

Abstract
Glucose-dependent insulinotropic polypeptide (GIP) potentiates glucose-stimulated insulin secretion, insulin biosynthesis, and β-cell proliferation and survival. In previous studies GIP was shown to promote β-cell survival by modulating the activity of multiple signaling modules and regulating gene transcription of pro- and anti-apoptotic bcl-2 family proteins. We have now evaluated the mechanisms by which GIP regulates the dynamic interactions between cytoplasmic bcl-2 family members and the mitochondria in INS-1 cells during apoptosis induced by treatment with staurosporine (STS), an activator of the mitochondria-mediated apoptotic pathway. STS induced translocation of bad and bimEL, activation of mitochondrial bax, release of mitochondrial cytochrome c, cleavage of caspase-3, and apoptosis. Each response was significantly diminished by GIP. Using selective enzyme inhibitors, overexpression of dominant-negative Akt, and Akt siRNA, it was demonstrated that GIP promoted β-cell survival via Akt-dependent suppression of p38 MAPK and JNK and that combined inhibition was sufficient to explain the entire pro-survival responses to GIP during STS treatment. This signaling pathway also explained the pro-survival effects of GIP on INS-1 cells exposed to two other promoters of stress: thapsigargin (endoplasmic reticulum stress) and etoposide (genotoxic stress). Importantly, we discovered that GIP suppressed p38 MAPK and JNK via Akt-mediated changes in the phosphorylation state of the apoptosis signal-regulating kinase 1 in INS-1 cells and human islets, resulting in inhibition of its activity. Inhibition of apoptosis by GIP is therefore mediated via a key pathway involving Akt-dependent inhibition of apoptosis signal-regulating kinase 1, which subsequently prevents the pro-apoptotic actions of p38 MAPK and JNK.
Type 2 diabetes mellitus results from a combination of insulin resistance and β-cell dysfunction (1–4), and it is now clear that increased apoptosis plays a major role in the β-cell defect (5). The initiation of β-cell apoptosis has been attributed to a number of contributing factors, including glucolipotoxicity (6, 7), ER2 and oxidative stress (8, 9), cytokine actions (10), and fibril formation of human islet amyloid polypeptide (11).
Several therapeutic strategies targeted at preserving β-cell mass are currently under examination (12). As a result of their insulinotropic activities (13–16), the incretin hormones glucagon-like peptide 1 and glucose-dependent insulinotropic polypeptide (GIP) have been targeted as therapeutics for type 2 diabetes mellitus, and two approaches have been taken, extending the half-lives of the peptides by inhibiting the enzyme responsible for their degradation, dipeptidyl peptidase IV (17), and the production of long acting incretin analogs (17, 18). Both hormones also exert powerful pro-survival effects on pancreatic β-cells (12, 19), and there is therefore considerable interest in establishing whether, and how, they might preserve β-cell mass.
Stimulation of β-cells with GIP results in the activation of protein kinase A (PKA) (20–22), exchange protein activated by cAMP 2 (EPAC2) (23), Erk1/2 (20, 21, 24), Ca2+-independent phospholipase A2 (25), phosphoinositide 3-kinase (20, 21, 26), and protein kinase B (Akt) (15, 20, 21). The roles of these signaling modules in GIP-mediated cell survival are unclear. GIP was shown to promote cell survival and inhibit the activation of caspase-3 and caspase-8 in streptozotocin-treated INS-1 cells (27), as well as to reduce the ER stress response in β-cells in a similar manner to glucagon-like peptide 1 (28). Similarly, GIP prevented the activation of caspase-9, poly(ADP-ribose) polymerase (21), and caspase-3 (29) and promoted the survival of glucose- and serum-starved INS-1 cells (21, 29) by suppressing the pro-apoptotic actions of p38 mitogen-activated protein kinase (MAPK) (29). The signaling pathway involved the production of cAMP but not activation of PKA, Erk1/2, or phosphoinositide 3-kinase. Subsequently it was shown that GIP promoted the survival of β-cells exposed to glucolipotoxic stress, and this was associated with reduced transcription of the key pro-apoptotic protein bax and increased transcription of the anti-apoptotic protein, bcl-2 (26). The mechanism underlying bax regulation was, at least in part, due to Akt phosphorylation of the transcription factor foxo-1 (26), thus inhibiting its nuclear transport. The regulation of bcl-2 expression was later shown to involve a complex interplay between PKA/cAMP-response element-binding protein (CREB), the CREB co-activator TORC2, and diminished AMP kinase activity (30).
In response to various stresses, there are multiple changes in the cellular distribution of pro- and anti-apoptotic bcl-2 family members that can result in mitochondrial release of cytochrome c, activation of caspase-3 and caspase-9, and induction of apoptosis (31–33). We postulated that GIP modulates the dynamic interactions between cytoplasmic bcl-2 family members and the mitochondria. In the current study, we examined the effects of GIP on such interactions in INS-1 cells exposed to staurosporine (STS), a rapid activator of the mitochondria-mediated apoptotic pathway. Additionally, we established that a major pathway by which GIP exerts its effects on β-cell survival is via an Akt-dependent (34) inhibition of the apoptosis signal-regulating kinase 1 (ASK1), resulting in suppression of p38 MAPK and Jun N-terminal kinase (JNK).
EXPERIMENTAL PROCEDURES
Cell Culture for INS-1 and MIN6 Cells and Human Islets
The INS-1 β-cell line (clone 832/13) was kindly provided by Dr. C. B. Newgard (Duke University Medical Center, Durham, NC). Cells were maintained in 11 mm glucose RPMI 1640 (Sigma) supplemented with 2 mm glutamine, 50 μm β-mercaptoethanol, 10 mm HEPES, 1 mm sodium pyruvate, 10% fetal bovine serum, 100 units/ml penicillin G-sodium, and 100 μg/ml streptomycin sulfate. Mouse insulinoma β-cells (MIN6) were cultured in 25 mm glucose Dulbecco's modified Eagle's medium (Sigma) supplemented with 10% fetal bovine serum, 100 units/ml penicillin G-sodium, and 100 μg/ml streptomycin sulfate. Human islets were provided by the Centre for Human Islet Transplant and β-cell Regeneration at the University of British Columbia and maintained in RPMI 1640 supplemented with 5 mm glucose, 0.25% HEPES (pH 7.4), 10% fetal bovine serum, 100 units/ml penicillin G-sodium, and 100 μg/ml streptomycin sulfate. Approximately 12 h prior to experiments, cells received fresh serum-starved medium (replaced with 0.1% bovine serum albumin) that contained low (3 mm) glucose. Reagents used in experiments were all from Calbiochem, except staurosporine and thapsigargin (Sigma).
Cell Lysis and ASK1 in Vitro Kinase Assay
Cells were lysed in 20 mm Tris (pH 7.5), 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1 μg/ml leupeptin supplemented with 1 mm phenylmethylsulfonyl fluoride and 1× Proteinase inhibitor mixture set III (Calbiochem #539134). For ASK1 kinase assays, endogenous ASK1 protein was immunoprecipitated from untransfected INS-1 cells with an anti-ASK1 antibody (Santa Cruz Biotechnology) conjugated to Protein A-Sepharose beads (Invitrogen). Beads from immunoprecipitates were washed twice in 500 μl of lysis buffer and twice in 500 μl of kinase buffer and subsequently resuspended in 60 μl of kinase buffer containing 200 μm ATP and 0.75 μg of a GST-tagged Mek6 fusion peptide (Cell Signaling Technology) that was used as a substrate for ASK1. Kinase reactions were performed at 30 °C for 30 min. Phosphorylation of GST-Mek6 was determined by Western analysis with anti-phospho-Mek 3/6 antibody (Cell Signaling Technology).
