

Abstract
Integrin αIIbβ3 is expressed in mast cells as well as in megakaryocytes/platelets. A recent study has shown that surface expression levels of integrin αVβ3 are elevated in integrin αIIb-deficient bone marrow-derived mast cells (BMMCs) as compared with wild-type (WT) counterparts, but the underlying mechanism remains obscure. Here we demonstrate by transducing integrin αIIb into integrin αIIb-deficient BMMCs that surface expression levels of integrin αVβ3 are inversely related to those of integrin αIIbβ3. Thus, competitive association of integrin β3 with integrin αIIb or integrin αV determines surface expression levels of integrin αIIbβ3 or αVβ3 in mast cells. We compared WT and integrin αIIb-deficient BMMCs as well as integrin αIIb-deficient BMMCs transduced with integrin αIIb(WT) or non-functional αIIb(D163A) mutant and found that enhancement of proliferation, degranulation, cytokine production, and migration of BMMCs through interaction with fibrinogen (FB) depended on integrin αIIbβ3. In addition, elevated surface expression of integrin αVβ3 failed to compensate for loss of FB-associated functions in integrin αIIb-deficient BMMCs while enhancing adhesion to vitronectin or von Willebrand factor. Importantly, integrin αIIb deficiency strongly suppressed chronic inflammation with the remarkable increase of mast cells induced by continuous intraperitoneal administration of FB, although it did not affect acute allergic responses or mast cell numbers in tissues in steady states. Interestingly, soluble FB promoted cytokine production of BMMCs in response to Staphylococcus aureus with FB-binding capacity, through integrin αIIbβ3-dependent recognition of this pathogen. Collectively, integrin αIIbβ3 in mast cells plays an important part in FB-associated, chronic inflammation and innate immune responses.
Introduction
Mast cells play a critical role in IgE-associated allergic disorders, but recent advances have delineated the involvement of mast cells in IgE-independent physiological and pathological processes, including certain innate immune responses. In fact, various stimuli, in addition to IgE and specific antigens, can activate mast cells to release a diverse array of preformed and newly synthesized pro-inflammatory mediators such as histamine, lipids, cytokines, and chemokines (1–4). Although mast cell numbers and activation in tissues are closely related to mast cell-mediated immunity, the underlying mechanism remains incompletely understood. As one of the key phenomena, mast cells interact with the extracellular matrix (ECM)2 through integrins composed of two subunits (α and β), thereby regulating mast cell functions. As previously reported (5–9), integrins α4β1 and α5β1, or integrin αVβ3, expressed in mast cells mediate binding to fibronectin (FN) or vitronectin (VN), respectively. Interestingly, integrin α4β7 is involved in intestinal homing of mast cell progenitors via interaction with mucosal vascular addressin cell adhesion molecule-1 (10). In view of the implication of mast cell integrins in innate immunity, integrin α2β1 expressed in peritoneal mast cells is required for the induction of inflammatory responses to infection (11). In addition, integrin αVβ6 is essential for nematode-induced mucosal mast cell hyperplasia and for expression of the granule chymase (12).
Integrin αIIb, also known as CD41, which forms a complex with integrin β3, is a well known marker of the megakaryocyte/platelet lineage. Integrin αIIbβ3 is required for normal platelet hemostatic function (13–17). Previously, we reported that integrin αIIbβ3 is also highly expressed in mast cells (9, 18). In addition, we demonstrated that mast cell interaction with fibrinogen (FB) via integrin αIIbβ3 enhances in vitro mast cell functions, by using a blocking Ab specific for integrin αIIb (9). On the other hand, higher surface expression levels of integrin αV and enhanced adhesion to VN were found in integrin αIIb-deficient BMMCs as compared with wild-type (WT) counterparts (18), suggesting that integrin αIIb and integrin αV counter-regulate their surface expression levels and functions in mast cells. Therefore, we attempted to carefully analyze the regulatory mechanisms by utilizing retroviral transduction with integrin αIIb WT or non-functional mutant into integrin αIIb-deficient BMMCs.
FB abundant in plasma contributes to blood clotting (19). In addition, FB as well as its degradation product fibrin are also present in ECM outside blood vessels, where they play important roles in inflammation and wound healing through recruitment and activation of inflammatory cells expressing FB-binding receptors (19–22). Interestingly, surface proteins that bind to FB are also expressed by several types of bacteria, such as Staphylococcus aureus and Streptococcus pyogenes, which modulate immune responses to bacterial infections (23–25).
In the present study, we showed that integrin αIIb expression levels regulate surface expression levels of integrin αVβ3 as well as integrin αIIbβ3 in mast cells and that mast cell functions augmented by interaction with FB are dependent on integrin αIIbβ3 and independent of integrin αVβ3. In accordance, integrin αIIb deficiency strongly suppressed FB-induced chronic inflammation. Notably, the interaction with soluble FB via integrin αIIbβ3 helps mast cells recognize and respond to S. aureus (Cowan I) with FB-binding capacity. Thus, integrin αIIbβ3 in mast cells modulates FB-associated, chronic inflammation and innate immune responses.
EXPERIMENTAL PROCEDURES
Mice
All experimental mice were sex- and age-matched (6–16 weeks old). Balb/c mice were purchased from Charles River Japan (Tokyo, Japan). Integrin aIIb−/− mice were generated as described previously (14) and backcrossed to Balb/c mice for at least six generations. Animal studies were performed according to the guidelines of the animal care committee of the Institute of Medical Science, University of Tokyo.
Antibodies and Other Materials
Source of antibodies (Abs) were as follows: anti-mouse integrin αIIbβ3 mAb (1B5) was a kind gift from Dr. B. S. Coller (Rockefeller University, New York, NY) (26). Anti-mouse integrin αV (8B3) and anti-mouse integrin β3 (8B11) mAbs were kind gifts from Drs. D. J. Gerber and S. Tonegawa (Picower Center, Massachusetts Institute of Technology, Boston, MA) (27). Anti-dinitrophenol (DNP) IgE (SPE-7) was from Sigma. Anti-trinitrophenol (TNP) IgE (C38-2), anti-mouse αIIb (MWReg30), anti-mouse αV (RMV-7 and H9.2B8), anti-mouse α4 (9C10), anti-mouse α5 (5H10–27), anti-mouse LFA1 (M17/4), anti-mouse β1 (Ha2/5), anti-mouse β2 (GAME-46), and anti-αVβ3 (2C9.G2) mAbs and other Abs were from BD Pharmingen. Cytokines such as mouse IL-3 and SCF were obtained from R&D Systems. Bovine serum VN and TNP-conjugated BSA (TNP-BSA) were from Sigma. Human plasma FB and von Willebrand factor (vWF) were from Chemicon. Formalin-fixed S. aureus Cowan I was purchased from Calbiochem.
