

Abstract
Multidrug (Mdr) transporters are membrane proteins that actively export structurally dissimilar drugs from the cell, thereby rendering the cell resistant to toxic compounds. Similar to substrate-specific transporters, Mdr transporters also undergo substrate-induced conformational changes. However, the mechanism by which a variety of dissimilar substrates are able to induce similar transport-compatible conformational responses in a single transporter remains unclear. To address this major aspect of Mdr transport, we studied the conformational behavior of the Escherichia coli Mdr transporter MdfA. Our results show that indeed, different substrates induce similar conformational changes in the transporter. Intriguingly, in addition, we observed that compounds other than substrates are able to confer similar conformational changes when covalently attached at the putative Mdr recognition pocket of MdfA. Taken together, the results suggest that the Mdr-binding pocket of MdfA is conformationally sensitive. We speculate that the same conformational switch that usually drives active transport is triggered promiscuously by merely occupying the Mdr-binding site.
Introduction
Mdr2 transporters are membrane proteins that expel a wide spectrum of chemically dissimilar drugs from the cell, thereby rendering it resistant to multiple drugs. They exist in all kingdoms of life and constitute a major mechanism underlying bacterial resistance to antibiotics and cancer resistance to chemotherapy. In addition to their clinical importance, Mdr transporters pose intriguing biochemical questions because of their multispecificity and their capacity for catalyzing coupled transport reactions with an extraordinarily broad spectrum of substrates.
Mdr transporters exist in many families of transport proteins that utilize various transport mechanisms (1, 2). Interestingly, however, Mdr transporters from different families share many similar mechanistic features (3, 4), suggesting that certain aspects of multispecific transport by transporters having different structures and families are similar. The research of recent years has shed light on the major biochemical properties of Mdr transporters. These transporters contain large substrate-binding pockets and can extract their substrates from the cytoplasm and/or the membrane. Additionally, it appears that hydrophobic and electrostatic interactions underlie multispecific drug binding (reviewed in Ref. 3). Nevertheless, the mechanism underlying transport and coupling remains to be elucidated.
Transporters function by alternating between conformational states; for efficient coupling the transporter must be able to conformationally respond to substrate binding. In the case of Mdr transporters, this situation is intriguing because they should be able to produce one or more transport-competent conformational responses that fit a variety of chemically and structurally unrelated substrates. Do all substrates induce the same conformational change? Or do dissimilar substrates induce different conformational changes, all of which facilitate transport? Although it was shown that substrate binding indeed induces conformational changes in Mdr transporters (5, 6), it is not understood how a single transporter can be “conformationally responsive” to the binding of a diverse group of compounds, such that all of them induce structural rearrangements in the protein that facilitate transport.
Here we studied this question by utilizing several approaches for detecting conformational changes in the Escherichia coli Mdr transporter MdfA, which serves as a model of secondary Mdr transport (7). MdfA is a member of the major facilitator superfamily, which constitutes the largest family of transporters (8) and is the most prominent family of bacterial drug transporters, many of which function as drug/H+ antiporters (3). The study reported here suggests that dissimilar substrates indeed induce similar conformational changes in MdfA. Additionally, we show that even nonsubstrate compounds can induce related conformational changes, provided that they are forced to bind at the putative Mdr recognition pocket of MdfA. Thus, the conformational changes are generated promiscuously. We conclude that MdfA has a sensitive conformational switch that can be triggered either by substrate binding or by attaching unrelated agents inside the pocket.
MATERIALS AND METHODS
Plasmids
Plasmids for overexpression of MdfA or various single cysteine mutants have been previously described (6, 9). pT7–5/Para/mdfA6HIS encoding the single cysteine mutant G410C was generated utilizing a standard PCR method with mutagenic oligonucleotide primers and a plasmid encoding Cys-less MdfA as a template. The plasmid was sequenced to verify that only the desired mutation was inserted.