Western Blot
Cell lysates were subjected to 10% (for ASK1) or 15% SDS-PAGE and electroblotted onto nitrocellulose membrane (Bio-Rad). Antibodies used to probe membranes were as follows. Santa Cruz Biotechnology was the supplier for anti-ASK1 (H-300, recognizes total human and rat protein). Cell Signaling Technology (Beverly, MA) was the supplier for anti-Akt (antibody 9272), anti-Akt1 (mouse mAb, 2H10, antibody 2967), anti-Akt2 (rabbit mAb, 5B5, antibody 2964), anti-Akt3 (antibody 4059), anti-β-actin (antibody 4967), anti-bad (antibody 9292), anti-bax (antibody 2772), anti-bcl-2 (antibody 2876), anti-bcl-XL (antibody 2762), anti-bim (antibody 2819), anti-β-tubulin (antibody 2146), anti-caspase-3 (rabbit mAb, 8G10, recognizes full-length and cleaved caspase-3), anti-COX IV (antibody 4844), anti-cytochrome c (rabbit mAb, 136F3), anti-GST (antibody 2622), anti-JNK (antibody 9252), anti-p38 MAPK (antibody 9212), anti-phospho-ASK1 (Ser83, antibody 3761, detects human isoform but not rat), anti-phospho-ASK1 (Ser845, antibody 3765, detects human isoform but not rat), anti-phospho-bad (Ser112, antibody 9291), anti-phospho-JNK (Thr183/Tyr185, mouse mAb, G9), anti-phospho-Mek 3/6 (Ser189/207, antibody 9231), and anti-phospho-p38 MAPK (Thr180/Tyr182, antibody 9211). Immunoreactive bands were visualized by enhanced chemiluminescence (Amersham Biosciences) using horseradish peroxidase-conjugated IgG secondary antibodies. For quantification of band density, films were analyzed using densitometric software (Eagle Eye, Stratagene).
Transfection of Plasmid Constructs and siRNA
The pcDNA3 construct encoding human ASK1 and a kinase-dead ASK1 containing a methionine mutation at lysine 709, which was used for the purpose of producing a dominant negative effect (Fig. 6D), were gifts from Dr. Hidenori Ichijo (Laboratory of Cell Signaling, The University of Tokyo (35)). The same ASK1 construct but containing an alanine mutation at serine 83, which prevented Akt-mediated phosphorylation of ASK1 (Fig. 6E), was a gift from Dr. Moses Chao (Skirball Institute for Biomolecular Medicine, New York University Medical Center (36)). The construct encoding kinase-dead Akt, used for the purpose of producing a dominant negative effect (Fig. 5C), was described previously (34). To transfect cells, DNA was incubated with LipofectamineTM LTX (Invitrogen) and PLUSTM reagent (Invitrogen) at a ratio of 2 μg of DNA/6.0 μl of Lipofectamine LTXTM/2.0 μl of PLUSTM in 200 μl of Opti-MEM I (Amersham Biosciences) for 30 min at room temperature and added to 2 × 106 cells in 1 ml of maintenance media without antibiotics. Media were replaced with maintenance media containing antibiotics after 6–16 h, and experiments were performed 36 h following transfection. The protocol for transfection of siRNA was adapted from Kibbey et al. (37). Briefly, Akt 1 siRNA (siRNA ID: SASI_Rn01_00063656, Sigma) and Akt 2 siRNA (siRNA ID: SASI_Rn01_00047688, Sigma) or scramble (control) siRNA (Cell Signaling Technology; catalogue no. 6568) were incubated with 10 μl of RNAifect Transfection reagent (Qiagen, catalogue no. 301605) in 200 μl of Opti-MEM I for 30 min at room temperature and then added to 1.5 × 106 cells in 1 ml of maintenance media without antibiotics (final concentrations of Akt 1/2 siRNA were 100 nm each, and scramble siRNA was 200 nm). Media were replaced with maintenance media with antibiotics after 6–16 h, and experiments were performed ∼72 h following transfection.
FIGURE 6.

GIP suppresses p38 MAPK and JNK activation via Akt-mediated inhibition of ASK1 in INS-1 cells and human islets. A, INS-1 cells transfected without or with GFP or human ASK1 were treated without or with STS (100 nm) ± 10 nm GIP for 4 h, and Western analysis was performed on total cell lysates with the indicated antibodies. B, untransfected INS-1 cells were treated without or with STS ± GIP for 4 h, and ASK1 in vitro kinase assays were performed on ASK1 protein that was immunoprecipitated with anti-ASK1 antibody. C, the mean ± S.E. change in ASK1 in vitro kinase activity relative to control is shown (n = 5). D, INS-1 cells transfected with GFP or dominant negative ASK1 (ASK1kinase-dead) were treated without or with STS (100 nm) for 4 h, and Western analysis was performed on total cell lysates with the indicated antibodies. E, INS-1 cells transfected with wild-type ASK1 (ASK1WT) or ASK1 containing a S83A mutation (ASK1S83A) were treated without or with STS (100 nm) ± 10 nm GIP for 4 h, and Western analysis was performed on total cell lysates with the indicated antibodies. F, human islets were treated without or with STS (100 nm) ± 10 nm GIP for 4 h, and Western analysis was performed on total cell lysates with the indicated antibodies. Shown in D and E are representative blots of three independent experiments. Anti-β-actin and anti-GST (GST-Mek 3/6) blots were internal controls. Note: anti-ASK1 antibody detects both human and rat ASK1 protein.
FIGURE 5.

GIP mediates anti-apoptotic signaling via dual suppression of p38 MAPK and JNK. A, INS-1 cells were treated without or with STS (100 nm) ± 10 nm GIP for 4 h in the absence or presence of Akt inhibitor IV (Akt IV, 400 nm), and Western analysis was performed on total cell lysates with the indicated antibodies. B, INS-1 cells transfected with GFP or dominant negative Akt (Akt-DN) were treated without or with STS (100 nm) ± 10 nm GIP for 4 h, and Western analysis was performed on total cell lysates with the indicated antibodies. C, INS-1 cells transfected with scramble or Akt 1 and 2 siRNA were treated without or with STS (100 nm) ± 10 nm GIP for 4 h, and Western analysis was performed on total cell lysates with the indicated antibodies. Of note, total protein levels of Akt were reduced 60–70% in cells transfected with Akt 1 and 2 siRNA. D, INS-1 cells were treated without or with STS ± p38 MAPK inhibitor (p38i, SB 203580, 5 μm) or JNK inhibitor (JNKi, SP 600125, 5 μm) for 4 h, and Western analysis was performed on total cell lysates with the indicated antibodies. E, INS-1 cells were treated without or with STS ± GIP, p38i, JNKi, or p38i and JNKi for 6 h, and cell death was determined (mean ± S.E. of cell death (n = 6); #, p < 0.05 versus STS alone). F, INS-1 cells were treated without or with STS ± p38i and JNKi for 4 h, and Western analysis was performed on mitochondrial and cytoplasmic fractions with the indicated antibodies. G, INS-1 cells were treated without or with STS ± p38i and JNKi for 4 h, and Western analysis was performed on mitochondrial fractions that were incubated with 10 mm bismaleimidohexane (BMH) for 30 min at room temperature. Shown in A–D, F, and G are representative blots of at least three independent experiments. Anti-β-actin and anti-COX IV blots were internal controls.
Mitochondrial/Cytosolic Fractionation and Bax Cross-linking
The cytosol and mitochondrial fractions of cells were obtained using the BioVision fractionation kit (Mountain View, CA) according to the manufacturer's protocol. Cross-linking of bax was performed using the method described by Kim et al. (38). Briefly, 10 mm bismaleimidohexane (Pierce) was incubated with isolated mitochondria for 30 min at room temperature. The reaction was terminated with SDS loading buffer, and levels of cross-linked bax were determined via Western analysis with anti-bax antibody.