Cells
To generate BMMCs with 95% purity (c-kit+/FcϵRI+ by flow cytometry), bone marrow cells from 6-week-old male mice were cultured for 5–8 weeks in the presence of 10 ng/ml IL-3 with or without 20 ng/ml SCF as described previously (9, 28).
DNA Constructs, Transfection, and Infection
To generate mouse integrin αIIb(D163A) mutant, two-step PCR mutagenesis was performed by using mouse integrin αIIb wild-type (WT) cDNA (provided by Dr. R. B. Basani, Children's Hospital of Philadelphia, PA) as a template. Retroviral transfection was as described in a previous study (29). Briefly, integrin αIIb(WT) or αIIb(D163A) mutant cDNA was subcloned into pMXs-IRES-puror (pMXs-IP) to generate pMXs-IP-integrin αIIb(WT) or αIIb(D163A), respectively. To generate recombinant retroviruses, pMXs-IP plasmids were transfected into PLAT-E packaging cells (30) with FuGENE 6 (Roche Diagnostics). Cells were infected with retroviruses in the presence of 10 μg/ml Polybrene. Selection with puromycin was started 48 h after infection.
Flow Cytometric Analysis
Cells were stained as described before (9, 28). Cells stained with the indicated Abs were analyzed with a FACSCalibur equipped with CellQuest software (BD Biosciences) and Flowjo software (Tree Star).
Adhesion Assay and Migration Assay
Adhesion assay was done as described (6, 9). In brief, 96-well plates were coated with 20 μg/ml FB, FN, VN, or vWF. BMMCs resuspended at 5 × 105 cells/ml were transferred into coated wells with or without stimulant for 1 h at 37 °C. After washing, cell adhesion was quantitated using CellTiter-GloTM (Promega, Madison, WI) and a Micro Lumat Plus luminometer (EG&G Berthold), according to the manufacturer's instructions. In assays using blocking Abs, BMMCs were preincubated with 20 μg/ml Abs for 1 h before adding the cells to the plate. Migration assays were carried out as described (7, 9), using 24-well Transwell chambers with 5-μm polycarbonate filters (Corning).
Measurement of Cytokines
The cells were transferred into FB-coated 96-well plates (1 × 104 cells/well) with or without stimulants. After incubating for 12 h at 37 °C, the supernatant of each well was collected, and the concentration of IL-6 or TNF-α was quantified by enzyme-linked immunosorbent assay with OptiEIA for IL-6 or TNF-α (BD Pharmingen) as described (9, 28).
β-Hexosaminidase Release Assay
β-Hexosaminidase release assay was as described before (31). Briefly, 5 × 104 cells of IgE-sensitized BMMCs in Tyrode buffer (10 mm HEPES buffer (pH 7.4), 130 mm NaCl, 5 mm KCl, and 5.6 mm glucose) containing 0.1% BSA, 1 mm CaCl2, and 0.5 mm MgCl2 were stimulated with the indicated concentration of TNP-BSA in BSA- or FB-coated 96-well plates for 1 h at 37 °C. Cell supernatants and total cell lysates solubilized with 1% Nonidet P-40 were collected, and β-hexosaminidase in the supernatants and cell lysates was quantified by spectrophotometric analysis of hydrolysis of p-nitrophenyl-N-acetyl-β-d-glucopyranoside (Sigma). The percentage of β-hexosaminidase release was calculated.
PCA Reactions
Passive cutaneous anaphylactic (PCA) reactions were performed as described (32–34). Briefly, anti-DNP IgE was intradermally injected into the ears of mice. After 24 h, 250 μg of DNP-BSA and 0.5% Evans blue dye was intravenously injected. The amounts of extravasated dye were measured after 30 min by extracting ears. In another type of experiment, mice received anti-DNP IgE intravenously. After 24 h, a skin reaction was elicited by applying 0.75% dinitrofluorobenzene acetone-olive oil solution to both sides of the ears. The reaction was assessed by measuring the ear thickness 1 h and 12 h after antigen challenge.
FB-induced Chronic Inflammation Model
200 μl of 0.5 mg/ml FB or PBS was intraperitoneally injected into WT or integrin αIIb−/− mice every 2 days. After 1 month, total peritoneal cells of the sacrificed mice were collected by using 3 ml of PBS. Total cell numbers were counted by using a hemocytometer; the percentages of mast cells (c-kit+/FcϵRI+), granulocytes (Gr-1high/CD11b+), and macrophages (F4/80+) were calculated by fluorescence-activated cell sorting analysis.
Responses to Fixed S. aureus Cowan I in BMMCs
Fixed S. aureus Cowan I (SA) purchased from Calbiochem was stained with Cell Tracker Orange (Molecular Probes). Five × 104 BMMCs suspended in 10% BSA/Tyrode buffer were incubated with SA in the presence or absence of 0.5 mg/ml soluble FB in BSA-coated 96-well plates for 2 h. The interaction between mast cells and SA was observed by using a fluorescence microscope. After the supernatant of each well was collected, bacterial cells were removed with a 0.22-μm filter. Cytokine concentrations in the supernatants were quantified by enzyme-linked immunosorbent assay.
Quantitation of Tissue Mast Cells
Tissue mast cells in ear skin, back skin, peritoneal wall, and intestine were quantified by light microscopy at ×400 by an observer who was unaware of the identity (i.e. mouse genotype) of the individual specimens, in Giemsa-stained sections, as previously described (28, 35, 36). Results were expressed as mast cells (mean ± S.E.) per mm2.
Statistical Analysis
Data are shown as the mean ± S.D. Statistical significance was determined by Student's t test, with p < 0.01 (**) and p < 0.05 (*) taken as being statistically significant.
