

Abstract
The phosphatase KdsC cleaves 3-deoxy-d-manno-octulosonate 8-phosphate to generate a molecule of inorganic phosphate and Kdo. Kdo is an essential component of the lipopolysaccharide envelope in Gram-negative bacteria. Because lipopolysaccharide is an important determinant of bacterial resistance and toxicity, KdsC is a potential target for novel antibacterial agents. KdsC belongs to the broad haloacid dehalogenase superfamily. In haloacid dehalogenase superfamily enzymes, substrate specificity and catalytic efficiency are generally dictated by a fold feature called the cap domain. It is therefore not clear why KdsC, which lacks a cap domain, is catalytically efficient and highly specific to 3-deoxy-d-manno-octulosonate 8-phosphate. Here, we present a set of seven structures of tetrameric Escherichia coli KdsC (ranging from 1.4 to 3.06 Å in resolution) that model different intermediate states in its catalytic mechanism. A crystal structure of product-bound E. coli KdsC shows how the interface between adjacent monomers defines the active site pocket. Kdo is engaged in a network of polar and nonpolar interactions with residues at this interface, which explains substrate specificity. Furthermore, this structural and kinetic analysis strongly suggests that the binding of the flexible C-terminal region (tail) to the active site makes KdsC catalytically efficient by facilitating product release.
The haloacid dehalogenase superfamily (HADSF)4 is considered one of the largest and most ubiquitous superfamilies of enzymes reported to date (1). The basic catalytic motif along with the overall protein fold shared by members of the HADSF seem to have evolved to facilitate phosphoryl transfer from a large number of quite diverse substrates. These enzymes, which occur in both eukaryotes and prokaryotes, are responsible for a wide range of important biological functions, such as the biosynthesis of important metabolic molecules (2), transport (3), signal transduction (4, 5), transcription (6), and DNA repair (7). However, the majority of enzymes comprising this superfamily remain uncharacterized, as does our understanding of the relationship between enzyme structure, substrate specificity, and catalytic efficiency.
Members of the HADSF contain a conserved mixed α/β Rossmann fold and a conserved nucleophilic aspartate that is essential for the chemistry (Fig. 1) (8). Based on additional structural elements that are inserted in the Rossmann fold, HADSF enzymes are divided into three subfamilies. Members of C1 and C2 subfamilies are usually monomeric and contain a structural insertion termed the cap domain (1, 9, 10). The cap domain acts as a “lid” covering the substrate in the active site thereby ensuring catalytic efficiency (9). In contrast, members of the C0 subfamily enzymes lack the cap domain and generally exhibit broad substrate specificity (9, 11). Interestingly, 3-deoxy-d-manno-octulosonate 8-phosphate phosphatase (KdsC, previously annotated as YrbI; EC 3.1.3.45), an archetypical HADSF enzyme from the C0 subfamily, displays a narrow substrate specificity (12, 13) and high catalytic efficiency (12). The origin of the highly efficient hydrolysis KdsC of its specific substrate, 3-deoxy-d-manno-octulosonate 8-phosphate (Kdo8P), is unknown.
FIGURE 1.

The conserved chemical mechanism of HAD phosphatases, shown here for E. coli KdsC.
KdsC, the third enzyme in the Kdo biosynthetic pathway, hydrolyzes Kdo 8-phosphate (Kdo8P) to Kdo and inorganic phosphate (Pi). Kdo is the key carbohydrate linker in the lipopolysaccharide of Gram-negative bacteria. Thus, any disruption in the Kdo biosynthetic pathway could reduce integrity of the lipopolysaccharide that is critical to bacterial survival, pathogenic potential (14), and antigenic response (15).