Cell Death Assays
Apoptosis was determined using the APOPercentageTM apoptosis assay kit according to the manufacturer's protocol (Biocolor, Northern Ireland). To determine total cell death, INS-1 cells were treated in media containing 500 ng/ml propidium iodide (Invitrogen) and 250 ng/ml Hoechst (Sigma), and cell death was measured by counting propidium iodide-positive nuclei, and total cell number was measured by counting Hoechst-positive nuclei. Propidium iodide and Hoechst were imaged with a Cellomics Arrayscan VTI (Thermo Fisher Scientific Inc.), and % cell death was calculated as the number of propidium iodide-positive cells/Hoechst-positive cells multiplied by 100.
Statistical Analysis
Data, expressed as mean ± S.E., were analyzed using the non-linear regression analysis program PRISM (GraphPad, San Diego, CA). Values of n in all cases represent individual experiments. Statistical significance of differences in mean value was tested using analysis of variance with the Newman-Keuls post hoc test. A p value of <0.05 was considered significant.
RESULTS
GIP Inhibits the Mitochondria-mediated Apoptotic Pathway in STS-treated INS-1 Cells
It was previously shown that GIP signaling protects STS-treated INS-1 cells from cell death in an Akt-dependent manner (34). To determine if this was due to GIP-mediated inhibition of the mitochondria-mediated apoptotic pathway, apoptosis assays and cytochrome c release studies were performed on INS-1 cells treated with 100 nm STS ± 10 nm GIP (STS ± GIP). Stimulation with GIP resulted in a significantly delayed onset and reduced levels of apoptosis (Fig. 1A), and this occurred in a concentration-dependent manner (EC50 = 515 ± 32 pm; Fig. 1B). In parallel experiments, the effects of 0–6 h STS treatment on release of mitochondrial cytochrome c into the cytoplasm and the cleavage of caspase-3 were studied. After 4 h, STS treatment caused a significant increase in both released mitochondrial cytochrome c (Fig. 1C) and cleaved caspase-3 levels (Fig. 1D). These responses were potently suppressed by the presence of GIP, indicating that its promotion of cell survival was mediated through suppression of mitochondrial-mediated apoptosis.
FIGURE 1.

GIP inhibits the mitochondria-mediated apoptotic pathway in STS-treated INS-1 cells. A, INS-1 cells were treated without or with STS (100 nm) ± 10 nm GIP for 0–8 h, and onset of apoptosis was determined (mean ± S.E. (n = 4); #, p < 0.05 versus DMSO control; $, p < 0.05 versus STS without GIP). B, INS-1 cells were treated without or with STS plus increasing concentrations of GIP (0–100 nm) for 6 h, and onset of apoptosis was determined (mean ± S.E. (n = 7); $, p < 0.05 versus DMSO control; #, p < 0.05 versus STS without GIP). In the upper right is the concentration-survival response with the calculated EC50 value. C, INS-1 cells were treated without or with STS ± 10 nm GIP for 0–4 h, and Western analysis was performed on mitochondrial or cytoplasmic protein fractions with the indicated antibodies. D, INS-1 cells were treated without or with STS ± 10 nm GIP for 0–6 h, and Western analysis was performed on whole cell lysates with indicated antibodies. Shown in C and D are representative blots of at least three independent experiments. Anti-β-actin and anti-COX IV were internal controls.
GIP-mediated Anti-apoptotic Signaling Requires the Production of cAMP but Does Not Involve Insulin Autocrine Signaling
GIP activates adenylate cyclase, with subsequent production of cAMP in β-cells (14). Promotion of STS-treated INS-1 cell survival by GIP was previously shown to involve EPAC2 but not PKA (34). To determine whether cAMP was required for GIP-mediated survival signaling, INS-1 cells were treated without or with STS ± GIP for 6 h in the absence or presence of the adenylate cyclase inhibitor, MDL-12,300A. The inhibitor had no additive effect on STS-induced cell death yet completely ablated the pro-survival effects of GIP (Fig. 2A), thus definitively showing that GIP-mediated survival requires the production of cAMP. GIP-mediated cAMP production promotes insulin release via activation of PKA (22) and EPAC2 (23). This raised the possibility that at least a component of the pro-survival effects of GIP could be mediated via an autocrine action of insulin. Although all experiments were performed with low glucose conditions, under which GIP does not stimulate insulin release (39), reports that low levels of insulin (≈200 pm) exert anti-apoptotic effects on β-cells (40, 41) made it necessary to exclude autocrine insulin signaling as a pathway by which GIP acted. This possibility was examined by treating INS-1 cells with or without STS with increasing concentrations (0–100 nm) of insulin for 6 h and comparing responses to those with GIP, forskolin, and IGF-I. Although significant protection of INS-1 cells was observed with GIP, forskolin, and IGF-I, insulin had no effect except with an extremely high level, which is capable of cross-reacting with the IGF-I receptor (Fig. 2B). Based on this finding, we considered it unlikely that insulin played a significant role in promoting INS-1 cell survival under GIP-treated conditions.
FIGURE 2.

GIP-mediated anti-apoptotic signaling requires the production of cAMP but does not involve insulin autocrine signaling. A, INS-1 cells were treated without or with STS (100 nm) ± 10 nm GIP for 6 h in the presence or absence of the adenylate cyclase inhibitor, MDL-12,300A (200 μm), and cell death was determined (mean ± S.E. (n = 5); #, p < 0.05 versus DMSO control; $, p < 0.05 versus STS without GIP; %, p < 0.05 versus STS plus GIP without MDL-12,300A). B, INS-1 cells were treated without or with STS (100 nm) plus increasing concentrations of insulin (0–100 nm), IGF-I (10 nm), GIP (10 nm), or forskolin (1 μm) for 6 h (mean ± S.E. (n = 8); #, p < 0.05 versus DMSO control; $, p < 0.05 versus STS without GIP; %, p < 0.05 versus STS plus GIP without MDL-12,300A).
GIP Dynamically Regulates Mitochondrial Levels of Bad and Bim and Activation of Mitochondrial Bax
Cell release of mitochondrial cytochrome c and initiation of the apoptotic program is coordinated by the bcl-2 family of proteins (31–33). Therefore, we examined whether GIP stimulation regulated mitochondrial levels of three pro-apoptotic proteins, bax, bad, and bimEL, and two anti-apoptotic proteins, bcl-2 and bcl-XL, in STS-treated INS-1 cells. Following treatment with STS ± GIP for 4 h, total or mitochondrial/cytoplasmic protein fractions were collected and analyzed via Western blot (Fig. 3). STS induced a significant increase in mitochondrial levels of bcl-XL, bad, and bimEL, and there was a trend for increased bax, but bcl-2 levels were unaltered. In the presence of GIP, levels of mitochondrial bax, bad, and bimEL were all similar to control levels. However, GIP had no effect on levels of mitochondria-associated bcl-XL. STS also induced a significant elevation in total bad protein levels that was ablated by GIP treatment. Bax is a key pro-apoptotic protein, and elevated levels of mitochondrial bad and bimEL and the associated release of cytochrome c indicated that bax was activated in STS-treated cells. Upon activation, bax undergoes a conformational shift and forms homodimers, and it has been established that this activated state can be measured using the bismaleimidohexane cross-linking method (38). Therefore, in parallel to the studies above, the functional state of bax was examined in mitochondrial samples collected from INS-1 cells treated without or with STS ± GIP. STS treatment resulted in a dramatic increase in cross-linked bax levels that were significantly diminished by GIP stimulation (Fig. 3E). This functional change in bax activity was shown to be directly linked to cell survival, because a bax channel blocker suppressed the onset of STS-induced cell death in a concentration-dependent manner (Fig. 3F). Collectively, these studies indicate that the protective effects of GIP on STS-treated INS-1 cells were largely mediated through inhibition of bax activity, which likely involves a signaling pathway that inhibits translocation of pro-apoptotic bad and bimEL proteins to the mitochondria.