Overexpression of MdfA and Preparation of Membranes
T&GE. coli cells harboring plasmid pUC18/Para/mdfA6HIS or pT7–5/Para/mdfA6HIS were grown at 37 °C in LB medium supplemented with ampicillin (200 μg/ml). Overnight cultures were diluted to 0.07 A600 units and grown to 1.0 A600 units, and the culture was then induced with 0.2% arabinose for 1 h. A typical 10-liter culture yielded ∼15 g (wet weight) of cells. Cell pellets were washed once in 150 ml of 50 mm potassium Pi (pH 7.3) supplemented with 2 mm MgSO4, and collected by centrifugation (15 min, 5,000 × g). Next, the cells were suspended in 90 ml of the same buffer containing 10 μg/ml DNase and 1 mm PMSF and passed three times through a liquidizer (Emulsiflex-C5; Avestin) (10,000 p.s.i.) for disruption. Cell debris was removed by centrifugation (30 min, 8,000 × g), and the membranes were collected by ultracentrifugation (1 h, 100,000 × g). The membranes were homogenized in 27 ml of urea buffer (20 mm Tris-HCl, pH 8, 0.5 m NaCl, 5 m urea, 10% glycerol, 28 mm β-mercaptoethanol, and 1 mm PMSF), incubated for 30 min at 4 °C, and collected by ultracentrifugation (2.5 h, 100,000 × g). The membranes were washed with 27 ml of membrane buffer (20 mm Tris-HCl, pH 8, 0.5 m NaCl, 10% glycerol, and 3.5 mm β-mercaptoethanol). Finally, the membranes were suspended by homogenization in 7 ml of the same buffer, and aliquots of 1 ml were snap-frozen in liquid nitrogen and stored at −80 °C.
Modification of Single Cysteine Mutants by Sulfhydryl Reagents
The membranes were thawed quickly at 37 °C and transferred to ice, collected by ultracentrifugation (1 h, 100,000 × g), resuspended, and homogenized in an equal volume of reaction buffer (20 mm Tris-HCl, pH 7, 0.5 m NaCl, 10% glycerol). Sulfhydryl reagents (final concentration, 2 mm) were added, and the suspension was sonicated (twice for 10 s using a probe sonicator) to allow distribution of the reagent on both sides of the membrane. The reaction was completed overnight by tilting at 4 °C. When light-sensitive reagents were used, the reaction and subsequent steps of the experiments were done in the dark to minimize bleaching.
Membrane Solubilization and MdfA Purification
The membranes were diluted to 2.7 ml in solubilization buffer (20 mm Tris-HCl, pH 8, 0.5 m NaCl, 10% glycerol, 0.1% β-dodecyl maltopyranoside (DDM), 5 mm imidazole) and solubilized by the addition of 0.33 ml of 10% DDM (final concentration, 1.1%). Insoluble material was discarded by ultracentrifugation (30 min, 100,000 × g), and the soluble fraction was mixed with solubilization buffer-equilibrated Talon beads (Clontech) (0.25 ml). Next, the mixture was agitated for 3 h at 4 °C, and the suspension was poured into a column. The column was then washed (2 × 2 ml of solubilization buffer). MdfA was eluted in 0.75 ml of elution buffer (20 mm Tris-HCl, pH 7.2, 0.12 m NaCl, 10% glycerol, 0.1% DDM, 100 mm imidazole) after initial discard of 0.125 ml. The protein was then dialyzed overnight against dialysis buffer (20 mm Tris-HCl, pH 7.2, 0.12 m NaCl, 10% glycerol, 0.01% DDM) at 4 °C. The protein concentration was determined spectrophotometrically by measuring A280.
Assessing the Degree of Chemical Modification Utilizing Maleimide-Polyethyleneglycol (Mal-PEG)
The proteins were denatured by 1% SDS to assist the exposure of cysteine residues. To test the degree of free cysteines, the protein was allowed to react with Mal-PEG (5000 kDa) for 2–3 h at room temperature. The products were analyzed by SDS-PAGE (12.5%), followed by Coomassie staining. The Mal-PEG-MdfA adduct appeared ∼20 kDa heavier than MdfA or MdfA modified by any of the test sulfhydryl reagents.