Putative homologs of Escherichia coli KdsC phosphatase (Fig. 2) are present in all Gram-negative bacteria with the known genomic sequence. Examples of these include YrbI from Haemophilus influenzae (16) and a putative phosphatase from Aquifex aeolicus (Protein Data Bank code 2p9j),5 which exhibit a tetrameric structure. The tetramer is stabilized by interactions of short β-hairpins, which are inserted in the core Rossmann fold. The active site of one monomer appears to be capped by an adjacent monomer (1). Specific contacts with the substrate, Kdo8P, or the products, Kdo and Pi, could not be deduced because neither reported structure contained the substrate or the products. Capturing substrate or products in the active site of KdsC crystal is challenging due to the high catalytic efficiency of the enzyme (12) and the lack of known tightly binding substrate or product analogs. During our initial efforts to obtain a crystal structure of substrate (Kdo8P)- or product (Kdo)-bound E. coli KdsC, we observed an unusual binding of the C-terminal eight-residue region of one monomer into the active site of a neighboring monomer. To our knowledge, this phenomenon has not been reported for any other HADSF protein. Remarkably, the deletion of this C-terminal region of E. coli KdsC resulted in a slower enzymatic turnover without affecting substrate binding (a kcat effect rather than a Km effect). We were able to capture the catalytic products in the crystal structure of this deletion mutant (KdsCΔ8) and thereby deduce the role of tetramerization in specific substrate recognition. Overall, six crystal forms of E. coli full-length KdsC and KdsCΔ8 with different combinations of divalent metals and catalytic products were obtained. The different conformations of KdsC revealed in the crystal structures combined with kinetic studies suggest a mechanistic role of the C terminus of KdsC in catalysis.
FIGURE 2.

Multiple sequence alignment of KdcC homologs. The secondary structure of E. coli KdsC is shown schematically above the alignment. Residues in the β2–β3-hairpin and outside of the hairpin that form the tetramerization interface are designated by the black bars and circles, respectively. Residues that are involved in coordination of Mg2+ and phosphate are shown by the open red circles and those that are involved in Kdo recognition are shown by the filled red circles. Four conserved HADSF motifs are designated. β-turns are designated as TT.
EXPERIMENTAL PROCEDURES
Cloning and Expression of KdsC
The E. coli kdsC was cloned into pT7-7 vector as described previously (12). The kdsCΔ8 (residues 1–180) was constructed by inserting a stop codon after amino acid 180 in the kdsC/pT7-7 construct using the QuikChange mutagenesis kit (Stratagene). Both E. coli KdsC and KdsCΔ8 were expressed and purified as described previously (12), with an additional size exclusion chromatographic step on an S-200 column (GE Healthcare) equilibrated with 10 mm HEPES, pH 7.2. The proteins were concentrated to 20 mg/ml for crystallization using Ultra-15 centrifugal filter unit (Millipore).
Crystallographic Experiments and Structure Determination
All crystal forms of KdsC and KdsCΔ8 were grown by hanging drop crystallizations at 20 °C. The drops contained a mixture of 1 μl of protein at 12–13 mg/ml in 10 mm HEPES, pH 7.2, with 1 μl of the crystallization solution. Kdo8P and divalent metals were present with the protein where specified in Table 1. For all crystal forms, the conditions for crystal growth and subsequent cryoprotection by flash freezing in liquid nitrogen are given in Table 1. The data were collected at 100 K at sector LS-CAT (21-ID) of the Advanced Photon Source at the Argonne National Laboratories. The data were processed with HKL2000 (17). Initially, the structure of KdsC was determined from 1.4 Å P1 crystal form data using the molecular replacement program PHASER (18) using a monomer of H. influenzae YrbI (16) as a search model. The structure was then iteratively built and refined at 1.4 Å using programs COOT (19) and REFMAC (20), respectively. Structures of the other crystal forms were determined using this refined structure as a molecular replacement search model and then built and refined analogously. For all crystal forms, the structure of each KdsC tetramer in an asymmetric unit was refined independently, and no noncrystallographic symmetry restraints were imposed, to avoid conformational bias. In the structure of the complex with of KdsCΔ8 with products, positions of Kdo and Pi were determined from the |Fo − Fc| map generated after refining the protein alone, prior to building the products, to avoid bias (see. Fig. 6C). The data and refinement statistics are summarized in Table 2. Solvent accessible surface area was calculated using program Surface Racer (21). The Protein Data Bank codes for the KdsC structures are given in Table 2.
TABLE 1.
Crystal growth and cryostabilization conditions

FIGURE 6.

A, |Fo − Fc| electron density map of the active site highlighting positions of Kdo and Pi, contoured at 3σ. The map is generated prior to modeling the Kdo and the Pi in the active site. B, a diagram of the specific interactions of Kdo and Pi in the KdsC active site. Interatomic distances shorter than 4 Å are shown by dashed lines. The phosphorus and the water in the active site are designated as P and W, respectively.
TABLE 2.
Data collection and refinement statistics

a The highest resolution shell values are shown in parentheses.
b These B factor values are affected by TLS refinement.
c Anisotropic B factor refinement.