FIGURE 3.

GIP dynamically regulates mitochondrial levels of bad and bimEL and activation of mitochondrial bax. A, INS-1 cells were treated without or with STS (100 nm) ± 10 nm GIP for 4 h, and Western analysis was performed on mitochondrial and cytoplasmic protein fractions and total cell lysates with indicated antibodies. B–D, for quantification, protein levels were normalized to β-tubulin (cytoplasmic, B), COX IV (mitochondrial, C), or β-actin (cell lysates, D) (mean ± S.E. changes in protein level relative to control; #, p < 0.05; n = 3–6). E, INS-1 cells were treated without or with STS ± 10 nm GIP for 4 h, and Western analysis was performed with the indicated antibodies on mitochondrial protein fractions that were incubated with 10 mm bismaleimidohexane (BMH) for 30 min at room temperature. Shown are representative blots of at least three independent experiments. Anti-COX IV was an internal control. F, INS-1 cells were treated without or with STS plus increasing concentrations of bax channel blocker (0–5 μm) for 6 h, and cell death was determined (mean ± S.E. of cell death (n = 5); #, p < 0.05 versus STS alone).
GIP-mediated Anti-apoptotic Signaling in STS-treated INS-1 Cells Does Not Require Transcriptional Changes
Given the rapid onset of STS-induced INS-1 cell death, it is unlikely that the effects of STS or GIP in this study involved changes in gene transcription but rather pro- and anti-apoptotic signals mediated via the existing bcl-2 family member proteins. However, because there was a significant elevation in bad protein levels in whole cell lysates (Fig. 3, A and D) a role for transcriptional changes could not be excluded, and INS-1 cells were therefore treated without or with STS ± GIP in the absence or presence of the mRNA translation inhibitor, cycloheximide (CHX) (Fig. 4). Treatment of cells with CHX and/or STS resulted in increased cell death in association with an elevation in bad protein, and stimulation with GIP significantly reduced these effects. Transcriptional changes were therefore not required for the activation of cell death by STS or the inhibition of cell death by GIP. Moreover, because STS ± inhibition of protein translation with CHX resulted in elevated bad protein levels, the increase in bad protein levels in STS-treated cells was likely due to a decrease in bad degradation.
FIGURE 4.
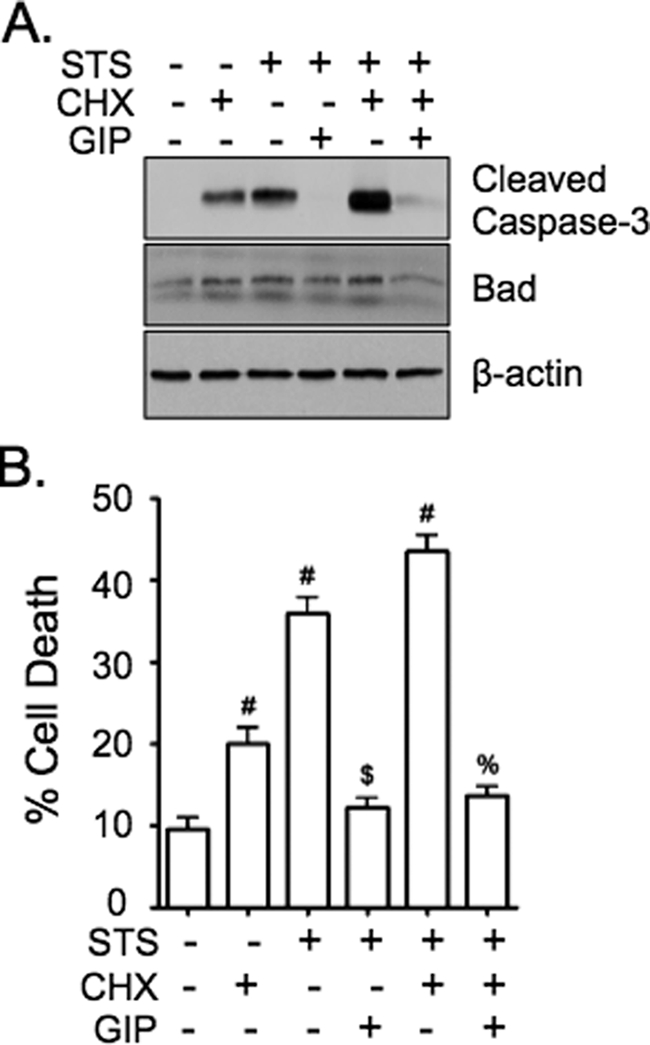
GIP-mediated anti-apoptotic signaling in STS-treated INS-1 cells does not require transcriptional changes. A and B, INS-1 cells were treated without or with STS (100 nm) ± 10 nm GIP for 4 h (A) or 6 h (B) in the presence or absence of the mRNA translation inhibitor, cycloheximide (CHX, 10 μg/ml), and Western analysis was performed on cell lysates with indicated antibodies (A) or cell death determined (B). In B, mean ± S.E. of cell death (n = 4); #, p < 0.05 versus DMSO control; $, p < 0.05 versus STS without GIP; %, p < 0.05 versus STS plus CHX without GIP.
GIP Mediates Anti-apoptotic Signaling via Dual Suppression of p38 MAPK and JNK
Because treatment with STS resulted in actions on bcl-2 family proteins that were reversed by GIP, attempts were made to identify the upstream signaling events involved. Both p38 MAPK and JNK are central pro-apoptotic proteins in β-cells (6–11, 42, 43) that can act on bax (44), bad (45, 46), and bimEL (47), and we showed previously that Akt is required for the survival effects of GIP in STS-treated INS-1 cells (34). Studies using selective inhibitors, overexpression of dominant negative Akt (AktDN), and Akt 1 and 2 siRNA were therefore used to determine if GIP promoted INS-1 cell survival via Akt-dependent inhibition of p38 MAPK and/or JNK. Firstly, INS-1 cells were treated without or with STS ± GIP for 4 h in the absence or presence of Akt inhibitor IV (Akti). Treatment with STS resulted in elevated levels of phosphorylated Mek 3/6 (upstream kinase of p38 MAPK), p38 MAPK, and JNK (active forms), and this was associated with a marked dephosphorylation of bad112 (active form (48, 49)) and elevated levels of cleaved caspase-3 (Fig. 5A). GIP stimulation almost ablated the effects of STS on Mek 3/6, p38 MAPK, JNK, bad, and caspase-3 in the absence, but not in the presence, of Akti. Next, parallel experiments were performed with INS-1 cells transfected with GFP or AktDN (Fig. 5B) as well as with cells transfected with scramble or Akt 1 and 2 siRNA (Fig. 5C; supplemental Fig. S1 demonstrates the selective knockdown of Akt 1 and 2 and not Akt 3). Similar to the effects of Akti, overexpression of AktDN and knockdown of Akt 1 and 2 clearly reduced the ability of GIP to suppress the effects of STS on p38 MAPK, JNK, Bad, and caspase-3. To determine if the effects of STS on phosphorylated bad112 and INS-1 cell survival were due to activation of p38 MAPK and/or JNK, INS-1 cells were treated without or with STS ± the p38 MAPK inhibitor, SB 203580 (p38i), and/or the JNK inhibitor, SP 600125 (JNKi) (Fig. 5, D and E); similarly, concentration-response studies were performed with these inhibitors as well as an additional p38 MAPK inhibitor, SB 202190, and the JNK inhibitor, JNK inhibitor VIII (supplemental Fig. S2). Inhibition of p38 MAPK and JNK prevented STS-induced dephosphorylation of bad112 and promoted the survival of INS-1 cells. Noteworthy was the fact that survival of cells treated with both inhibitors (p38i + JNKi) was equivalent to that achieved with GIP (Fig. 5E). To additionally validate the findings observed in INS-1 cells, the effects of STS ± GIP and STS ± p38i and JNKi were determined in an alternative β-cell line, mouse insulinoma (MIN6) cells (supplemental Fig. S3). Treatment of MIN6 cells with STS also induced phosphorylation of p38 MAPK and JNK and dephosphorylation of bad112 and elevated levels of cleaved caspase-3 and cell death, all of which were diminished by GIP (supplemental Fig. S3, A and B). Similarly, p38i and JNKi suppressed the dephosphorylation of Bad112 as well as increased levels of cleaved caspase-3 and cell death (supplemental Fig. S3, C and D). To validate whether the effects of GIP on bcl-2 family proteins were due to inhibition of p38i and JNKi, INS-1 cells were next treated without or with STS ± both p38i and JNKi, and mitochondrial/cytoplasmic fractions were collected for analysis of levels of mitochondrial bimEL, bax, and bad (Fig. 5F). Treatment of INS-1 cells with p38i and JNKi prevented STS-induced elevations in bimEL, bax, and bad, similar to that found in cells treated with GIP (see Fig. 3, A–C). Moreover, STS-induced activation of mitochondrial bax was also prevented by treatment of INS-1 cells with p38i and JNKi (Fig. 5G). Together, these studies indicate that the suppression of p38 MAPK and JNK by GIP can fully explain the anti-apoptotic actions of GIP in STS-treated INS-1 cells.
