of Glu216 and Glu231 will destabilize the interaction of the N and P domains with the A domain and stabilize the ATP-mediated interaction of the N to P domain. The result of this competition is the greatly accelerated rate of E2(2K) → E1Na by ATP bound to E2(2K) with a low affinity and stabilization of E1 by ATP bound with high affinity. The explanation of this effect of ATP implies that the slow rate of E2(2Rb) → E1Na, without ATP, is limited, at least partially, by the high activation energy required to break these electrostatic interactions. Because the total strength of N-A and P-A domain interactions represents the balance of attractive and repulsive forces, one can presume that removal of the Asp369–Glu214 repulsion in the D369N/D369A mutants allows the A domain to approach the N and P domains more closely and increase their interaction energy via the Arg544–Glu216 and Arg685–Glu231 proximities. This will, in turn, increase further the activation energy required to break them and slow the rate of E2(2Rb) → E1Na compared with the wild type. Fig. 12C also shows that Arg560 and Arg678 interact with the oxygens on the β-phosphate and on the sugar moiety of the ATP analogue, respectively. Therefore, another implication is that ADP should be as effective as ATP in repulsing the A domain and accelerating the E2(2K) → E1Na transition. Acceleration of E2(2K) → E1Na by ADP, as for ATP, has indeed been inferred from effects of ADP on K+-K+ exchange reactions (48), proteolytic digestion (10), and demonstrated directly using an iodoacetamidofluorescein-labeled Na+,K+-ATPase (49). Finally, it is of interest that for Ca2+-ATPase, although the two arginines (Arg560 and Arg678) are conserved, the residue at position Glu216 of Na+,K+-ATPase is Val185 in Ca2+-ATPase, and the residue at position Glu231 is Asp203 in Ca2+-ATPase. Thus, only one of the salt bridges can be formed in the E2P or E2 conformations. Although it has been known for many years that ATP activates the E2 → E1 transition of Ca2+-ATPase (50), ATP accelerates the rate of E2 ↔ E1 in Ca2+-ATPase much less (20-fold at pH 6.0 and less at higher pH (51)) than it accelerates the rate of E2(2K) → E1Na of Na+,K+-ATPase (100–150-fold) (11, 49, 52).
The lack of effect of the D369N and D369A mutants on the rate of E1 → E2(2Rb) indicates that, in these conditions, the limiting step is not affected by neutralization of the charge on Asp369 but is the same as in the wild type. The result in Fig. 7B shows that the rate of this transition depends on the nature of the bound cation and that with K+ being considerably faster than that with bound Rb+. Thus, the experiment indicates that the limiting process is the movement of trans-membrane segments with occluded K+ or Rb+ ions. The rate of these movements should be the same in wild type and D369N/D369A mutants.
Effects of Phosphate and Vanadate
The finding (Fig. 10) that phosphate induced the E1 → E2·P transition in the D369N/D369A mutants even without added Mg2+ ions, by contrast with the wild type, can be understood by reference to the structure in Fig. 12A. The Mg2+ ion interacts with the MgF42−, with the carboxyl groups of Asp369 and Asp710, with the carbonyl of Thr371, and via a water molecule with Gly213. Binding of Mg2+ ions to Asp710 has also been inferred by mutation work (53). The MgF42− also interacts strongly with Asp369, Lys698, and Thr610 and is very close to Glu214 (2.4 Å). Because Mg2+ is not required for the E1 to E2·P transition in the D369N/D369A mutants, an important role of the Mg2+ in the wild type appears to be neutralization of the negative charge on the Asp369 with bound phosphate and enabling the A domain to approach the P domain. Without charge neutralization between the MgF42− (HPO42−) and Glu214, which are only 2.4 Å distant from each other, there should be strong repulsion between P and A domains. Bound Mg2+ will neutralize the negative charge on the MgF42− and reduce the charge repulsion with Glu214. The structure also clarifies two related previous findings. First, D369A binds ouabain weakly even without added Mg2+ ions, although binding to wild type is absolutely dependent on Mg2+ ions (19). Ouabain binding stabilizes the E2 conformation, and again, one would infer that Mg2+ is required in the wild type, but not in D369A, to allow Glu214 (A domain) to approach Asp369 (P domain). Second, Fe2+-dependent oxidative cleavages in the active site, characteristic of the E2 conformation, are largely suppressed in the D369N mutant (20). Because Fe2+ substitutes for Mg2+ ions in catalyzing phosphate transfer, the result is consistent with suboptimal binding of Fe2+ to the D369N mutant. It is interesting that the D369N/D369A mutants behave differently with vanadate, compared with phosphate, in that Mg2+ ions are still required to stabilize the E2 conformation. Vanadate is a phosphate transition state analogue, binds much more tightly than phosphate, and could be expected to show different interactions. Indeed, Asp710, which is a Mg2+-ligating residue, has been shown to be much more important for vanadate compared with phosphate binding in E2 (53).