Phosphatase Assays
The substrate for KdsC, Kdo8P, was obtained by enzymatic incubation of arabinose 5-phosphate with phosphoenolpyruvate in the presence of E. coli 3-deoxy-d-manno-2-octulosonic acid 8-phosphate synthase followed by its purification on analytical grade macroporous anion exchange resin anion exchange and P2 desalting columns. The KdsC phosphatase activity was measured using the discontinuous colorimetric assay as described in Ref. 12. Briefly, full-length KdsC (42 nm) and KdsCΔ8 (4 μm) were incubated with a large excess of Kdo8P (1–800 μm) for 1 min at 37 °C in a reaction mixture containing 100 mm HEPES, pH 7.0, 1 mm Mg2+, and NaCl (as specified). The amount of accumulated phosphate at these conditions represented the steady-state rate V of the phosphatase activity. The values of kcat and Km were then obtained from fitting these measured values of V as a function of [Kdo8P] to the following Michaelis-Menten rate law using KaleidaGraph (Synergy Software, Reading, PA),
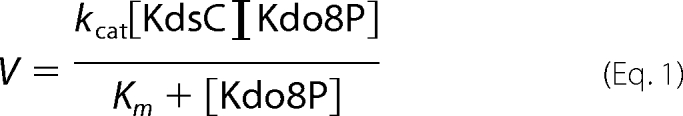 |
where [KdsC] and [Kdo8P] are initial concentrations of the protein and the substrate, respectively.
RESULTS
KdsC Structure and Its Tetrameric Organization
To understand the mechanistic details of KdsC phosphatase activity, we crystallized E. coli KdsC in the absence or presence of Kdo8P and divalent metals. We obtained six different crystal forms that yielded seven structures depicted in Fig. 3. The structure of E. coli KdsC bound to its catalytic metal Mg2+, determined at 1.4 Å, reveals the conserved features of the KdsC family, which are shared by all structures reported here (Fig. 3). The monomers of KdsC have a Rossmann fold and are organized in a tetramer with approximate 4-fold symmetry (Fig. 4A). The C terminus of each monomer is located near the active site of an adjacent monomer (Fig. 4A). A central 8-stranded intermolecular β-barrel formed by a β-hairpin insert from each of the four monomers stabilizes the tetramer (Fig. 4, A and B). The hairpin contacts are dominated by hydrophobic interactions among conserved residues Leu-41, Tyr-43, Leu-51, and Ala-53 (Fig. 2) and bury 880 Å2 of solvent accessible surface area per monomer at the β-barrel interface. Each active site cleft is located in a monomer-monomer interface. The disposition of conserved catalytic residues is similar in all four monomer-monomer interfaces. Three aspartate residues (the carboxylate of Asp-32, the backbone carbonyl oxygen of Asp-34 from HADSF Motif I, and the carboxylate of Asp-125 from HADSF Motif IV) and three water molecules coordinate Mg2+ (Fig. 4C). This coordination is similar to that of the Co2+ ion contacts seen in the H. influenzae YrbI structure (16). This organization of the active site is in excellent agreement with the proposed catalytic mechanism (1) (Fig. 1).
FIGURE 3.

A representative view of all seven tetrameric KdsC structures. The conformation of the active site (open or closed) for each monomer is specified. The positions of Mg2+, Ca2+, and Cl− ions are shown as the red, purple, and orange spheres, respectively. Kdo8P (blue), Kdo (blue), and the phosphate (green) are shown as stick models. Captions designate the contents of each crystal forms, their space groups, and resolutions (for more details, see Tables 1 and 2). The compounds present in the crystallization mixture but not observed in the protein structure are written in parentheses.
FIGURE 4.

A, structure of the KdsC tetramer showing the relative disposition of the β-hairpins responsible for tetramerization. The Mg2+ (red) and the Cl− (orange) located in the catalytic cleft are shown as spheres. B, the side view of the tetramer with one monomer highlighted. C, the 1.4 Å |2Fo − Fc| electron density map of the active site contoured at 1σ. D, conformational differences between the open (magenta) and the closed (blue) conformations of the active site. The Kdo8P binding site is highlighted with a shadow. The tail of an adjacent monomer is colored black. res, residues.