RESULTS
Surface Expression Levels of Integrin αV Are Elevated in Integrin αIIb-deficient BMMCs
To investigate the role of integrin αIIb in mast cells, bone marrow cells from WT and integrin αIIb−/− mice were cultured in the presence of IL-3 for 5 weeks to generate comparable numbers of morphologically pure (>95%) mast cells. BMMCs from WT and integrin αIIb−/− Balb/c mice exhibited similar levels of FcϵRI and c-kit on their cell surfaces as determined by flow cytometry (Fig. 1A). In addition, proliferative responses to IL-3 as well as apoptosis induced by growth factor (IL-3) deprivation were comparable between both BMMCs (Fig. 1, C and D). Thus, integrin αIIb deficiency did not affect Balb/c mice-derived mast cell development and growth in suspension culture, as previously reported in C57BL/6 mice (18). Moreover, when IgE-sensitized BMMCs were stimulated with the indicated doses of antigen, we found comparable levels of β-hexosaminidase release and cytokine (IL-6 and TNF-α) production (Fig. 1, E and F, and data not shown). This also suggested that integrin αIIb deficiency did not modulate FcϵRI signaling in suspension culture of mast cells. However, in keeping with previous findings (18), we confirmed the striking differences between the two cell types: surface expression levels of integrin αV and integrin αVβ3 were 10-fold higher in integrin αIIb-deficient BMMCs as compared with WT counterparts, despite comparable expression levels of other integrins such as integrins α4, α5, and β1, and no detectable expression of integrin β2 in either BMMC (Fig. 1E). Collectively, these results led us to postulate that integrin αIIb deficiency influenced mast cell functions through interaction with ECM.
FIGURE 1.

Functional analysis of WT and integrin αIIb-deficient BMMCs in in vitro suspension culture. A and B, surface expression levels of FcϵRI and c-kit as well as several integrins such as integrin αIIb, αV, αVβ3, α4, α5, β1, and β2 in WT and integrin αIIb-deficient BMMCs. Mean fluorescent intensities of staining were indicated. Data are representative of three independent experiments. C, in vitro growth curves of bone marrow cells derived from WT and integrin αIIb-deficient mice. Numbers of trypan blue-excluding cells in bone marrow cell cultures in IL-3-containing medium were counted weekly. Data are representative of three independent experiments. All data points correspond to the mean ± S.D. D, IL-3 deprivation-induced apoptosis of WT and integrin αIIb-deficient BMMCs. Percentage of annexin V-positive cells after 48 h was measured by flow cytometric analysis. Data represent three independent experiments. All data points correspond to the mean ± S.D. E and F, after IgE-sensitized WT and integrin αIIb-deficient BMMCs were stimulated with the indicated concentrations of antigen for 50 min or 24 h, the amounts of β-hexosaminidase (E) or IL-6 (F), respectively, released into medium were measured. Data represent three independent experiments. All data points correspond to the mean ± S.D.
Surface Expression Levels of Integrin αV Are Inversely Correlated with Those of Integrin αIIb
We next investigated the mechanism by which surface expression levels of integrin αV were elevated in integrin αIIb-deficient BMMCs. As previously reported (18), mRNA levels of integrin αV and integrin β3 were comparable between WT and integrin αIIb-deficient BMMCs (data not shown), suggesting the post-translational regulation of surface expression levels of integrin αV in BMMCs. Because integrin β3 forms a complex with integrin αIIb or integrin αV, we hypothesized that integrin αV competed with integrin αIIb in the association with integrin β3. To test this, integrin αIIb-deficient BMMCs were retrovirally transduced with integrin αIIb WT or mock. Notably, flow cytometric analysis demonstrated that transduction with integrin αIIb(WT) strongly down-regulated surface expression of integrin αV in integrin αIIb-deficient BMMCs (Fig. 2A). In addition, integrin αIIb(D163A) mutant (37), which lost the capacity to bind to FB, was transduced into integrin αIIb-deficient BMMCs. Consistent with a previous report (37), surface expression levels of integrin αIIb(D163A) mutant were weaker than those of integrin αIIb(WT) in the transduced cells. In proportion to less induction of integrin αIIb(D163A), surface expression levels of integrin αV in integrin αIIb(D163A) mutant-transduced cells were less down-regulated as compared with those in integrin αIIb(WT)-transduced BMMCs (Fig. 2A). Furthermore, similar experiments were performed using murine T cell lymphoma cell line BW5147, which originally expressed integrin αVβ3 but not integrin αIIbβ3. As shown in Fig. 2B, transduction with integrin αIIb(WT) into BW5147 cells down-regulated surface expression of integrin αV to a greater degree as compared with transduction with integrin αIIb(D163A) mutant. Collectively, surface expression levels of integrin αV were inversely related to those of integrin αIIb, and even non-functional integrin αIIb competed with integrin αV for integrin β3.
FIGURE 2.

Elevated surface expression levels of integrin αV in integrin αIIb-deficient BMMCs were reduced by transduction with integrin αIIb. A and B, integrin αIIb-deficient BMMCs (A) or BW5147 cells (B) were transduced with integrin αIIb(WT), αIIb(D163A) mutant, or mock. Surface expression levels of integrin αIIb or integrin αV in these transfectants were analyzed by flow cytometry. Mean fluorescent intensities of staining were indicated. Data represent three independent experiments.
Reduced Adhesion to FB and Enhanced Adhesion to VN and vWF in Integrin αIIb-deficient BMMCs
Next, we examined the effects of integrin αIIb deficiency on mast-cell adhesion to ECM proteins such as FN, FB, VN, and vWF. IgE stimulation-dependent adhesion to FB was strongly suppressed in integrin αIIb-deficient BMMCs, whereas adhesion to VN or vWF was drastically enhanced in integrin αIIb-deficient BMMCs, presumably because of increased surface expression of integrin αVβ3 in integrin αIIb-deficient BMMCs when compared with WT BMMCs (Fig. 3A). On the other hand, integrin αIIb deficiency did not significantly affect the adhesion to FN (Fig. 3A). In addition, we examined the inhibitory effect of pretreatment with blocking Abs against integrin αIIbβ3 or integrin αVβ3 on mast-cell adhesion to ECM proteins, confirming that, in WT BMMCs, the binding to FB, VN, or vWF was dependent on integrin αIIbβ3, integrin αVβ3, or both (Fig. 3B). Similar experiments were also performed with regard to integrin αIIb-deficient BMMCs, demonstrating that pretreatment with blocking Abs against integrin αV dampened IgE stimulation-dependent strong adhesion to VN or vWF as well as weak adhesion to FB, whereas it did not affect the adhesion to FN (Fig. 3B). In contrast, pretreatment with blocking Ab for integrin αIIb did not reduce the adhesive property at all (Fig. 3B). Collectively, these results suggested that elevated surface expression levels of integrin αVβ3 in αΙΙb-deficient BMMCs enhanced the adhesion to VN or vWF, but it did not compensate for the defective adhesion to FB owing to integrin αIIbβ3 deficiency.