The scheme in Fig. 13 can explain simply the finding that, for the D369N/D369A mutants, the rate of conversion of E1 → E2·P or E1 → E2·vanadate showed a fast component, by contrast to the slow rate of conversion for the wild type (Fig. 11 and Table 5). Both phosphate and vanadate are assumed to bind much more tightly to E2 than to E1. By the left-hand path, E2·P or E2·vanadate are stabilized by preferential binding of phosphate or vanadate to E2 and prevention of the back reaction, E2 → E1. The slow observed rate of fluorescence change is presumably limited by the E1 → E2 transition without bound phosphate or vanadate. This is assumed to be the path for the wild type. By contrast, the right-hand path may apply to the D369N/D369A mutants. Although phosphate and vanadate bind weakly to E1, neutralization of the charge on the Asp369 allows a fast conformational change from E1·P → E2 → P or E1·vanadate → E2·vanadate, i.e. bound phosphate or vanadate induce the fast component observed in Fig. 11. Again, it is interesting that the effect with D369N is stronger than with D369A, at least with vanadate. The slow component may correspond to the fraction of molecules, which reach the E2·P or E2·vanadate conformation by the slow left-hand route E1 → E2 → E2·P.
FIGURE 13.
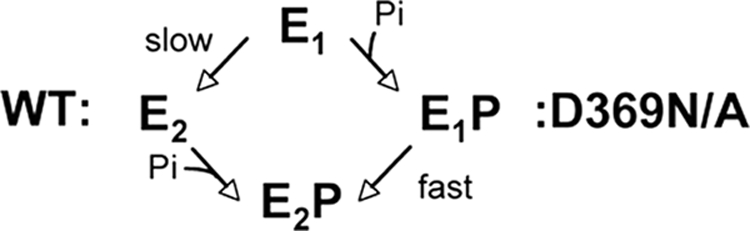
Scheme showing alternative pathways for conformational change from E1 to E2P.
Role of Electrostatic Interactions of Asp369 in the Normal Catalytic Cycle
Bound Mg2+ ions are already known to play a crucial role in reducing negative charge repulsion, between the γ-phosphate of bound ATP and the aspartate, enabling close proximity necessary for phosphorylation. D369N/D369A mutants of Na+,K+-ATPase and D351A/D351N/D351T mutants of Ca2+-ATPase have a very high ATP affinity, which is the result of removal of electrostatic repulsion between the γ-phosphate and the aspartate (19, 22, 23). In the wild type, binding of Mg2+ ions to the Asp369 (Na+,K+-ATPase) or Asp351 (Ca2+-ATPase) and the γ-phosphate reduces this repulsion. Indeed, in the case of Ca2+-ATPase ATP binds only weakly in the absence of Mg2+ ions (22).