Binding of C-terminal Tail to the Active Site and Associated Conformational Changes
The crystals of Mg2+-bound KdsC contain two tetramers per asymmetric unit. These two tetramers are observed in two distinct conformations (referred to as structures a and b in Fig. 3). All four monomers of one tetramer are in one conformation, termed closed (Fig. 3, structure a). In the closed conformation, the eight C-terminal residues (residues 181–188; termed the tail) of an adjacent monomer occlude the active site cleft of a neighboring monomer and are ordered (Fig. 5A). In this conformation, the active site contains a chloride ion from the crystallization solution bound 4.4 Å away from the magnesium, suggesting a potential location for the scissile phosphate (Fig. 4B; see below). All four monomers of the other tetramer are in a different conformation, which is called open (Fig. 3, structure b). In the open conformation, the C-terminal tail of the adjacent monomer is disordered, thus exposing the active site to solvent (Fig. 5B). In this conformation, the active site does not contain an anion. Because the anion binding coincides with the conformational change, the tail binding to the active site is likely cooperative with anion binding. In a tetramer of KdsC that lacks Mg2+, obtained by crystallization in the absence of any divalent metals, all four active sites are in the closed conformation and all contain a Cl− (Fig. 3, structure g). This further supports the cooperativity of the tail and anion binding to the active site, independent of divalent metal binding.
FIGURE 5.

A cutaway view of the active site cleft in the closed (A) and open (B) conformations. The structures and the |2Fo − Fc| electron density map contoured at 1σ (shown by the violet mesh) correspond to structures b and a from Fig. 3, respectively. The C-terminal tail and the chloride (orange sphere) are shown by the arrows.
In addition to the tail and anion binding, the open-closed conformational transition is characterized by large concerted conformational changes of two loops (defined as loops I and II in Fig. 2) surrounding the active site (Fig. 4D). These conformational changes aid in snugly fitting the tail into the active site cleft of the neighboring monomer. The backbones of the loops undergo a movement of several Å, e.g. the Cα atom of Arg-78 of loop 1 moves by 3.01 Å, and the Cα atom of Ser-100 of loop 2 moves by 4.13 Å. Moreover, side chains of several residues in these two loops, most notably those of Thr-76, Arg-78, and Lys-102, change conformations upon the movement of the loops. The Cδ atom of Arg-78 moves by 5.66 Å.
In all structures of full-length E. coli KdsC reported here, we observe one or the other conformation of the tail and the two loops. The loops are invariably in the closed conformation when the tail occludes the active site and in the open conformation when the tail is disordered. No intermediate conformation of the tail or the loops is observed in any of the structures. Because different tetramer packing arrangements are observed in different crystal forms, with most forms containing more than one tetramer per asymmetric unit, we conclude that the open-closed conformational transition is not a crystal packing artifact.
Attempts to Capture Kdo8P in the Active Site
In an effort to obtain a structure of the KdsC tetramer bound to the substrate or products, we performed co-crystallization of KdsC in the presence of a large excess of Kdo8P at room temperature and at 4 °C. In these experiments, in addition to Mg2+, we tested Ca2+ as a cofactor because Ca2+ does not support efficient catalytic turnover (12). In a separate experiment, to slow down catalytic turnover, crystals of KdsC were soaked in a large excess of Kdo8P for 30 s to 5 min at a low temperature (4 °C) and then flash frozen. Unfortunately, neither Kdo8P nor Kdo were trapped in the crystals of KdsC in any of these experiments, presumably because of the turnover, even at these conditions. In tetramers of full-length KdsC, crystallized in the presence Mg2+ and then soaked with Kdo8P at 4 °C (Fig. 3, structure c), the four monomers adopt alternating closed and open conformations. Although Mg2+ is bound to all four active sites, Cl− and the tail are bound only in the two sites in the closed conformation.
In co-crystals of KdsC with Kdo8P and Ca2+, all four active sites of tetramers are in the closed conformation and contain a phosphate product, in place of a chloride (Fig. 3, structure e). The phosphate product appears to be trapped in the active site by the tail. In KdsC crystals that were grown in the presence of Ca2+ but not soaked with Kdo8P, a Cl− instead of the phosphate was bound in all four active sites of each tetramer (Fig. 3, structure f). All four active sites were in the closed conformation, as expected.