Binding of [3H]TPP
The binding assays were performed essentially as described (9) with the following modifications. Purified protein (typically 30 μg) was mixed with nickel-nitrilotriacetic acid beads (150 μl) in 5 ml of buffer A (20 mm Tris-HCl, pH 8, 0.5 m NaCl, 10% glycerol, 5 mm imidazole, 0.1% DDM) and gently agitated for 30 min at 4 °C. The unbound material (supernatant) was discarded by brief centrifugation (2 min, 700 × g). The beads were resuspended in 1.5 ml of buffer C (20 mm Tris-HCl, pH 7, 0.5 m NaCl, 0.1% DDM), divided into 100-μl aliquots, and mixed with 100 μl of substrate containing solutions (in buffer C) to yield a final concentration of 10 nm [3H]TPP (2 Ci/mmol) either in the presence or absence of 1 mm cold TPP or 0.5 mm chloramphenicol (Cm). The mixture was incubated by tilting for 10 min at 4 °C. 180 μl of the resin was then transferred to a Promega Wizard minicolumn on top of a microcentrifuge tube and centrifuged at 10,000 × g for 20 s. Unbound (flow-through) material was discarded, and the resin was resuspended in 100 μl of buffer C containing 350 mm imidazole. The radioactivity of this suspension was measured by liquid scintillation. The amount of [3H]TPP bound to the resin in the absence of MdfA was subtracted from all measurements. The amount of bead-bound protein was determined for each of the differently modified proteins for comparison, by analyzing the eluates by SDS-PAGE, Coomassie staining, and densitometry.
Fluorescence Measurements
For spectra measurements, MdfA was diluted to 1 μm in binding buffer (20 mm Tris-HCl, pH 7.2, 0.12 m NaCl, 10% glycerol, 0.1% DDM), transferred to a quartz cuvette, and preheated to 30 °C in the fluorimeter (Varian Cary Eclipse fluorescence spectrophotometer). The emission spectra were recorded using excitation wavelengths of 280 or 375 nm for tryptophan or mBBr fluorescence, respectively, in the presence or absence of 50 μm TPP. The inner filter effect introduced by the slight absorption of TPP was corrected as described (10).
For TPP binding experiments, MdfA was diluted in binding buffer to 0.1–0.25 μm. A prewarmed MdfA sample (2 ml at 30 °C) was continuously stirred in a quartz cuvette throughout the experiment. The fluorescence was measured continuously, and the sample was titrated by the addition of 10-μl TPP aliquots (in binding buffer) to yield the indicated final concentrations and incubated with TPP for 1 min, and the fluorescence was measured. Excitation/emission wavelengths for tryptophan and mBBr fluorescence were 280/340 and 375/460 nm, respectively. The TPP-induced inner filter effect was corrected in the tryptophan fluorescence measurements as described (10). The concentration of Cm was kept tightly constant to avoid correcting for Cm light absorption. The measurements were done in triplicate, and fluorescence of buffer/TPP solutions containing no MdfA was subtracted. The results were fitted to the following binding function using the nonlinear regression software LABfitTM,
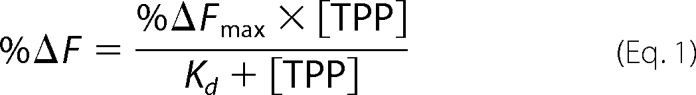 |
where %ΔF is the percentage of fluorescence change (quenching or increase), %ΔFmax represents %ΔF in a saturating TPP concentration, and Kd is the dissociation constant.
Limited Proteolysis of MdfA by Proteinase K (PK)
20 μl of MdfA (0.15 μg/ml in binding buffer) were mixed with 3 μl of the substrate to yield 50 μm TPP or 1 mm in the case of other substrates/compounds. To initiate proteolysis, 3 μl of 5 mg/ml PK (in 20 mm Tris-HCl, pH 8, 2 mm CaCl2) were added and mixed. Proteolysis was typically allowed to proceed for 10 min or several hours at 4 °C. The reaction was quenched by the addition of 2 μl of 200 mm PMSF followed by incubation at room temperature (10 min). The cleaved protein was analyzed by 12.5% SDS-PAGE and Coomassie staining.