GIP Suppresses p38 MAPK and JNK Activation via Akt-mediated Inhibition of ASK1 in INS-1 Cells and Human Islets
We then sought to identify the link between Akt signaling and dual inhibition of p38 MAPK and JNK. ASK1 is an upstream MAPK kinase kinase that phosphorylates (activates) both Mek 3/6 and Mek 4/7, which then phosphorylate their downstream targets, p38 MAPK and JNK, respectively (35, 50). Akt has been shown to phosphorylate ASK1 at the serine 83 residue (ASK183 (36, 51)) in several cell lines, resulting in inactivation, and this was associated with decreased phosphorylation at threonine 845 (ASK1845 (50, 51)) a site that is important for its activation. The ability of GIP to inhibit ASK1 in INS-1 cells via elevating phosphorylated levels of ASK183 and preventing STS-induced increases in phosphorylated levels of ASK1845 was therefore evaluated. Because currently available antibodies directed against the phosphorylated ASK183 and ASK1845 epitope recognize the human protein but not those from rat, INS-1 cells were transfected with a pcDNA3 vector that expressed the wild-type human ASK1 protein. Cells were then treated without or with STS ± GIP, and levels of phosphorylated ASK183 and ASK1845 were determined via Western blot. In untransfected INS-1 cells, endogenous (rat) ASK1 protein was detected, but, as expected, there were no detectable levels of phosphorylated ASK183 or ASK1845 (Fig. 6A). In comparison, the human ASK1 protein was efficiently expressed in transfected cells (Fig. 6A). In STS-treated cells the levels of phosphorylated human ASK183 were markedly reduced, and phosphorylated human ASK1845 levels were increased relative to untreated cells (control), whereas GIP treatment neutralized the effects of STS on the phosphorylation state of human ASK1 (Fig. 6A). Although the phosphorylation state of endogenous ASK1 could not be detected, to ensure that GIP regulated endogenous ASK1 activity in INS-1 cells, untransfected INS-1 cells were treated without or with STS ± GIP, then endogenous ASK1 protein was immunoprecipitated and ASK1 enzyme activity was determined using an in vitro kinase assay. STS treatment of INS-1 cells caused a significant increase in the enzyme activity of ASK1, and this was blunted in cells treated with GIP (Fig. 6, B and C). To verify that the effects of STS on p38 MAPK and JNK were mediated by ASK1, INS-1 cells were transfected with pcDNA3 vector expressing GFP (control) or kinase-dead ASK1 (dominant negative) and treated without or with STS. Although there was no detectable effect in cells not treated with STS, the effects of STS on p38 MAPK, JNK, and caspase-3 were reduced in INS-1 cells expressing kinase-dead ASK1 relative to those expressing GFP (Fig. 6D). To then verify that the protective effects of GIP on STS-treated INS-1 cells was due to Akt-mediated phosphorylation of ASK183, INS-1 cells were transfected with pcDNA3 vector expressing wild-type ASK1 (ASK1WT) or ASK1 containing an alanine mutation at serine 83 (ASK1S83A) and treated without (control) or with STS ± GIP. As validation of this mutant, Western blot analysis with anti-ASK183 antibody showed detectable levels of phosphorylated ASK183 in INS-1 cells transfected with ASK1WT but not ASK1S83A (Fig. 6E). However, more importantly, the ability of GIP to suppress the effects of STS on p38 MAPK, JNK, and caspase-3 was clearly reduced in INS-1 cells expressing ASK1S83A relative to those expressing ASK1WT (Fig. 6E). Lastly, to establish that phosphorylation of transfected human ASK183 and ASK1845 equated to effects of GIP on endogenous ASK1 in β-cells, responses to GIP were examined in human islets treated without or with STS ± GIP (Fig. 6F). Treatment of human islets with STS resulted in decreased levels of phosphorylated ASK183, increased levels of phosphorylated ASK1845 and Mek 3/6, as well as increases in levels of cleaved caspase-3. However, these effects were ablated in the presence of GIP, indicating a similar role in regulating ASK1 activity in islets to that observed with INS-1 cells.
GIP-mediated Suppression of p38 MAPK and JNK Also Promotes the Survival of INS-1 Cells Exposed to ER and Genotoxic Stress
It was next determined whether suppression of p38 MAPK and JNK is also involved in GIP signaling responses to other pro-apoptotic stimuli. INS-1 cells were treated without or with 100 nm STS (mitochondrial stress), 500 nm thapsigargin (ER stress), or 5 μm etoposide (genotoxic stress) ± GIP for 4 h, followed by Western analysis. In the presence of STS, thapsigargin, or etoposide phosphorylated p38 MAPK and JNK levels were increased, phosphorylated bad112 levels were decreased, and cleaved caspase-3 increased; each of these effects was greatly reduced with GIP treatment (Fig. 7A). In parallel experiments extended to 6 h, levels of cell death were ∼30% in cells treated with STS, thapsigargin, or etoposide, and GIP significantly reduced these levels with all apoptosis-promoting agents (Fig. 7B). Treatment of INS-1 cells without or with STS, thapsigargin, or etoposide ± p38i or JNKi showed that both inhibitors reduced cell death (Fig. 7C). Collectively this indicates that inhibition of p38 MAPK and JNK is a key component of GIP-mediated anti-apoptotic signaling in cells exposed to stress.
FIGURE 7.

GIP-mediated suppression of p38 MAPK and JNK also promotes the survival of INS-1 cells exposed to ER and genotoxic stress. A, INS-1 cells were treated without or with STS (100 nm), thapsigargin (500 nm), or etoposide (5 μm) ± 10 nm GIP for 4 h, and Western analysis was performed on total cell lysates with the indicated antibodies. Shown are representative blots of at least three independent experiments. Anti-β-actin blot was an internal control. B, INS-1 cells were treated without or with STS, thapsigargin, or etoposide ± GIP for 6 h, and cell death was determined (mean ± S.E. of cell death (n = 6); #, p < 0.05 as indicated). C, INS-1 cells were treated without or with STS, thapsigargin, or etoposide ± p38i (5 μm) or JNKi (5 μm) for 6 h, and cell death was determined (mean ± S.E. of cell death (n = 6); #, p < 0.05 versus STS only; $, p < 0.05 versus thapsigargin only; %, p < 0.05 versus etoposide only).