To explore a possible mechanistic role of the C-terminal tail, we expressed and purified a mutant of KdsC (KdsCΔ8) lacking the eight C-terminal residues. Co-crystallization of KdsCΔ8 with substrate Kdo8P was carried out in the presence of Mg2+ or Ca2+. In a tetramer of KdsCΔ8 crystallized in the presence of Mg2+ and Kdo8P, the active sites are observed in two different states (Fig. 3, structure d). Mg2+ is coordinated to the aspartate triad in all four active sites. Whereas all four sites also contain a bound Pi product, only two sites contain the other product, Kdo. The presence of Kdo moiety is apparent in the |Fo − Fc| electron density map generated without ligands in the active site (Fig. 6A). The quality of this electron density map alone is not sufficiently high to resolve the orientation of the sugar unambiguously, due to some movement of the Kdo in the active site or to the moderate resolution (2.5 Å) of the data. However, the position of Kdo was derived with a high degree of certainty from both the electron density map and considerations of chemical bonding and catalysis (Fig. 6B). Notably, the distance between the phosphorus and the O8 of Kdo is greater than 2.2 Å, indicating that the hydrolysis of Kdo8P has occurred. The phosphate occupies the same site as the Cl− or the phosphate in other structures discussed above. The oxygen atoms on the phosphate product form bonds with the Mg2+, the side chains of Lys-102 and Thr-76, and the main chain amide of Gly-77 (Fig. 6B). These phosphate-interacting residues are located in loops I and II. In addition, the phosphate forms a hydrogen bond with the main chain nitrogen of Val-33.
The Kdo product is locked in the active site by an extensive set of specific interactions with active site residues. These residues belong both to the monomer that contributes the catalytic nucleophile and to an adjacent monomer in the tetramer. All of Kdo moieties are engaged in interactions with the protein, as shown in Fig. 6B. The carboxylate of Kdo forms a completely buried salt bridge with a universally conserved Arg-86 and a hydrogen bond with the main chain nitrogen of Val-56. Both Arg-86 and Val-56 belong to the adjacent monomer. The O4 and O5 of Kdo interact with the guanidinium group of the universally conserved Arg-78 (in loop I), which is precisely positioned with the aid of Glu-50. The O2 of Kdo interacts with the side chain of Arg-63 of the adjacent monomer (an Arg or a Lys in other bacteria) and the O4 forms a water-mediated hydrogen bond with the side chain hydroxyl of Thr-89. The side chain of Asp-34 forms a hydrogen bond with the C8 hydroxyl of Kdo generated upon the Pi cleavage. The side chains of Val-56 and Leu-90 of the adjacent monomer, which are solvent-exposed in the unbound structure, contact the C8 and C3 carbons of Kdo, respectively.
The interactions of Kdo in the active site resemble the interactions of the bound tail in the closed conformation of the full-length KdsC. In the tail-bound KdsC, the C-terminal carboxyl of KdsC (Ile-188) forms a salt bridge with Arg-86, and the hydroxyl of Ser-187 reaches into the catalytic pocket to make a water-mediated hydrogen bond with Mg2+ (Fig. 7A). The backbone and the side chain positions of Ser-187 and Ile-188 are partially isosteric with bound Kdo. Specifically, the Kdo carboxyl in the product-bound active site occupies a position similar to that of the C-terminal carboxyl of the tail in the closed active site, both of which are engaged in a salt bridge with Arg-86 (Fig. 7B).
FIGURE 7.

A, a close-up view of the tail-bound active site (the closed conformation). The active site residues marked in green belong to the neighboring monomer in A and B. The tail is shown in magenta, and the terminal carboxyl is shown in black. B, the structure of the product-bound active site of KdsC. The Kdo is in blue, and the Pi is in light green.
The Protein Conformational Changes Induced by Binding of Kdo
As we have demonstrated, loops I and II contain residues responsible for substrate specificity. The conformations of these specificity loops in the Kdo-bound active site of KdsCΔ8 (Fig. 7B) are practically identical to those in the closed (tail- and anion-bound) active site of the full-length KdsC (Fig. 7A), as defined above. As a consequence of the large concerted movements of loops I and II to ensure Kdo recognition, solvent gets excluded from the products almost completely (by 91.5% relative to unbound Kdo).
Effects of Deleting the C-terminal Tail and Changing Salt Concentration on Catalysis
As demonstrated above, the C-terminal residues specifically interact with residues of the active site pocket in the tail-bound state, thereby partially occluding the active site. In addition, in the closed state, the active site pocket is invariably occupied by either a phosphate product or its anionic in vitro surrogate chloride. These two observations suggest a potential role of the tail in the catalytic mechanism. To test this hypothesis, we measured the steady-state rate of phosphatase activity of KdsC (Fig. 8). Indeed, the observed rate constant of steady-state Kdo8P phosphatase turnover (kcatobs) for KdsCΔ8 is 100-fold smaller than that for full-length KdsC at 200 mm NaCl (Table 3). It is also observed that the maximum rate of hydrolysis of KdsCΔ8 is independent of salt concentration, whereas that of full-length KdsC increases with decreasing concentration of NaCl (Fig. 9). The effects of tail deletion both on the magnitude and the salt concentration dependence of the kinetics suggest that the tail is likely to be involved in the rate-limiting step during turnover. The full-length KdsC and KdsCΔ8 exhibit similar Km values that do not depend significantly on salt concentration (Table 3), indicating that substrate binding is not affected by deletion of the tail. This observation argues against the mechanistic role of the tail in the substrate binding step.