FIGURE 3.

Enhanced adhesion to VN or vWF and deteriorated adhesion to FB in integrin αIIb-deficient BMMCs. A, WT and integrin αIIb-deficient BMMCs were incubated with or without 5 μg/ml SPE-7 IgE in FN-, FB-, VN-, or vWF-coated plates. Percentage of adherent cells was measured. Data are representative of three independent experiments. All data points correspond to the mean ± S.D. ** (p < 0.01) and * (p < 0.05) indicate statistical differences. B, pretreatment with blocking Ab for integrin αIIb or integrin αV inhibited to various degrees the adhesion of WT- or integrin αIIb-deficient BMMCs stimulated by 5 μg/ml IgE in FN-, FB-, VN-, or vWF-coated plates. Percentage of inhibition was measured. Data are representative of three independent experiments. All data points correspond to the mean ± S.D. C, WT or integrin αIIb-deficient BMMCs in the upper wells were attracted by 100 ng/ml SCF in the lower wells through BSA-, FB-, FN-, or VN-coated Transwells. Migrated cells were counted. Data represent three independent experiments. All data points correspond to the mean ± S.D. ** (p < 0.01) indicates statistical differences.
Enhancement of Migration, Proliferation, Degranulation, and Cytokine Production of BMMCs through Interaction with FB Is Dependent on Integrin αIIb
We next examined the effect of integrin αIIb deficiency on mast-cell functions through interaction with FB. As previously reported, SCF induced migration of WT BMMCs when the lower membranes of the Transwells were pre-coated with FB, FN, or VN. Comparison of the migrating cell numbers between WT and integrin αIIb-deficient BMMCs revealed that integrin αIIb deficiency strongly diminished or enhanced the migration of BMMCs through interaction with FB or VN, respectively, whereas it did not affect mast cell migration through interaction with FN (Fig. 3C). These results also suggested that, in integrin αIIb-deficient BMMCs, both the adhesive and migratory ability were altered toward integrin αVβ3, whereas there was little interaction with FB, a specific ligand for integrin αIIbβ3. Moreover, it was found in WT, but not integrin αIIb-deficient, BMMCs that SCF-stimulated mast-cell proliferation was accelerated in FB-coated plates as compared with BSA-coated plates (Fig. 4A). Similarly, when stimulated by IgE plus antigen, WT, but not integrin αIIb-deficient, BMMCs enhanced β-hexosaminidase release and cytokine (IL-6 and TNF-α) production through interaction with FB (Fig. 4, B–D). Altogether, integrin αIIbβ3 plays crucial roles in enhancing mast-cell functions through interaction with FB.
FIGURE 4.

Enhanced proliferation, degranulation, and cytokine production of WT, but not integrin αIIb-deficient, BMMCs through interaction with FB. A, cell numbers of WT or integrin αIIb-deficient BMMCs stimulated by 10 ng/ml IL-3 plus 100 ng/ml SCF for 5 days in BSA- or FB-coated plates. B, β-hexosaminidase release of IgE-sensitized WT or integrin αIIb-deficient BMMCs stimulated by 30 ng/ml TNP-BSA for 60 min in BSA- or FB-coated plates. C and D, IL-6 (C) and TNF-α (D) production of IgE-sensitized WT or integrin αIIb-deficient BMMCs stimulated by 30 ng/ml TNP-BSA for 16 h in BSA- or FB-coated plates. All data are representative of four independent experiments. All data points correspond to the mean ± S.D. * (p < 0.05) indicates statistical differences.
Transduction with Integrin αIIb(WT), but Not Integrin αIIb(D163A) Mutant, into Integrin αIIb-deficient BMMCs Recovered Mast Cell Functions through Interaction with FB
To further reduce the possibility that enhanced expression levels of integrin αVβ3 modulated mast-cell functions through interaction with FB, we performed similar experiments on adhesion and cytokine production in integrin αIIb-deficient BMMCs transduced with integrin αIIb(WT), integrin αIIb(D163A) mutant, or mock. As depicted in Fig. 5A, transduction with integrin αIIb(WT), but not integrin αIIb(D163A) mutant, augmented the adhesion to FB as compared with transduction with mock. On the other hand, transduction with integrin αIIb(D163A) as well as integrin αIIb(WT) diminished the adhesion to VN, with the degree of the former being a little lower than that of the latter, which was consistent with surface expression levels of integrin αVβ3 in integrin αIIb-deficient BMMCs transduced with integrin αIIb(WT) and (D163A) mutant (Fig. 2A). Moreover, transduction with integrin αIIb(WT), but not integrin αIIb(D163A), induced the enhancement of cytokine production through interaction with FB in integrin αIIb-deficient BMMCs (Fig. 5B). Collectively, these results definitively confirmed that enhanced mast cell functions through interaction with FB were dependent on integrin αIIbβ3 but not integrin αVβ3.
FIGURE 5.