RESULTS
Site-specific Labeling of MdfA by mBBr Stimulates Substrate Binding
We have previously employed a genetic screen to study how MdfA interacts with its substrates (6). This assay identified residues that potentially participate in Mdr recognition. When viewed on a three-dimensional model of the transporter (11), most of the genetically identified residues appear to line a central pathway in MdfA, proposed to constitute an Mdr recognition pocket (Fig. 1). Several of the genetically identified sites have been studied in more detail, and residue Ala147 was found important for interaction of the transporter with Cm (6). To investigate substrate binding further, we labeled the putative pocket by a fluorescence probe covalently bound at position 147, utilizing a functional Cys-less mutant with a single cysteine inserted at this position (MdfA-A147C). To this end, we used the cysteine-reactive fluorescent compound mBBr (12), which also proved useful for studying integral membrane proteins (13). Labeling was accomplished by incubating isolated membranes with mBBr, and the mBBr-MdfA adduct was purified (Fig. 2A). The efficiency of mBBr labeling was analyzed using a second sulfhydryl reagent, Mal-PEG, which increases the mass of the protein upon covalent binding. When mixed with the unlabeled MdfA-A147C mutant, Mal-PEG reacted with the protein and generated a single, higher molecular weight adduct, consistent with the presence of one reactive cysteine in the protein (Fig. 2A, lane 2). In contrast, when MdfA-A147C was prelabeled by mBBr, the reaction with Mal-PEG was blocked (Fig. 2A, lane 4), suggesting that the cysteine at position 147 was already occupied by mBBr. Thus, a large fraction of MdfA-A147C appears to be labeled by mBBr. To determine whether the modified protein is functional, we compared the substrate binding capabilities of the purified labeled and unlabeled MdfA-A147C. Surprisingly, we observed that the mBBr-labeled transporter binds more TPP (an MdfA substrate) than the unlabeled protein (Fig. 2B, gray bars). Because MdfA in our experiments is nearly pure (Fig. 2A), this indicates that the modification by mBBr stimulated TPP binding by MdfA.
FIGURE 1.
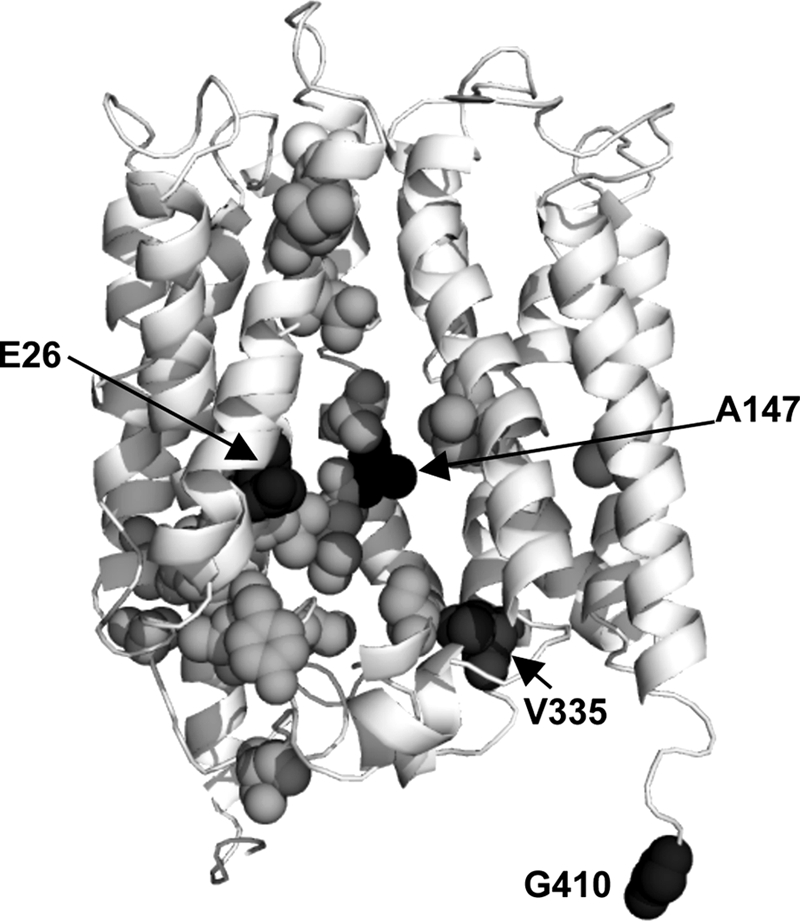
Putative substrate binding residues viewed on the three-dimensional model of MdfA. The ribbon representation of MdfA (11) shows Glu26, and those residues that were studied here at position 147, 335, and 410 (black spheres). The residues shown as gray spheres are putatively involved in substrate recognition by MdfA (6).