We propose here an additional role of charge interactions of Asp369 in the normal catalytic cycle, namely as triggers of E1P(3Na) → E2P and E2(2K) → E1Na conformational transitions. As discussed in the Introduction, dissociation of ADP from E1P(3Na) is necessary but may not itself be sufficient to trigger the spontaneous conformational change to E2P. The lack of requirement of Mg2+ ions for the phosphate-induced conformational change (Fig. 10), and the fast phosphate- or vanadate-induced E1 → E2·P or E1 → E2·vanadate transitions, observed for the D369N/D369A mutants (Fig. 11), suggest that the behavior of the mutants resembles that of normal wild type E1P(3Na) with bound Mg2+ ions. Thus E1P(3Na) → E2P may be triggered by neutralization of the charge on Asp369 resulting from binding the divalent cation, Mg2+, together with the covalently bound phosphate (which is a mixture of -C-O-PO32− and -C-O-POH3− at physiological pH). The interconversion between conformational states has been described as “fairly stochastic,” i.e. one in which the energy barriers between states are comparable with the thermal energy (2, 3). In the stochastic process of approach, binding, and disengagement of A to and from P and N domains, the lack of repulsion between Asp369-phosphate/Mg2+ and Glu214, as well as attraction of the bound Mg2+ ion to the A domain via Gly213 and a water molecule, should favor approach of the A domain to the P and N domains. E2P, with Mg2+ ions tightly bound, may then be stabilized via the Arg685–Glu231 and Arg544–Glu216 salt bridges between A and P and A and N domains, respectively. After dephosphorylation of E2P, phosphate/Mg2+ dissociates, and the negative charge on Asp369 should favor disengagement of the N-A and P-A domains in E2(2K) due to repulsion of the Glu214 in the A domain. Dissociation of A from P and N domains in E2(2K) and transition to E1 is then strongly accelerated by ATP, which breaks the Arg685–Gly231 and Arg544–Glu216 salt bridges, as discussed above.
This work was supported by Grant 538/04 from the Israel Science Foundation.
Throughout this paper, conformational equilibria are denoted by the symbol ↔, and unidirectional conformational transitions are denoted by the symbol →.
Habeck, M., Cerri, E., Katz, A., Karlish, S. J. D., and Apell, H. J. (2009) Biochemistry 48, in press.
The value of KC for the wild type with either Rb+ or K+ is lower than the KC inferred for native membrane-bound renal Na+,K+-ATPase (KC ≈ 1000) (13) and shows that the KC of the detergent-soluble recombinant wild type enzyme is poised less strongly toward E2. This will be described in a separate paper (T. Belogus and S. J. D. Karlish, manuscript in preparation). It does not affect the conclusions on the effects of the mutations.
The electrostatic potential Δψ is calculated from the relationship Δψ ≈ e·k/D·r, where e is the electronic unit charge (1.6.10−19 C); k is the Coulomb constant (9·109 N·m2·C−2); D is the dielectric constant in the site, assumed to have a value of 20; and r is the distance (in meters). The Nernst equation: lnKCΔψ = 0 = lnKC + (ZF/RT)Δψ− or logKC,Δψ = 0/KC = Δψ/57 (in mV).
- FITC
- fluorescein 5′-isothiocyanate
- DDM
- n-dodecyl-β-d-maltopyranoside
- C12E8
- octaethylene glycerol monododecyl ether
- SOPS
- 1-stearoyl-2-oleoyl-sn-glycero-3-[phosphor-l-serine]
- AMPPCP
- adenosine 5′-(β,γ-methylenetriphosphate)
- Mes
- 4-morpholineethanesulfonic acid
- MOPS
- 4-morpholinepropanesulfonic acid
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- PDB
- Protein Data Bank.