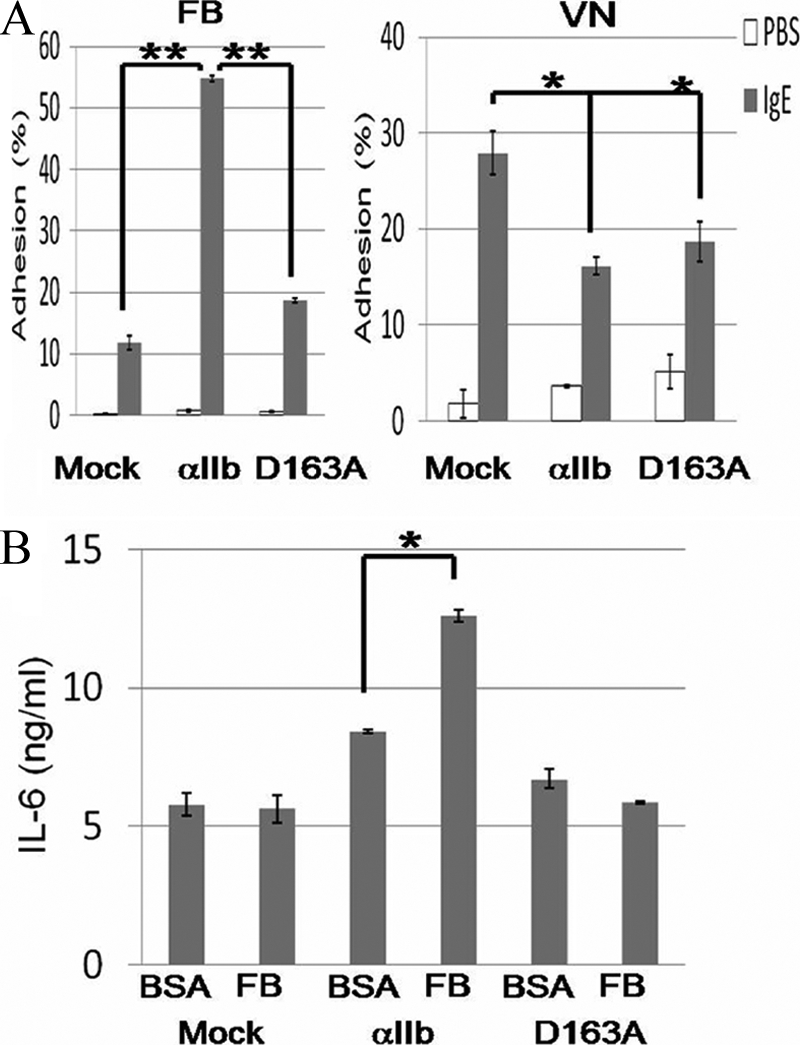
Transduction with integrin αIIb(WT) enhanced or suppressed the adhesion to FB or VN, respectively, in integrin αIIb-deficient BMMCs. A, integrin αIIb-deficient BMMCs transduced with integrin αIIb(WT), αIIb(D163A) mutant, or mock were incubated with 5 μg/ml SPE-7 IgE for 60 min in FB- or VN-coated plates. The percentage of adherent cells was measured. Data are representative of three independent experiments. All data points correspond to the mean ± S.D. ** (p < 0.01) and * (p < 0.05) indicate statistical differences. B, integrin αIIb-deficient BMMCs transduced with integrin αIIb(WT), αIIb(D163A) mutant, or mock were incubated with 5 μg/ml SPE-7 IgE for 16 h in FB-coated plates. The amounts of IL-6 released into medium were measured. Data are representative of three independent experiments. All data points correspond to the mean ± S.D. * (p < 0.05) indicates statistical differences.
Integrin αIIb Deficiency Affected Neither Tissue Mast Cell Numbers in Steady States nor Mast Cell-mediated Acute Allergic Reactions
Because enhanced proliferation and migration of BMMCs through interaction with FB were suppressed by integrin αIIb deficiency, we compared the quantity of tissue mast cells in WT and in integrin αIIb−/− mice. Microscopic analysis demonstrated that mast-cell numbers in the ear skin, back skin, peritoneum wall, and small intestine were not different in these mice (Table 1). Based on this, we addressed the question of whether tissue FB extravasated by acute inflammation modulated mast cell-associated allergic reactions of WT and integrin αIIb−/− mice. However, no significant difference of two types of PCA reaction was observed in these mice (data not shown), despite enhanced in vitro degranulation and cytokine production of mast cells through integrin αIIbβ3-dependent interaction with FB (Fig. 4, B–D). These results indicated that integrin αIIbβ3 was not involved in tissue mast-cell numbers and distributions in steady states or IgE-mediated acute allergic responses.
TABLE 1.
Numbers of mast cells in ear skin, back skin, peritoneum wall, and small intestine
Numbers of mast cells per ten randomly selected high power fields were determined under light microscopy. Results are the mean values ± S.E. for four mice/group. WT, wild type; KO, knockout.
| Tissue | WT | KO |
|---|---|---|
| Ear skin | 112 ± 11.7 | 109 ± 3.2 |
| Back skin | 29.7 ± 23 | 41.3 ± 13 |
| Peritoneum wall | 9.3 ± 5.0 | 10 ± 2 |
| Small intestine | 5.3 ± 3.3 | 6 ± 3.0 |
Integrin αIIb Deficiency Suppressed Peritoneal Chronic Inflammation with a Remarkable Increase of Mast Cells Induced by Repetitive Intraperitoneal FB Administration
We next asked whether integrin αIIb deficiency influenced chronic inflammation with extravascular FB and fibrin deposition. To explore the direct effects of FB, we adopted FB-induced chronic inflammation models where FB was administered into peritoneal cavities every other day. After 1 month, we counted total peritoneal cell numbers and estimated cell populations by flow cytometric analysis. In steady states before the stimulation, we found no significant differences in total peritoneal cell numbers or in mast cell numbers between WT and integrin αIIb−/− mice (Fig. 6A and data not shown). Interestingly, repetitive intraperitoneal injection of FB, but not PBS as a control, induced severe chronic inflammation with a remarkable increase of mast cells as well as total inflammatory cells in the peritoneal cavities of WT mice (Fig. 6, A and B). Thus, an FB-induced chronic inflammation model was established. Intriguingly, integrin αIIb deficiency strongly suppressed the number of mast cells as well as the total number of inflammatory cells in the peritoneal cavities (Fig. 6, A and B), although the percentages of granulocytes and macrophages were not significantly different in the WT and integrin αIIb−/− mice (Fig. 6B, right panel). Considering the in vitro roles of mast cell integrin αIIβ3-FB interaction, these results strongly suggested that in vivo FB-induced chronic inflammation was largely dependent on integrin αIIbβ3 in mast cells, although the effect of few, if any, platelets in the peritoneal cavities on this phenomenon was not completely ruled out. In addition, we found comparable numbers of inflammatory cells in WT and integrin αIIb-deficient mice 24 h after single dose of FB injection (data not shown). Therefore, FB-induced chronic inflammation required continuous administration of FB. Collectively, integrin αIIbβ3 in mast cells played an important role in FB-mediated chronic, but not acute, inflammatory responses.
FIGURE 6.