FIGURE 2.

Purification and TPP binding activity of mBBr-labeled MdfA-A147C. A, membranes from cells overexpressing MdfA-A147C were incubated in the presence or absence of mBBr. MdfA was purified to homogeneity and treated as described under “Materials and Methods” with (lanes 2 and 4) or without (lanes 1 and 3) Mal-PEG, separated by SDS-PAGE, and stained by Coomassie. Mal-PEG adduct is indicated by an arrow. B, [3H]TPP binding by mBBr labeled or unlabeled MdfA-A147C with or without a saturating concentration of Cm or excess of cold TPP.
Previous work on MdfA showed that it can interact with two substrates, Cm and TPP, simultaneously and that binding of Cm stimulates TPP binding to MdfA (9). To test the potential relevance to our results with the mBBr-labeled protein, we measured the effect of Cm on TPP binding by the purified, labeled, and unlabeled MdfA-A147C proteins. Indeed, as observed previously with wild-type MdfA, Cm also stimulated the binding of TPP to MdfA-A147C but to a lesser extent, possibly because cysteine in this position slightly impairs the interaction with chloramphenicol (6). In contrast, however, Cm had merely a minor effect on TPP binding to the mBBr-labeled protein (Fig. 2B). This slight effect might reflect stimulation of a small fraction of the protein that was not labeled by mBBr. The diminished effect of Cm on TPP binding indicates that the stimulatory potential of Cm had been exploited by mBBr labeling at this site. In addition, it is also likely that Cm binding was blocked by the covalently bound mBBr. Notably, the effects of mBBr and Cm were quantitatively similar (Fig. 2B), suggesting that mBBr labeling and Cm binding affect the transporter similarly (see below).
mBBr Mimics the Effect of Cm on MdfA Only When Covalently Bound at the Putative Mdr Recognition Pocket
As demonstrated previously, the stimulatory effect of Cm on TPP binding is due to an increased affinity of the binary complex MdfA-Cm for TPP (9). To further characterize and compare the effects of Cm and mBBr on MdfA-A147C, we measured the affinity for TPP by utilizing two fluorimetric assays. With the unlabeled MdfA-A147C, we used intrinsic fluorescence measurements, based on a reduction (of ∼10%) in fluorescence upon TPP binding (Fig. 3A). With the mBBr-labeled transporter, we utilized mBBr fluorescence, which increases (by ∼30%) upon TPP binding (Fig. 3C). Notably, by measuring mBBr fluorescence, we could specifically examine the labeled protein molecules because unlabeled molecules are invisible. The results of these studies show that both Cm binding and mBBr labeling increase the affinity for TPP by ∼2.5-fold and that Cm has no influence on the mBBr-labeled protein (Fig. 3, B and D), consistent with the results of direct binding experiments (Fig. 2B). Thus, affinity measurements imply that mBBr labeling affects MdfA by a mechanism similar to that of Cm binding. Because mBBr and Cm are structurally dissimilar (see Fig. 5A), it seems unlikely that the stimulation is mediated through direct interaction of Cm or mBBr with TPP. Instead, the increased TPP affinity probably results from a conformational change in MdfA that is induced by Cm binding or mBBr labeling.