DISCUSSION
A key feature in the pathogenesis of type 2 diabetes mellitus is an inadequate insulin response for the prevailing blood glucose level due to β-cell dysfunction and increased apoptotic β-cell death (2, 4, 5). Currently it is not possible to quantify β-cell mass in vivo, but evidence has been presented for improvements in “functional β-cell mass” with a number of therapeutics, including thiazolidinediones (12), interleukin-1 receptor antagonists (10, 52), and agents that mimic or enhance incretin actions (12, 15, 19, 53). The development of therapies with demonstrated ability to prevent islet loss and preserve β-cell mass is one of the primary goals in diabetes research, and pre-clinical studies have demonstrated that incretin analogs and mimetics have the potential to fulfill such a role (13–15, 17–19).
GIP has been shown to promote β-cell survival by modulating the activity of multiple signaling modules (16, 21, 26, 29, 30, 34) and regulating gene transcription of anti-apoptotic bcl-2 and pro-apoptotic bax (26, 30). Disruption of the signaling pathway involved in regulating bcl-2 expression resulted in a reduced ability of GIP to suppress caspase-3 activation (30). Therefore GIP and glucagon-like peptide 1 appear to play important roles in regulating the amounts of mitochondria-associated bcl-2 family members at levels that are appropriate for maintaining cell integrity under unstressed conditions. As with other cells, the β-cell is exposed to various stresses requiring responses to counteract the redistribution of pro-apoptotic bcl-2 proteins from the cytoplasm to the mitochondria and pore formation by bax and bak oligomers (6–11, 32, 33, 42, 54–57). In the current study, we examined how GIP regulates the interactions of pro- and anti-apoptotic bcl-2 family proteins with the mitochondria and thus suppresses apoptosis induced in INS-1 β-cells by staurosporine, an apoptotic agent that induces oxidative stress and activates the mitochondria-mediated apoptotic pathway. In INS-1 β-cells, STS has been shown to promote the generation of reactive oxygen species and mitochondrial lipid peroxidation, and the loss of mitochondrial membrane potential (58). In the current study, cytochrome c was found to be released from mitochondria within 4–6 h of STS treatment, caspase-3 was activated, and apoptosis was initiated (Fig. 1). In the presence of GIP, all of these processes were suppressed in a concentration-dependent manner (Fig. 1). Previous studies have demonstrated that GIP stimulates insulin secretion from the β-cell via a pathway that involves activation of PKA (20–22) and EPAC2 (23). However, although cAMP has been previously implicated in GIP-mediated pro-survival effects (29, 34), the current results clearly demonstrate a dependence upon adenylate cyclase activation for this process, and a contribution from autocrine insulin signaling appears unlikely (Fig. 2). From the studies on bcl-2 family members, it is evident that STS induced translocation of bad and bimEL to the mitochondria as well as the activation of mitochondrial bax (Fig. 3, A–E). The level of mitochondria-associated bcl-2 remained fairly constant during STS treatment, whereas that of bcl-XL increased. This suggests a role for bcl-2 in regulating the pro-apoptotic proteins bound under unstressed conditions, with bcl-XL contributing during an apoptotic stimulus. GIP almost completely counteracted the STS-induced increase in association of bimEL, bax, and bad with the mitochondria (Fig. 3), and, because CHX treatment had no effect on bad levels, caspase-3 activation, or protection against cell death, transcriptional changes did not contribute significantly (Fig. 4). The reduction in dimerization of bax in STS-treated cells induced by GIP treatment (Fig. 3F) is likely a key component of GIP-mediated survival, because it has been established that bax or bak oligomerization is critical for the initiation of mitochondria-associated apoptosis (31–33).
How does GIP activation of adenylate cyclase prevent the apoptotic effects of STS? Recently, we demonstrated that GIP activates Akt (protein kinase B) through a non-canonical pathway involving EPAC2 (34), and the inhibitor Akt IV (Akti) blocked GIP-induced Akt activation. In the current studies, STS treatment of INS-1 cells was shown to increase levels of phosphorylated Mek 3/6, an upstream kinase, and phosphorylated (active) forms of both p38 MAPK and JNK. All of the phosphorylation events, plus caspase-3 activation, were greatly reduced by GIP treatment, in an Akti-sensitive manner (Fig. 5A). Furthermore, overexpression of AktDN or knockdown of both Akt 1 and 2 produced similar results to Akti treatment (Fig. 5, B and C), clearly demonstrating that Akt is central to the pro-survival actions of GIP in STS-treated INS-1 cells. Inhibitor studies showed that p38 MAPK and JNK mediated STS-induced dephosphorylation of bad112 (Fig. 5D), the mitochondrial translocation of bad and bimEL, and mitochondrial release of cytochrome c (Fig. 5F) as well as the activation of mitochondrial bax (Fig. 5G) and onset of cell death (Fig. 5E and supplemental Fig. S2). Because combined inhibition of p38 MAPK and JNK promoted a similar level of survival to that observed in INS-1 cells treated with GIP (Fig. 5E) and exerted similar actions on mitochondrial bax activation (Fig. 5E), we conclude that this pathway was central to GIP-promoted survival of STS-treated INS-1 cells. Similar results were found in cells exposed to ER-stress (thapsigargin) and genotoxic stress (etoposide) (Fig. 7, A and B), indicating that antagonism of the p38 MAPK and JNK pathways is a common mechanism by which GIP acts. Because bimEL and bad have well established pro-apoptotic actions, and bad has previously been shown to play an important role in β-cells (59, 60), the effects of GIP on their cellular distribution likely played an important role in reducing STS-induced INS-1 cell death. However, the precise mechanisms whereby p38 MAPK and JNK promoted activation of bax could not be determined in the current study. Both kinases phosphorylate bax at threonine 167 (44), and, in ceramide-treated A549 cells, PP2A was shown to dephosphorylate bax at serine 184 (61), an Akt target site that, when phosphorylated, inactivates the pro-apoptotic actions of bax (62). These events could therefore result in bax activation and mitochondrial translocation, and their involvement could be an interesting avenue for future β-cell research.
The link between GIP signaling and suppression of p38 MAPK and JNK was found to involve Akt-mediated suppression of ASK1 in INS-1 cells and human islets. Although human and rodent β-cells have been shown to exhibit differences in proliferative responses (63), the current studies indicate that the core incretin-mediated anti-apoptotic pathways are conserved. However, further studies are needed to establish whether ASK1 plays a central role in cell death associated with human diabetes. In this study, stimulation with GIP elevated levels of phosphorylated ASK1 at the Akt target site, serine 83 (Figs. 6, A and F), phosphorylation of which has been shown to inhibit ASK1 activation (36) as well as the downstream activation of p38 MAPK and JNK (35, 50). Similarly, GIP prevented phosphorylation of the threonine 845 ASK1 activation site (Fig. 6, A and F) (50, 51). Consistent with this, GIP prevented STS-induced elevations in ASK1 in vitro kinase activity (Fig. 6, B and C) and phosphorylation of the downstream ASK1 target, Mek 3/6 (Figs. 5A and 6F). Of interest was that dominant negative ASK1 suppressed STS-induced elevations in levels of phosphorylated p38 MAPK and JNK, as well as cleaved caspase-3 (Fig. 6D), and that INS-1 cells expressing ASK1 protein that lacked the Akt phosphorylation site were resistant to the effects of GIP on p38 MAPK, JNK, and caspase-3 (Fig. 6E). This demonstrated that the effect of STS and GIP on INS-1 cell death/survival was relayed via signals sent to ASK1. To our knowledge, this is the first study that identifies a key role for ASK1 in β-cells. In other cell types such as lymphocytes, cardiac tissue, neurons, and endothelial cells, ASK1 has been shown to operate as a redox sensor that, upon exposure to excessive levels of reactive oxygen species, activates the mitochondria-mediated apoptotic pathway via activation of p38 MAPK and JNK (50, 64). In agreement with such a role, sustained activation of p38 MAPK and JNK and the onset of apoptosis were significantly diminished in mouse embryonic fibroblasts from ASK1 knockout mice that were exposed to H2O2 and tumor necrosis factor-α (65). Stressors that activate ASK1 include oxidative stress, ER stress, Ca2+ overload, and receptor-mediated inflammatory signals, and the underlying mechanism that coordinates ASK1 activation involves a high molecular weight complex described as the “ASK1 signalosome” (50, 64). Bound to this complex is the anti-oxidative protein, thioredoxin, the reduced form of which binds the N-terminal region of ASK1, preventing ASK1 signaling. When intracellular reactive oxygen species levels exceed the anti-oxidative capacity of the cell, and thus convert thioredoxin to the oxidized form, it dissociates from ASK1 and is no longer inhibitory. ASK1 then commences to activate Mek 3/6 and Mek 4/7, which then activate p38 MAPK and JNK, respectively. Furthermore, even if thioredoxin is released from ASK1, an additional regulatory mechanism exists for ASK1 in which Akt phosphorylates serine 83 and prevents ASK1 activation (36), as found under GIP-stimulated conditions in the current study. This constitutes an intriguing scenario in which, even in the presence of elevated intracellular stress, extracellular signals can relay instructions to the cell, via Akt, resulting in the interception of ongoing pro-apoptotic signals, and it highlights the complex balance between life and death signaling in the cell.