FIGURE 8.

The dependence of the phosphatase rate (V/[KdsC]) of full-length E. coli KdsC on substrate concentration ([Kdo8P]) at [NaCl] = 0 mm (circles) and [NaCl] = 200 mm (squares). Error bars represent the S.D. of three independent measurements. The theoretical curves represent the best nonlinear regression fit of the data to the Michaelis-Menten rate law (see Equation 1 under “Experimental Procedures”) with the values of kcat and Km given in Table 3.
TABLE 3.
Kinetic parameters of Kdo8P hydrolysis by Escherichia coli KdsC
| Protein | Km | kcat | kcat/Km |
|---|---|---|---|
| μm | s−1 | μm−1s−1 | |
| E. coli KdsC (no NaCl) | 71.5 ± 9.4 | 456 ± 17 | 6.37 |
| E. coli KdsCΔ8 | 99.4 ± 8.9 | 2.66 ± 0.08 | 0.0268 |
| E. coli KdsC (200 mm NaCl) | 147.8 ± 4.8 | 145 ± 2 | 0.98 |
FIGURE 9.

The dependence of the relative phosphatase rate, V, (relative to the value of V measured at [NaCl] = 0 mm, V[NaCl] = 0) on salt concentration for full-length E. coli KdsC (black bars) and KdsCΔ8 (white bars). The rate in all cases is measured at [Kdo8P] = 1 mm ≫ Km, i.e. V closely approximates Vmax.
DISCUSSION
In this study, we investigated the enigmatic origins of substrate specificity and catalytic efficiency of KdsC. The position of the divalent cation in the structures of E. coli KdsC reported here and in the earlier reported structure of a KdsC homolog (YrbI) from H. influenzae (16) indicates the general location of the phosphatase active site. However, the position of substrate Kdo8P in the active site has not been clear. Deleting the eight C-terminal residues of KdsC enabled us to capture both Kdo and Pi products in the active site, likely as a result of slow product release of this deletion mutant. This product-bound KdsCΔ8 structure reveals structural features that explain the role of tetramerization in defining substrate specificity. Substrate recognition in KdsC is achieved not only by the monomer containing the catalytic aspartate residue but also by its neighboring monomer. In contrast, the substrate specificity in other families of the haloacid dehalogenase superfamily is ensured by large rigid body movements of the cap domain that seal off the substrate in the active site and provide specificity determinants located on the cap itself (1). A tetrameric organization similar to that of KdsC was recently reported for a moderately homologous and functionally divergent HADSF phosphatase KDN-9-P (BT1713) from Bacteroides thetaiotaomicron (11).
The active site cleft of KdsC is lined with mostly polar and charged residues, which dictate the specificity of KdsC to its hydrophilic substrate Kdo8P. All accessible moieties of Kdo are engaged in specific interactions with residues in the active site pocket (Fig. 6B). Interestingly, two well conserved hydrophobic residues, Val-56 and Leu-90 of an adjacent monomer (Fig. 2), that are solvent-exposed in the unbound structures of KdsC form hydrophobic interactions with the C-8 and C-3 methylene groups of Kdo, respectively. Another interaction with the adjacent monomer that appears to define the substrate specificity is a salt bridge between the Kdo carboxyl and the guanidinium group of Arg-86. A mutation of the homologous Arg-64 to an alanine in BT1713, where it forms a structurally similar salt bridge with the carboxyl of N-acetylneuraminate, abolished the phosphatase activity of BT1713 (11).
The comparison of the structures of KdsC tetramers, in all crystal forms, revealed concerted conformational changes of two regions of the protein: the C-terminal tail and loops I/II surrounding the active site. The flexible C-terminal tail undergoes a dramatic conformational change between a disordered state, in which the tail is not observed in the electron density, and a defined conformation bound in the active site (also occupied by an anion) of a neighboring monomer. Strictly speaking, this disordered state of the tail is an ensemble of states in which the tail adopts several or a continuum of conformations. Thus, in this paper, the term “open conformation” also implies this conformational ensemble. The tail docking and product binding appear to drive similar movements of loops I and II that contain several conserved residues involved in recognition of Kdo8P. One of these residues, Arg-78 located in loop I (Fig. 7), also provides affinity for the tail presumably to displace the Kdo product upon completion of catalysis.