FIGURE 3.

Comparison of the effects of mBBr and Cm on TPP binding to MdfA. A and C, effect of saturating TPP concentrations on the intrinsic fluorescence of unlabeled MdfA-A147C (A) or the mBBr fluorescence of the mBBr-MdfA-A147C (C). B, TPP binding by unlabeled MdfA-A147C, as measured by intrinsic fluorescence with and without Cm. D, TPP binding by mBBr-MdfA-A147C, as measured by mBBr fluorescence with and without Cm. The experiment was performed in triplicate, and the data were fitted to a binding equation using nonlinear regression software. The indicated dissociation constants were determined. E and F, direct measurements of [3H]TPP binding to Cys-less MdfA in the presence of increasing mBBr concentrations (E) and in the presence or absence of 500 μm Cm, mBBr, or excess cold TPP (F).
FIGURE 5.

Effects of various sulfhydryl reagents on Cys-less MdfA or MdfA-A147C. A, structures of Cm, TPP, and the tested sulfhydryl reagents shown as adducts. P-s indicates the sulfur atom of the cysteine residue of the protein. B, [3H]TPP binding by Cys-less MdfA in the presence of substrates or free sulfhydryl reagents. C, proteolysis of Cys-less MdfA by PK in the presence of substrates or free sulfhydryl reagents. The experiment was performed as described in the legend to Fig. 4 (for 24 h). D, modification of MdfA-A147C by the indicated sulfhydryl reagents and assessment by allowing all purified adducts to react with Mal-PEG (see the legend to Fig. 2A). E, [3H]TPP binding by MdfA-A147C either unmodified or modified by the indicated reagents with or without Cm or excess of cold TPP. F, proteolysis of unmodified or modified MdfA-A147C versions by PK in the absence or presence of Cm. 10-min time points indicate the fast part of the cleavage kinetics and are shown as controls.
The question of whether mBBr must be covalently bound to induce stimulation of TPP binding was investigated utilizing a functional Cys-less version of MdfA (6). Because this mutant does not contain cysteines, it cannot be labeled by mBBr. Fig. 3E shows that free mBBr did not stimulate TPP binding to Cys-less MdfA. In contrast, the free compound inhibited TPP binding, suggesting that mBBr and TPP might compete. Direct binding competition experiments revealed a kI of ∼34 μm for mBBr (Fig. 3E). Thus, for mBBr-induced stimulation of TPP binding to occur, mBBr must be covalently attached to residue A147C. Moreover, saturating the transporter with free mBBr did not prevent Cm from stimulating TPP binding to Cys-less MdfA (Fig. 3F), indicating that free mBBr interacts with a site that differs from position 147. Therefore, we conclude that the Cm-mimicking effect (Fig. 2B) is forced by covalent attachment of mBBr at position 147.
Substrate-induced Conformational Changes Measured by Limited Proteolysis
Previous studies (9) and those presented above suggest that MdfA responds to Cm or covalently bound mBBr by changing its conformation. To examine this notion further, we utilized a limited proteolysis approach in which detergent-solubilized MdfA was proteolyzed by PK in the absence or presence of substrates or covalent modifications. The PMSF-sensitive cleavage of Cys-less MdfA by PK proceeded through several steps (Fig. 4A). The initial cleavage was relatively rapid (completed by <10 min) and resulted in a ∼10% shorter protein, as judged by its mobility in SDS-PAGE (Fig. 4A, lane 3). This product was designated CF1 (cleavage fragment 1) (Fig. 4A). The cleavage event that followed was slower (hours) and generated a shorter proteolytic product, designated CF2 (Fig. 4A, lanes 4–11). The addition of the substrate TPP changed the proteolysis pattern by significantly increasing the amount of CF2, with no effect on the amount of CF1 (Fig. 4A, lane 5), indicating that TPP affects the availability of proteolytic sites for PK in CF2. This effect most likely results from a substrate-induced conformational change in MdfA (see “Discussion”). Kinetic trials showed that TPP increased the accumulation of CF2 without affecting the rate of CF1 degradation (Fig. 4B, compare lanes 1–7 with lanes 8–14), implying that TPP binding stabilizes CF2 against cleavage by PK. Notably, although both CF1 and CF2 were sometimes affected by substrates and chemical modifications, we focused on the behavior of CF2 because the appearance and disappearance of CF1 were less reproducible. To assess the specificity of this phenomenon, we tested how other substrates affect the proteolysis of MdfA by PK (Fig. 4A). The results show that Cm, ethidium, and tetracycline had similar but more moderate effects on the proteolysis compared with TPP (lanes 6–8). In contrast, compounds that are not substrates, such as lactose, spectinomycin, and nalidixic acid, did not have any effect compared with the control sample with no addition (compare lanes 9–11 with lane 4). Thus, the apparent stabilization of CF2 is mediated by various substrates that may influence the conformation of MdfA similarly.