Repetitive injection of FB into peritoneal cavities induced chronic inflammation more severely in WT mice in comparison to integrin αIIb-deficient mice. A, peritoneal mast cell numbers of WT and integrin αIIb-deficient mice before (left panel) and after (right panel) FB injection. B, total peritoneal cell numbers (left panel) and cell populations (right panel) of WT and integrin αIIb-deficient mice after continuous intraperitoneal inoculation of FB or PBS for 1 month (n = 5/genotype). All data points correspond to the mean ± S.D. ** (p < 0.01) indicates statistical differences.
Soluble FB Enhanced Cytokine Production of WT, but Not Integrin αIIb-deficient BMMCs, in Response to S. aureus Cowan I with FB-binding Capacity
As previously reported, mast cells adhered to soluble FB as well as plate-coated FB via integrin αIIbβ3. Because soluble FB is bound by certain types of bacteria such as S. aureus (Cowan I), the immune cells expressing FB-binding receptors are thought to modulate the immune responses to these pathogens (23–25). We then investigated whether soluble FB influenced the response of mast cells to S. aureus (Cowan I). When WT or integrin αIIb-deficient BMMCs were incubated with S. aureus (Cowan I) for 2 h in the presence of soluble FB, fluorescent microscopic analysis demonstrated that WT, but not integrin αIIb-deficient, BMMCs were completely surrounded by aggregated S. aureus (Cowan I) probably through interaction with soluble FB (Fig. 7A). On the other hand, BMMCs were not apparently covered with S. aureus (Cowan I) in the absence of soluble FB. These results suggested that integrin αIIbβ3-dependent interaction of BMMCs with S. aureus (Cowan I) via soluble FB probably helped mast cells recognize this pathogen. Moreover, IL-6 released into each supernatant was quantified by enzyme-linked immunosorbent assay, demonstrating that soluble FB-induced enhancement of IL-6 production was observed only in WT, but not integrin αIIb-deficient, BMMCs in response to S. aureus (Cowan I) (Fig. 7B). To examine the specificity of this phenomenon, similar experiments were performed using Escherichia coli without FB-binding capacity. As shown in Fig. 7B, soluble FB-dependent enhancement of IL-6 production of BMMCs stimulated by E. coli was not observed irrespective of integrin αIIb expression, suggesting that soluble FB induced the enhancement of cytokine production of BMMCs in response to bacteria with, but not without, FB-binding capacity. Because Toll-like receptors primarily play an important part in the recognition of and response to bacteria (3), we also asked if soluble FB or immobilized FB enhanced cytokine production of both BMMCs stimulated by LPS, a Toll-like receptor 4 agonist. As depicted in Fig. 7 (B and C), soluble FB did not affect IL-6 production of either WT or integrin αIIb-deficient BMMCs stimulated by LPS, whereas immobilized FB enhanced IL-6 production of WT, but not integrin αIIb-deficient, BMMCs stimulated by LPS. These results suggested the synergism of Toll-like receptor 4 signaling and integrin αIIbβ3 signaling through interaction with immobilized FB, but not soluble FB. Altogether, soluble FB enhances the cytokine production of BMMCs in responses to S. aureus (Cowan I), probably because mast cell-soluble FB-S. aureus (Cowan I) complex formation promoted the quick and tight recognition of this pathogen by mast cells.
FIGURE 7.

Soluble FB enhanced cytokine production of WT, but not integrin αIIb-deficient, BMMCs in response to S. aureus with FB-binding capacity. A, WT or integrin αIIb-deficient BMMCs were incubated with heat-killed S. aureus labeled by Cell Tracker Orange in the presence or absence of 500 μg/ml soluble FB for 2 h. WT, but not integrin αIIb-deficient, BMMCs were covered with SA aggregates in the presence of soluble FB (arrowhead). B, WT or integrin αIIb-deficient BMMCs were incubated with 100 ng/ml LPS, 100 μg/ml heat-killed E. coli, 100 μg/ml S. aureus, or PBS as control in the presence or absence of 500 μg/ml soluble FB for 8 h. The ratio of the amounts of IL-6 released in the presence of soluble FB to those of IL-6 in the absence of soluble FB was measured. Data are representative of three independent experiments. All data points correspond to the mean ± S.D. ** (p < 0.01) indicates statistical differences. C, WT or integrin αIIb-deficient BMMCs were incubated with the indicated concentrations of LPS in FB- or BSA-coated plates. Data represent three independent experiments. All data points correspond to the mean ± S.D. * (p < 0.05) indicate statistical differences.
DISCUSSION
In a previous study, we found that integrin αIIbβ3 is highly expressed in mast cells, in addition to the megakaryocyte/platelet lineage and a subset of hematopoietic progenitors (9, 14, 18). Experiments using blocking Abs specific for integrins demonstrated that adhesion to FB, VN, or vWF was mediated through integrin αIIbβ3, integrin αVβ3, or both, respectively (9). In the follow-up study, we first paid attention to the interesting results shown by Berlanga O et al. that integrin αIIb-deficient BMMCs displayed extremely higher surface expression levels of integrin αVβ3 as compared with WT counterparts (18). Because counter-regulation of integrin αIIbβ3 and integrin αVβ3 on their surface expression levels might affect in vivo functions of integrin αIIbβ3 in mast cells, we attempted to delineate the underlying mechanism. Our hypothesis that integrin αIIb competed with integrin αV in heterodimerization with integrin β3 in mast cells was illustrated by experimental results as follows: retroviral transduction with integrin αIIb(WT) into integrin αIIb-deficient BMMCs reduced surface expression of integrin αVβ3 at levels comparable to those in WT BMMCs (Fig. 2A). In addition, transduction with integrin αIIb(D163A) mutant led to less reduction in surface expression levels of integrin αVβ3 together with less induction in those of integrin αIIbβ3 in integrin αIIb-deficient BMMCs. Thus, surface expression levels of integrin αVβ3 were conversely related to those of integrin αIIbβ3 in mast cells (Fig. 2A). Notably, this phenomena was true for BW5147 cells transduced with integrin αIIb(WT) or αIIb(D163A) mutant (Fig. 2B). However, integrin αIIb deficiency did not affect surface expression levels of integrin αVβ3 in platelets (data not shown). These results suggested that regulatory mechanisms on surface expression levels of integrin αVβ3 differed between mast cells and platelets. One possible explanation is as follows: integrin αIIb deficiency might fail to influence surface expression levels of integrin αVβ3 if integrin β3 expression were sufficient in platelets, whereas it might promote the association of integrin αV with integrin β3 if integrin β3 expression were insufficient in mast cells. Further examination is necessary to fully understand the mechanism. Importantly, all the functional analyses (Figs. 2–4) showed that higher surface expression levels of integrin αVβ3 in integrin αIIb-deficient BMMCs enhanced adhesion to VN or vWF but did not compensate for the loss of mast-cell functions through interaction with FB. Based on this, we compared in vivo mast cell functions between WT and integrin αIIb-deficient mice.