The crystal structure of KdsC soaked with Kdo8P in presence of Ca2+ (Fig. 3, structure e) revealed the positions of the Pi 4.4 Å away from the catalytic Mg2+. The similarity in the coordination of Pi and that of the sulfate in the H. influenzae KdsC (YrbI) structure (16) lends support to structural conservation of residues mediating the Pi and divalent metal coordination in the KdsC family. Indeed, Lys-102 is universally conserved, Thr-76 is highly conserved (with a few exceptions of a functionally similar Ser), and Gly-77 is replaced with a Ser or an Ala only in a few bacterial species (Fig. 2). These three conserved residues are all located in flexible loops I and II. In contrast, the Mg2+-coordinating triad of aspartate residues (Asp-32, -34, and -125) exhibit very little or no movement, thus preserving the geometry required for catalysis. The side chain of Asp-32, the conserved nucleophile in the HADSF, appears to alternate between two rotamers, as judged from the partial occupancy of the two rotamers in electron density in the product-bound KdsCΔ8 structure (data not shown). This may reflect destabilization of the catalytic center to allow product dissociation.
Several structural and biochemical observations strongly indicate an unexpected regulatory role of the C-terminal region of KdsC, likely in Kdo product release. The products are almost fully excluded from solvent and hence from possible interactions with the tail due to the active site closure. Therefore, the tail is unlikely to play an essential role in the covalent bond breaking step of the mechanism. Our steady-state kinetic analysis demonstrates that the enzymatic turnover of the KdsCΔ8 is much slower than that of full-length KdsC, whereas Km is similar for these two proteins. These observations indicate the tail is unlikely to play a role in the substrate binding step either. Furthermore, Vmax of the full-length protein is dependent on salt concentration, whereas that of KdsCΔ8 is not. This observation strongly suggests that the tail is involved in the rate-limiting step of the turnover. This rate-limiting step in the enzymatic turnover (with rate constant kcatobs) must involve solvation/desolvation of the tail upon the tail's conformational change. One plausible and simple mechanistic interpretation of these data is that the desolvation of the tail occurs as the tail binds into the active site to release the Kdo product. During the initial stages of tail binding, the C-terminal carboxyl may compete with the Kdo carboxyl for forming the salt bridge with Arg-86, thereby weakening the association of the Kdo product with the active site. Once Kdo is displaced, the tail stays transiently bound in the active site, which is still occupied by Pi. The slow release of Pi in the presence of Ca2+ could explain the previously observed dramatic loss of catalytic efficiency when Ca2+ is used instead of Mg2+ (12).
The multiple crystal structures of the KdsC-ligand complexes (Fig. 3) mimic various conformational intermediates of the KdsC reaction pathway (Fig. 10). Together with biochemical observations, these structures strongly suggest a mechanistic model, in which the likely slow Kdo release, driven by the C-terminal tail, is followed by Pi release. Finally, the tail docking was observed to occur in a pairwise manner, where a diagonally opposed pair of active sites switches between the open and closed conformations in a concerted manner. Such pairwise occurrence of different states is seen in all crystal forms (Fig. 3) and therefore unlikely to be a crystal packing artifact. Moreover, in the KdsCΔ8-product structure (Fig. 3, structure d), both Kdo and Pi are bound in two diagonal sites, whereas only Pi is bound in the other two sites. Therefore, the mechanistic steps are likely to occur in a pairwise fashion (Fig. 10). The reason for this pairwise transition is not known at this time.
FIGURE 10.

The proposed mechanism of the enzymatic turnover of KdsC. The letters a–g correspond to the structures in Fig. 3, which serve as models of the respective states. The Mg2+ and Cl− ions are shown as the red and orange circles, respectively. Based on structures a and b in Fig. 3 that are obtained from the same crystal, we propose an equilibrium between the open and the closed (the tail-/Cl−-bound) states of the active site, depicted in the shaded rectangle.