REFERENCES
- 1.Olesen C., Picard M., Winther A. M., Gyrup C., Morth J. P., Oxvig C., Møller J. V., Nissen P. (2007) Nature 450, 1036–1042 [DOI] [PubMed] [Google Scholar]
- 2.Toyoshima C. (2008) Arch. Biochem. Biophys. 476, 3–11 [DOI] [PubMed] [Google Scholar]
- 3.Toyoshima C. (2009) Biochim. Biophys. Acta 1793, 941–946 [DOI] [PubMed] [Google Scholar]
- 4.Garty H., Karlish S. J. (2006) Annu. Rev. Physiol. 68, 431–459 [DOI] [PubMed] [Google Scholar]
- 5.Morth J. P., Pedersen B. P., Toustrup-Jensen M. S., Sørensen T. L., Petersen J., Andersen J. P., Vilsen B., Nissen P. (2007) Nature 450, 1043–1049 [DOI] [PubMed] [Google Scholar]
- 6.Shinoda T., Ogawa H., Cornelius F., Toyoshima C. (2009) Nature 459, 446–450 [DOI] [PubMed] [Google Scholar]
- 7.Post R. L., Hegyvary C., Kume S. (1972) J. Biol. Chem. 247, 6530–6540 [PubMed] [Google Scholar]
- 8.Karlish S. J., Yates D. W., Glynn I. M. (1978) Biochim. Biophys. Acta 525, 252–264 [DOI] [PubMed] [Google Scholar]
- 9.Jorgensen P. L. (1975) Biochim. Biophys. Acta 401, 399–415 [DOI] [PubMed] [Google Scholar]
- 10.Schuurmans Stekhoven F. M., Swarts H. G., Zhao R. S., de Pont J. J. (1986) Biochim. Biophys. Acta 861, 259–266 [DOI] [PubMed] [Google Scholar]
- 11.Karlish S. J., Yates D. W. (1978) Biochim. Biophys. Acta 527, 115–130 [DOI] [PubMed] [Google Scholar]
- 12.Skou J. C., Esmann M. (1980) Biochim. Biophys. Acta 601, 386–402 [DOI] [PubMed] [Google Scholar]
- 13.Karlish S. J. (1980) J. Bioenerg. Biomembr. 12, 111–136 [DOI] [PubMed] [Google Scholar]
- 14.Dupont Y. (1976) Biochem. Biophys. Res. Commun. 71, 544–550 [DOI] [PubMed] [Google Scholar]
- 15.Dupont Y., Leigh J. B. (1978) Nature 273, 396–398 [DOI] [PubMed] [Google Scholar]
- 16.Pick U., Karlish S. J. (1982) J. Biol. Chem. 257, 6120–6126 [PubMed] [Google Scholar]
- 17.Kuntzweiler T. A., Wallick E. T., Johnson C. L., Lingrel J. B. (1995) J. Biol. Chem. 270, 16206–16212 [DOI] [PubMed] [Google Scholar]
- 18.Ohtsubo M., Noguchi S., Takeda K., Morohashi M., Kawamura M. (1990) Biochim. Biophys. Acta 1021, 157–160 [DOI] [PubMed] [Google Scholar]
- 19.Pedersen P. A., Rasmussen J. H., Jørgensen P. L. (1996) Biochemistry 35, 16085–16093 [DOI] [PubMed] [Google Scholar]
- 20.Strugatsky D., Gottschalk K. E., Goldshleger R., Bibi E., Karlish S. J. (2003) J. Biol. Chem. 278, 46064–46073 [DOI] [PubMed] [Google Scholar]
- 21.Karlish S. J. (2003) Ann. N.Y. Acad. Sci. 986, 39–49 [DOI] [PubMed] [Google Scholar]
- 22.McIntosh D. B., Woolley D. G., MacLennan D. H., Vilsen B., Andersen J. P. (1999) J. Biol. Chem. 274, 25227–25236 [DOI] [PubMed] [Google Scholar]
- 23.Marchand A., Winther A. M., Holm P. J., Olesen C., Montigny C., Arnou B., Champeil P., Clausen J. D., Vilsen B., Andersen J. P., Nissen P., Jaxel C., Møller J. V., le Maire M. (2008) J. Biol. Chem. 283, 14867–14882 [DOI] [PubMed] [Google Scholar]
- 24.Skou J. C., Esmann M. (1981) Biochim. Biophys. Acta 647, 232–240 [DOI] [PubMed] [Google Scholar]
- 25.Kapakos J. G., Steinberg M. (1986) J. Biol. Chem. 261, 2084–2089 [PubMed] [Google Scholar]
- 26.Kapakos J. G., Steinberg M. (1986) J. Biol. Chem. 261, 2090–2095 [PubMed] [Google Scholar]