First, integrin αIIb deficiency did not affect mast-cell numbers in tissues under normal conditions (Table 1). This seems reasonable, given that FB is not abundant outside blood vessels under normal conditions. Second, integrin αIIb deficiency did not affect two types of PCA estimated by ear dye extravasation or swelling (data not shown), which was reported to be mast cell-dependent (32–34). These results indicate that extravascular FB and fibrin accompanied by acute inflammation are unable to enhance mast-cell functions. On the other hand, recent advances demonstrate that FB is a central regulator of the inflammatory response as well as of hemostasis. Analysis of gene-targeted mice expressing a mutant form of FB, lacking the integrin αMβ2-binding motif, demonstrated that the high affinity engagement of FB by integrin αMβ2 in neutrophils and macrophages was critical for inflammatory responses (38, 39). Therefore, we speculated that FB extravasated at acute inflammatory sites activated neutrophils and macrophages via integrin αMβ2 but failed to enhance mast-cell functions via integrin αIIbβ3. In contrast, we found striking differences between WT and integrin αIIb−/− mice in FB-induced chronic inflammation: integrin αIIb deficiency strongly suppressed the increase of total inflammatory cells with mastocytosis in the peritoneal cavities. However, administration of single dose FB did not lead to any significant difference of initial inflammatory responses in these mice 24 h after inoculation (data not shown), confirming the negligible role of integrin αIIb in acute inflammation. Taking into consideration that platelets are absent in the peritoneal cavities and that integrin αVβ3 did not significantly affect in vitro mast-cell functions through interaction with FB, we concluded that FB-induced chronic inflammation depended on integrin αIIbβ3 in mast cells. The relevant mechanism might be as follows: FB activates macrophages and granulocytes via integrin αMβ2 to produce inflammatory cytokines and chemokines in the initial phase, leading to the gradual recruitment, proliferation, and activation of mast cells in the presence of FB. Alternatively, activated mast cells also produce a diverse array of chemical mediators, accelerating chronic inflammation. Thus, FB-mediated inflammation appears to be augmented with the increase of mast cells in tissues. This scenario may explain in part why mast cell numbers increase in a variety of chronic inflammatory diseases such as atopic dermatitis and asthma that are thought to cause continuous extravasation of FB in tissues. Further analysis of WT and integrin αIIb-deficient mice using different types of chronic inflammation models will be required to delineate the role of mast cell integrin αIIbβ3 in chronic inflammatory diseases.
Another important finding in this study was that soluble FB enhanced IL-6 production of WT, but not integrin αIIb-deficient, BMMCs in response to S. aureus (Cowan I) with FB-binding capacity. On the other hand, soluble FB failed to enhance IL-6 production of WT BMMCs stimulated by E. coli harboring no FB-binding capacity, LPS, or bacterial lipopeptide (Fig. 7B and data not shown). Because WT, but not integrin αIIb-deficient, BMMCs apparently kept a strong contact with aggregated S. aureus (Cowan I) in the presence of soluble FB, integrin αIIbβ3-dependent recognition of S. aureus (Cowan I) in mast cells may augment the innate response to this pathogen. Considering that soluble FB facilitates the interaction of platelets with S. aureus by bridging clumping factor A in S. aureus and integrin αIIbβ3 in platelets (23–25), a similar mechanism probably occurs in mast cells: the complex formation of mast cell integrin αIIbβ3-FB-S. aureus (Cowan I) promotes quick and tight recognition of this pathogen by mast cells, thereby enhancing innate immune responses. Collectively, these results suggested that integrin αIIbβ3 plays an important part in the innate responses of mast cells to certain types of bacteria with FB-binding capacity.
In conclusion, the integrin αIIbβ3-dependent interaction of mast cells with FB augments FB-associated chronic inflammation or innate responses to FB-binding bacteria. Elucidation of the in vivo function of integrin αIIbβ3 in mast cells will lead to new approaches in the prevention of and therapy for the relevant inflammatory and infectious diseases.
Acknowledgments
We thank Drs. B. S. Coller, V. L. Woods, D. J. Gerber, and S. Tonegawa for providing Abs. We thank Dr. R. Basani for providing plasmid. We are grateful to Dr. Dovie Wylie for her excellent language assistance.
This work was supported by grants from the Ministry of Education, Science, Technology, Sports and Culture and the Ministry of Health and Welfare, Japan.
- ECM
- extracellular matrix
- BMMCs
- bone marrow-derived mast cells
- FB
- fibrinogen
- FN
- fibronectin
- SA
- fixed S. aureus Cowan I
- PCA
- passive cutaneous allergic reaction
- SCF
- stem cell factor
- VN
- vitronectin
- vWF
- von Willebrand factor
- WT
- wild type
- Ab
- antibody
- mAb
- monoclonal antibody
- IL-3
- interleukin-3
- BSA
- bovine serum albumin
- PBS
- phosphate-buffered saline
- LPS
- lipopolysaccharide
- TNP
- trinitrophenol
- DNP
- dinitrophenol
- WT
- wild type
- KO
- knockout.