In summary, we have shown that GIP promotes β-cell survival in STS (mitochondrial stress)-treated INS-1 cells and human islets via Akt-dependent inhibition of ASK1, resulting in the dual suppression of p38 MAPK and JNK. This restrains the mitochondrial translocation of bad and bimEL and activation of mitochondrial bax, thus preventing the release of cytochrome c, activation of caspase-3, and onset of apoptosis. Because GIP induced similar effects in INS-1 cells exposed to thapsigargin (ER stress) and etoposide (genotoxic stress), the Akt/ASK1 pathway appears to be a key component of GIP anti-apoptotic signaling.
Supplementary Material
Acknowledgments
We thank Dr. C. B. Newgard (Duke University Medical Center, Durham, NC) for kindly providing us with INS-1 cells (clone 832/13), Dr. Hidenori Ichjo for providing the wild-type and kinase-dead ASK1 constructs, and Dr. Moses Chao for providing the ASK1 construct containing an Ser833Ala mutation.
This work was supported by funding from the Canadian Institutes of Health Research and the Canadian Foundation for Innovation (to C. H. S. M.), the Michael Smith Foundation for Health Research (MSFHR) (to C. H. S. M. and G. W.), and scholarships from the Natural Sciences and Engineering Research Council of Canada and MSFHR (to S. B. W.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- ER
- endoplasmic reticulum
- GIP
- glucose-dependent insulinotropic polypeptide
- PKA
- protein kinase A
- EPAC2
- exchange protein activated by cAMP 2
- Erk
- extracellular signal-regulated kinase
- MAPK
- mitogen-activated protein kinase
- STS
- staurosporine
- ASK1
- apoptosis signal-regulating kinase 1
- JNK
- Jun N-terminal kinase
- mAb
- monoclonal antibody
- CHX
- cycloheximide
- siRNA
- small interference RNA
- Akti
- Akt inhibitor IV
- p38i
- p38 MAPK inhibitor (SB 203580)
- JNKi
- JNK inhibitor (SP 600125)
- WT
- wild type
- DN
- dominant negative.
REFERENCES
- 1.Petersen K. F., Shulman G. I. (2006) Am. J. Med. 119, S10–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leahy J. L. (2005) Arch. Med. Res. 36, 197–209 [DOI] [PubMed] [Google Scholar]
- 3.Weir G. C., Bonner-Weir S. (2004) Diabetes 53, Suppl. 3, S16–S21 [DOI] [PubMed] [Google Scholar]
- 4.Kahn S. E. (2003) Diabetologia 46, 3–19 [DOI] [PubMed] [Google Scholar]
- 5.Rhodes C. J. (2005) Science 307, 380–384 [DOI] [PubMed] [Google Scholar]
- 6.Poitout V., Robertson R. P. (2008) Endocr. Rev. 29, 351–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans J. L., Goldfine I. D., Maddux B. A., Grodsky G. M. (2002) Endocr. Rev. 23, 599–622 [DOI] [PubMed] [Google Scholar]
- 8.Scheuner D., Kaufman R. J. (2008) Endocr. Rev. 29, 317–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eizirik D. L., Cardozo A. K., Cnop M. (2008) Endocr. Rev. 29, 42–61 [DOI] [PubMed] [Google Scholar]
- 10.Donath M. Y., Størling J., Berchtold L. A., Billestrup N., Mandrup-Poulsen T. (2008) Endocr. Rev. 29, 334–350 [DOI] [PubMed] [Google Scholar]
- 11.Haataja L., Gurlo T., Huang C. J., Butler P. C. (2008) Endocr. Rev. 29, 303–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wajchenberg B. L. (2007) Endocr. Rev. 28, 187–218 [DOI] [PubMed] [Google Scholar]
- 13.Ahrén B., Schmitz O. (2004) Horm. Metab. Res. 36, 867–876 [DOI] [PubMed] [Google Scholar]
- 14.Drucker D. J. (2006) Cell Metab. 3, 153–165 [DOI] [PubMed] [Google Scholar]
- 15.Kim W., Egan J. M. (2008) Pharmacol. Rev. 60, 470–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McIntosh C. H., Widenmaier S., Kim S. J. (2009) Vitam. Horm. 80, 409–471 [DOI] [PubMed] [Google Scholar]
- 17.Drucker D. J., Nauck M. A. (2006) Lancet 368, 1696–1705 [DOI] [PubMed] [Google Scholar]
- 18.Green B. D., Flatt P. R. (2007) Best Pract. Res. Clin. Endocrinol. Metab. 21, 497–516 [DOI] [PubMed] [Google Scholar]
- 19.Salehi M., Aulinger B. A., D'Alessio D. A. (2008) Endocr. Rev. 29, 367–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trümper A., Trümper K., Trusheim H., Arnold R., Göke B., Hörsch D. (2001) Mol. Endocrinol. 15, 1559–1570 [DOI] [PubMed] [Google Scholar]
- 21.Trümper A., Trümper K., Hörsch D. (2002) J. Endocrinol. 174, 233–246 [DOI] [PubMed] [Google Scholar]
- 22.Ding W. G., Gromada J. (1997) Diabetes 46, 615–621 [DOI] [PubMed] [Google Scholar]
- 23.Kashima Y., Miki T., Shibasaki T., Ozaki N., Miyazaki M., Yano H., Seino S. (2001) J. Biol. Chem. 276, 46046–46053 [DOI] [PubMed] [Google Scholar]
- 24.Ehses J. A., Pelech S. L., Pederson R. A., McIntosh C. H. (2002) J. Biol. Chem. 277, 37088–37097 [DOI] [PubMed] [Google Scholar]
- 25.Ehses J. A., Lee S. S., Pederson R. A., McIntosh C. H. (2001) J. Biol. Chem. 276, 23667–23673 [DOI] [PubMed] [Google Scholar]
- 26.Kim S. J., Winter K., Nian C., Tsuneoka M., Koda Y., McIntosh C. H. (2005) J. Biol. Chem. 280, 22297–22307 [DOI] [PubMed] [Google Scholar]
- 27.Pospisilik J. A., Martin J., Doty T., Ehses J. A., Pamir N., Lynn F. C., Piteau S., Demuth H. U., McIntosh C. H., Pederson R. A. (2003) Diabetes 52, 741–750 [DOI] [PubMed] [Google Scholar]
- 28.Yusta B., Baggio L. L., Estall J. L., Koehler J. A., Holland D. P., Li H., Pipeleers D., Ling Z., Drucker D. J. (2006) Cell Metab. 4, 391–406 [DOI] [PubMed] [Google Scholar]
- 29.Ehses J. A., Casilla V. R., Doty T., Pospisilik J. A., Winter K. D., Demuth H. U., Pederson R. A., McIntosh C. H. (2003) Endocrinology 144, 4433–4445 [DOI] [PubMed] [Google Scholar]