- 27.Stürmer W., Bühler R., Apell H. J., Läuger P. (1991) J. Membr. Biol. 121, 163–176 [DOI] [PubMed] [Google Scholar]
- 28.Bühler R., Stürmer W., Apell H. J., Läuger P. (1991) J. Membr. Biol. 121, 141–161 [DOI] [PubMed] [Google Scholar]
- 29.Farley R. A., Tran C. M., Carilli C. T., Hawke D., Shively J. E. (1984) J. Biol. Chem. 259, 9532–9535 [PubMed] [Google Scholar]
- 30.Karlish S. J., Beaugé L. A., Glynn I. M. (1979) Nature 282, 333–335 [DOI] [PubMed] [Google Scholar]
- 31.Rephaeli A., Richards D., Karlish S. J. (1986) J. Biol. Chem. 261, 6248–6254 [PubMed] [Google Scholar]
- 32.Hegyvary C., Jorgensen P. L. (1981) J. Biol. Chem. 256, 6296–6303 [PubMed] [Google Scholar]
- 33.Smirnova I. N., Faller L. D. (1993) J. Biol. Chem. 268, 16120–16123 [PubMed] [Google Scholar]
- 34.Pick U., Karlish S. J. (1980) Biochim. Biophys. Acta 626, 255–261 [DOI] [PubMed] [Google Scholar]
- 35.Orlowski S., Champeil P. (1993) FEBS Lett. 328, 296–300 [DOI] [PubMed] [Google Scholar]
- 36.Rabon E. C., Bassilian S., Sachs G., Karlish S. J. (1990) J. Biol. Chem. 265, 19594–19599 [PubMed] [Google Scholar]
- 37.Cohen E., Goldshleger R., Shainskaya A., Tal D. M., Ebel C., le Maire M., Karlish S. J. (2005) J. Biol. Chem. 280, 16610–16618 [DOI] [PubMed] [Google Scholar]
- 38.Haviv H., Cohen E., Lifshitz Y., Tal D. M., Goldshleger R., Karlish S. J. (2007) Biochemistry 46, 12855–12867 [DOI] [PubMed] [Google Scholar]
- 39.Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., Pease L. R. (1989) Gene 77, 51–59 [DOI] [PubMed] [Google Scholar]
- 40.Lifshitz Y., Petrovich E., Haviv H., Goldshleger R., Tal D. M., Garty H., Karlish S. J. (2007) Biochemistry 46, 14937–14950 [DOI] [PubMed] [Google Scholar]
- 41.Lifshitz Y., Lindzen M., Garty H., Karlish S. J. (2006) J. Biol. Chem. 281, 15790–15799 [DOI] [PubMed] [Google Scholar]
- 42.Glynn I. M., Karlish S. J. (1990) Annu. Rev. Biochem. 59, 171–205 [DOI] [PubMed] [Google Scholar]
- 43.Jorgensen P. L., Nielsen J. M., Rasmussen J. H., Pedersen P. A. (1998) Biochim. Biophys. Acta 1365, 65–70 [DOI] [PubMed] [Google Scholar]
- 44.Montigny C., Arnou B., Marchal E., Champeil P. (2008) Biochemistry 47, 12159–12174 [DOI] [PubMed] [Google Scholar]
- 45.Toyoshima C., Mizutani T. (2004) Nature 430, 529–535 [DOI] [PubMed] [Google Scholar]
- 46.Jacobsen M. D., Pedersen P. A., Jorgensen P. L. (2002) Biochemistry 41, 1451–1456 [DOI] [PubMed] [Google Scholar]
- 47.McIntosh D. B. (1992) J. Biol. Chem. 267, 22328–22335 [PubMed] [Google Scholar]
- 48.Kaplan J. H., Kenney L. J. (1982) Ann. N.Y. Acad. Sci. 402, 292–295 [DOI] [PubMed] [Google Scholar]
- 49.Steinberg M., Karlish S. J. (1989) J. Biol. Chem. 264, 2726–2734 [PubMed] [Google Scholar]
- 50.Scofano H. M., Vieyra A., de Meis L. (1979) J. Biol. Chem. 254, 10227–10231 [PubMed] [Google Scholar]
- 51.Mintz E., Mata A. M., Forge V., Passafiume M., Guillain F. (1995) J. Biol. Chem. 270, 27160–27164 [DOI] [PubMed] [Google Scholar]
- 52.Clarke R. J., Humphrey P. A., Lüpfert C., Apell H. J., Cornelius F. (2003) Ann. N.Y. Acad. Sci. 986, 159–162 [DOI] [PubMed] [Google Scholar]
- 53.Pedersen P. A., Jorgensen J. R., Jorgensen P. L. (2000) J. Biol. Chem. 275, 37588–37595 [DOI] [PubMed] [Google Scholar]