REFERENCES
- 1.Kawakami T., Galli S. J. (2002) Nat. Rev. Immunol. 2, 773–786 [DOI] [PubMed] [Google Scholar]
- 2.Kalesnikoff J., Galli S. J. (2008) Nat. Immunol. 9, 1215–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marshall J. S. (2004) Nat. Rev. Immunol. 4, 787–799 [DOI] [PubMed] [Google Scholar]
- 4.Kawakami T., Kitaura J. (2005) J. Immunol. 175, 4167–4173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinashi T., Springer T. A. (1994) Blood 83, 1033–1038 [PubMed] [Google Scholar]
- 6.Kitaura J., Eto K., Kinoshita T., Kawakami Y., Leitges M., Lowell C. A., Kawakami T. (2005) J. Immunol. 174, 4495–4504 [DOI] [PubMed] [Google Scholar]
- 7.Kitaura J., Kinoshita T., Matsumoto M., Chung S., Kawakami Y., Leitges M., Wu D., Lowell C. A., Kawakami T. (2005) Blood 105, 3222–3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bianchine P. J., Burd P. R., Metcalfe D. D. (1992) J. Immunol. 149, 3665–3671 [PubMed] [Google Scholar]
- 9.Oki T., Kitaura J., Eto K., Lu Y., Maeda-Yamamoto M., Inagaki N., Nagai H., Yamanishi Y., Nakajima H., Nakajina H., Kumagai H., Kitamura T. (2006) J. Immunol. 176, 52–60 [DOI] [PubMed] [Google Scholar]
- 10.Gurish M. F., Tao H., Abonia J. P., Arya A., Friend D. S., Parker C. M., Austen K. F. (2001) J. Exp. Med. 194, 1243–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edelson B. T., Li Z., Pappan L. K., Zutter M. M. (2004) Blood. 103, 2214–2220 [DOI] [PubMed] [Google Scholar]
- 12.Knight P. A., Wright S. H., Brown J. K., Huang X., Sheppard D., Miller H. R. (2002) Am. J. Pathol. 161, 771–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shattil S. J., Newman P. J. (2004) Blood 104, 1606–1615 [DOI] [PubMed] [Google Scholar]
- 14.Emambokus N. R., Frampton J. (2003) Immunity 19, 33–45 [DOI] [PubMed] [Google Scholar]
- 15.Eto K., Murphy R., Kerrigan S. W., Bertoni A., Stuhlmann H., Nakano T., Leavitt A. D., Shattil S. J. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 12819–12824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kieffer N., Fitzgerald L. A., Wolf D., Cheresh D. A., Phillips D. R. (1991) J. Cell Biol. 113, 451–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suehiro K., Smith J. W., Plow E. F. (1996) J. Biol. Chem. 271, 10365–10371 [DOI] [PubMed] [Google Scholar]
- 18.Berlanga O., Emambokus N., Frampton J. (2005) Exp. Hematol. 33, 403–412 [DOI] [PubMed] [Google Scholar]
- 19.Mosesson M. W. (2005) J. Thromb. Haemost. 3, 1894–1904 [DOI] [PubMed] [Google Scholar]
- 20.Tang L., Jennings T. A., Eaton J. W. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 8841–8846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drew A. F., Liu H., Davidson J. M., Daugherty C. C., Degen J. L. (2001) Blood 97, 3691–3698 [DOI] [PubMed] [Google Scholar]
- 22.Szaba F. M., Smiley S. T. (2002) Blood 99, 1053–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fitzgerald J. R., Foster T. J., Cox D. (2006) Nat. Rev. Microbiol. 4, 445–457 [DOI] [PubMed] [Google Scholar]
- 24.Loughman A., Fitzgerald J. R., Brennan M. P., Higgins J., Downer R., Cox D., Foster T. J. (2005) Mol. Microbiol. 57, 804–818 [DOI] [PubMed] [Google Scholar]
- 25.Fitzgerald J. R., Loughman A., Keane F., Brennan M., Knobel M., Higgins J., Visai L., Speziale P., Cox D., Foster T. J. (2006) Mol. Microbiol. 59, 212–230 [DOI] [PubMed] [Google Scholar]
- 26.Lengweiler S., Smyth S. S., Jirouskova M., Scudder L. E., Park H., Moran T., Coller B. S. (1999) Biochem. Biophys. Res. Commun. 262, 167–173 [DOI] [PubMed] [Google Scholar]
- 27.Gerber D. J., Pereira P., Huang S. Y., Pelletier C., Tonegawa S. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 14698–14703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kitaura J., Song J., Tsai M., Asai K., Maeda-Yamamoto M., Mocsai A., Kawakami Y., Liu F. T., Lowell C. A., Barisas B. G., Galli S. J., Kawakami T. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 12911–12916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morita S., Kojima T., Kitamura T. (2000) Gene Ther. 7, 1063–1066 [DOI] [PubMed] [Google Scholar]
- 30.Kitamura T., Koshino Y., Shibata F., Oki T., Nakajima H., Nosaka T., Kumagai H. (2003) Exp. Hematol. 31, 1007–1014 [PubMed] [Google Scholar]
- 31.Furumoto Y., Nunomura S., Terada T., Rivera J., Ra C. (2004) J. Biol. Chem. 279, 49177–49187 [DOI] [PubMed] [Google Scholar]
- 32.Hata D., Kawakami Y., Inagaki N., Lantz C. S., Kitamura T., Khan W. N., Maeda-Yamamoto M., Miura T., Han W., Hartman S. E., Yao L., Nagai H., Goldfeld A. E., Alt F. W., Galli S. J., Witte O. N., Kawakami T. (1998) J. Exp. Med. 187, 1235–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inagaki N., Goto S., Nagai H., Koda A. (1986) Int. Arch. Allergy Appl. Immunol. 81, 58–62 [DOI] [PubMed] [Google Scholar]
- 34.Nagai H., Sakurai T., Inagaki N., Mori H. (1995) Biol. Pharm. Bull. 18, 239–245 [DOI] [PubMed] [Google Scholar]
- 35.Wong M. X., Roberts D., Bartley P. A., Jackson D. E. (2002) J. Immunol. 168, 6455–6462 [DOI] [PubMed] [Google Scholar]
- 36.Artis D., Humphreys N. E., Potten C. S., Wagner N., Müller W., McDermott J. R., Grencis R. K., Else K. J. (2000) Eur. J. Immunol. 30, 1656–1664 [DOI] [PubMed] [Google Scholar]
- 37.Honda S., Tomiyama Y., Shiraga M., Tadokoro S., Takamatsu J., Saito H., Kurata Y., Matsuzawa Y. (1998) J. Clin. Invest. 102, 1183–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flick M. J., LaJeunesse C. M., Talmage K. E., Witte D. P., Palumbo J. S., Pinkerton M. D., Thornton S., Degen J. L. (2007) J. Clin. Invest. 117, 3224–3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flick M. J., Du X., Witte D. P., Jirousková M., Soloviev D. A., Busuttil S. J., Plow E. F., Degen J. L. (2004) J. Clin. Invest. 113, 1596–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]