- 30.Kim S. J., Nian C., Widenmaier S., McIntosh C. H. (2008) Mol. Cell. Biol. 28, 1644–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reed J. C. (2006) Cell Death Differ. 13, 1378–1386 [DOI] [PubMed] [Google Scholar]
- 32.Shi Y. (2002) Mol. Cell 9, 459–470 [DOI] [PubMed] [Google Scholar]
- 33.Harwood S. M., Yaqoob M. M., Allen D. A. (2005) Ann. Clin. Biochem. 42, 415–431 [DOI] [PubMed] [Google Scholar]
- 34.Widenmaier S. B., Sampaio A. V., Underhill T. M., McIntosh C. H. (2009) J. Biol. Chem. 284, 10764–10773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ichijo H., Nishida E., Irie K., ten Dijke P., Saitoh M., Moriguchi T., Takagi M., Matsumoto K., Miyazono K., Gotoh Y. (1997) Science 275, 90–94 [DOI] [PubMed] [Google Scholar]
- 36.Kim A. H., Khursigara G., Sun X., Franke T. F., Chao M. V. (2001) Mol. Cell. Biol. 21, 893–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kibbey R. G., Pongratz R. L., Romanelli A. J., Wollheim C. B., Cline G. W., Shulman G. I. (2007) Cell Metab. 5, 253–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim H., Rafiuddin-Shah M., Tu H. C., Jeffers J. R., Zambetti G. P., Hsieh J. J., Cheng E. H. (2006) Nat. Cell Biol. 8, 1348–1358 [DOI] [PubMed] [Google Scholar]
- 39.Kim S. J., Choi W. S., Han J. S., Warnock G., Fedida D., McIntosh C. H. (2005) J. Biol. Chem. 280, 28692–28700 [DOI] [PubMed] [Google Scholar]
- 40.Beith J. L., Alejandro E. U., Johnson J. D. (2008) Endocrinology 149, 2251–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson J. D., Bernal-Mizrachi E., Alejandro E. U., Han Z., Kalynyak T. B., Li H., Beith J. L., Gross J., Warnock G. L., Townsend R. R., Permutt M. A., Polonsky K. S. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 19575–19580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim W. H., Lee J. W., Gao B., Jung M. H. (2005) Cell. Signal. 17, 1516–1532 [DOI] [PubMed] [Google Scholar]
- 43.Sumara G., Formentini I., Collins S., Sumara I., Windak R., Bodenmiller B., Ramracheya R., Caille D., Jiang H., Platt K. A., Meda P., Aebersold R., Rorsman P., Ricci R. (2009) Cell 136, 235–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim B. J., Ryu S. W., Song B. J. (2006) J. Biol. Chem. 281, 21256–21265 [DOI] [PubMed] [Google Scholar]
- 45.Grethe S., Coltella N., Di Renzo M. F., Pörn-Ares M. I. (2006) Biochem. Biophys. Res. Commun. 347, 781–790 [DOI] [PubMed] [Google Scholar]
- 46.Grethe S., Pörn-Ares M. I. (2006) Cell. Signal. 18, 531–540 [DOI] [PubMed] [Google Scholar]
- 47.Cai B., Chang S. H., Becker E. B., Bonni A., Xia Z. (2006) J. Biol. Chem. 281, 25215–25222 [DOI] [PubMed] [Google Scholar]
- 48.Zha J., Harada H., Yang E., Jockel J., Korsmeyer S. J. (1996) Cell 87, 619–628 [DOI] [PubMed] [Google Scholar]
- 49.Yang E., Zha J., Jockel J., Boise L. H., Thompson C. B., Korsmeyer S. J. (1995) Cell 80, 285–291 [DOI] [PubMed] [Google Scholar]
- 50.Takeda K., Noguchi T., Naguro I., Ichijo H. (2008) Annu. Rev. Pharmacol. Toxicol. 48, 199–225 [DOI] [PubMed] [Google Scholar]
- 51.Zhang R., Luo D., Miao R., Bai L., Ge Q., Sessa W. C., Min W. (2005) Oncogene 24, 3954–3963 [DOI] [PubMed] [Google Scholar]
- 52.Larsen C. M., Faulenbach M., Vaag A., Vølund A., Ehses J. A., Seifert B., Mandrup-Poulsen T., Donath M. Y. (2007) N. Engl. J. Med. 356, 1517–1526 [DOI] [PubMed] [Google Scholar]
- 53.McIntosh C. H. (2008) Front. Biosci. 13, 1753–1773 [DOI] [PubMed] [Google Scholar]
- 54.Hanke J. (2001) Cells Tissues Organs 169, 113–124 [DOI] [PubMed] [Google Scholar]
- 55.Laybutt D. R., Kaneto H., Hasenkamp W., Grey S., Jonas J. C., Sgroi D. C., Groff A., Ferran C., Bonner-Weir S., Sharma A., Weir G. C. (2002) Diabetes 51, 413–423 [DOI] [PubMed] [Google Scholar]
- 56.Shimabukuro M., Wang M. Y., Zhou Y. T., Newgard C. B., Unger R. H. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 9558–9561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Allen D. A., Yaqoob M. M., Harwood S. M. (2005) J. Nutr. Biochem. 16, 705–713 [DOI] [PubMed] [Google Scholar]
- 58.Seleznev K., Zhao C., Zhang X. H., Song K., Ma Z. A. (2006) J. Biol. Chem. 281, 22275–22288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Danial N. N., Walensky L. D., Zhang C. Y., Choi C. S., Fisher J. K., Molina A. J., Datta S. R., Pitter K. L., Bird G. H., Wikstrom J. D., Deeney J. T., Robertson K., Morash J., Kulkarni A., Neschen S., Kim S., Greenberg M. E., Corkey B. E., Shirihai O. S., Shulman G. I., Lowell B. B., Korsmeyer S. J. (2008) Nat. Med. 14, 144–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Danial N. N., Gramm C. F., Scorrano L., Zhang C. Y., Krauss S., Ranger A. M., Datta S. R., Greenberg M. E., Licklider L. J., Lowell B. B., Gygi S. P., Korsmeyer S. J. (2003) Nature 424, 952–956 [DOI] [PubMed] [Google Scholar]
- 61.Xin M., Deng X. (2006) J. Biol. Chem. 281, 18859–18867 [DOI] [PubMed] [Google Scholar]
- 62.Xin M., Deng X. (2005) J. Biol. Chem. 280, 10781–10789 [DOI] [PubMed] [Google Scholar]
- 63.Parnaud G., Bosco D., Berney T., Pattou F., Kerr-Conte J., Donath M. Y., Bruun C., Mandrup-Poulsen T., Billestrup N., Halban P. A. (2008) Diabetologia 51, 91–100 [DOI] [PubMed] [Google Scholar]
- 64.Matsuzawa A., Ichijo H. (2008) Biochim. Biophys. Acta 1780, 1325–1336 [DOI] [PubMed] [Google Scholar]
- 65.Tobiume K., Matsuzawa A., Takahashi T., Nishitoh H., Morita K., Takeda K., Minowa O., Miyazono K., Noda T., Ichijo H. (2001) EMBO Rep. 2, 222–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.