The combination of the structural and biochemical observations reported herein reveals the origin of substrate specificity and strongly suggests a series of conformational changes that drive the catalytic cycle of KdsC. The C-terminal tail of an adjacent monomer performs an important mechanistic function in ensuring the catalytic efficiency at the active site likely through facilitating the product release. Both substrate specificity and catalytic efficiency of KdsC are ultimate consequences of the tetramerization of KdsC, which occurs through a short β-hairpin insertion found in place of the missing cap domain present in other HAD enzymes.
Acknowledgments
We thank Dr. Spencer Anderson and the staff of sector Life Sciences Collaborative Access Team at the Advanced Photon Source of the Argonne National Laboratories for assistance with collection of the diffraction data. We also thank Brett Ehrmann for assistance with structure analysis.
This work was supported, in whole or in part, by National Institutes of Health Grant AI61531 (to R. W. W.). This work was also supported by the start-up funds from the College of Pharmacy, University of Michigan (to O. V. T.).
The atomic coordinates and structure factors (codes 3HYC, 3I6B, 2R8E, 2R8X, 2R8Y, and 2R8Z) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
H. Yang, I. Chen, Y. Agari, A. Ebihara, A. Shinkai, S. Kuramitsu, S. Yokoyama, J. P. Rose, and B. C. Wang, unpublished data.
- HADSF
- haloacid dehalogenase superfamily
- Kdo8P
- 3-deoxy-d-manno-octulosonate 8-phosphate.
REFERENCES
- 1.Allen K. N., Dunaway-Mariano D. (2004) Trends Biochem. Sci. 29, 495–503 [DOI] [PubMed] [Google Scholar]
- 2.Lahiri S. D., Zhang G., Dunaway-Mariano D., Allen K. N. (2002) Biochemistry 41, 8351–8359 [DOI] [PubMed] [Google Scholar]
- 3.Toyoshima C., Nakasako M., Nomura H., Ogawa H. (2000) Nature 405, 647–655 [DOI] [PubMed] [Google Scholar]
- 4.Diaz A. R., Stephenson S., Green J. M., Levdikov V. M., Wilkinson A. J., Perego M. (2008) J. Biol. Chem. 283, 2962–2972 [DOI] [PubMed] [Google Scholar]
- 5.Kim Y., Gentry M. S., Harris T. E., Wiley S. E., Lawrence J. C., Jr., Dixon J. E. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 6596–6601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghosh A., Shuman S., Lima C. D. (2008) Mol. Cell 32, 478–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deshpande R. A., Wilson T. E. (2004) Biochemistry 43, 8579–8589 [DOI] [PubMed] [Google Scholar]
- 8.Cho H., Wang W., Kim R., Yokota H., Damo S., Kim S. H., Wemmer D., Kustu S., Yan D. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 8525–8530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lahiri S. D., Zhang G., Dai J., Dunaway-Mariano D., Allen K. N. (2004) Biochemistry 43, 2812–2820 [DOI] [PubMed] [Google Scholar]
- 10.Burroughs A. M., Allen K. N., Dunaway-Mariano D., Aravind L. (2006) J. Mol. Biol. 361, 1003–1034 [DOI] [PubMed] [Google Scholar]
- 11.Lu Z., Wang L., Dunaway-Mariano D., Allen K. N. (2009) J. Biol. Chem. 284, 1224–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu J., Woodard R. W. (2003) J. Biol. Chem. 278, 18117–18123 [DOI] [PubMed] [Google Scholar]
- 13.Kuznetsova E., Proudfoot M., Gonzalez C. F., Brown G., Omelchenko M. V., Borozan I., Carmel L., Wolf Y. I., Mori H., Savchenko A. V., Arrowsmith C. H., Koonin E. V., Edwards A. M., Yakunin A. F. (2006) J. Biol. Chem. 281, 36149–36161 [DOI] [PubMed] [Google Scholar]
- 14.Raetz C. R., Whitfield C. (2002) Annu. Rev. Biochem. 71, 635–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen H. P., Seto N. O., MacKenzie C. R., Brade L., Kosma P., Brade H., Evans S. V. (2003) Nat. Struct. Biol. 10, 1019–1025 [DOI] [PubMed] [Google Scholar]
- 16.Parsons J. F., Lim K., Tempczyk A., Krajewski W., Eisenstein E., Herzberg O. (2002) Proteins 46, 393–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 18.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) J. Appl. Cryst. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 20.Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 21.Tsodikov O. V., Record M. T., Jr., Sergeev Y. V. (2002) J. Comput. Chem. 23, 600–609 [DOI] [PubMed] [Google Scholar]
- 22.Deleted in proof