FIGURE 4.

Proteolysis of MdfA by PK. A, 3 μg of Cys-less MdfA were incubated in the absence (lane 1) or presence of PK without or with various compounds. At the indicated times, PMSF was added to stop the reaction. CF1 and CF2 designate the cleavage fragments that were observed on Coomassie-stained SDS-PAGE. Time points at 0 and 10 min indicate the fast part of the cleavage kinetics and are shown as controls. B, kinetics of proteolysis of Cys-less MdfA in the absence (lanes 1–7) or presence (lanes 8–14) of TPP. At the indicated times, the reaction was stopped by PMSF. C, proteolysis of MdfA-A147C and mBBr-MdfA-A147C by PK in the absence or presence of TPP or Cm. D, proteolysis of Cys-less MdfA by PK in the absence or presence of TPP, Cm, or free mBBr.
Next, we tested the impact of TPP and Cm on the PK-catalyzed cleavage of mutant MdfA-A147C and a similar picture emerged, with both substrates stabilizing the CF2 form: TPP strongly and Cm moderately (Fig. 4C, compare lanes 3 and 4 with lane 2). Interestingly, however, when MdfA-A147C was labeled by mBBr, the covalent modification per se influenced CF2 cleavage just like substrates (Fig. 4C, lane 6), reinforcing the notion that mBBr labeling affects MdfA by inducing a substrate-like conformational change. The addition of Cm or TPP to mBBr-MdfA-A147C did not significantly improve the stability of CF2 (Fig. 4C, lanes 6–8). Notably, the effect of mBBr on the proteolysis was more similar to the effect of TPP compared with Cm. Taking into account the results of the TPP binding experiments (Figs. 2 and 3), we suggest that both Cm binding and the covalent attachment of mBBr at position 147 induce a high TPP affinity conformation in MdfA. The PK cleavage experiments suggest that the effect of Cm on MdfA-A147C is moderate compared with that of TPP or covalently bound mBBr, possibly reflecting a reduced affinity or response of this mutant to Cm. This finding is also in line with the reduced stimulation imposed by Cm on TPP binding to this mutant as compared with Cys-less MdfA (compare Fig. 2B with Fig. 3F), suggesting, as noted above, that the A147C mutation itself partially disrupts the interaction of MdfA with Cm (6).
In direct binding assays, we observed that free mBBr has an inhibitory effect on TPP binding to Cys-less MdfA (Fig. 3E), indicating that it might be a substrate and that in its free form it occupies a site that differs from 147. To test this notion further, we studied the effect of free mBBr on the proteolysis of the nonreactive Cys-less transporter. The results indicate that free mBBr also stabilizes the CF2 fragment against PK (Fig. 4D), lending support to the suggestion that it is a true substrate of MdfA.
The Substrate-like Conformational Response to Modification at Position 147 Is Not Restricted to mBBr
Our results suggest that forced attachment of mBBr in the putative Cm-binding site triggers a substrate-induced conformational change in MdfA. Can this effect also be reproduced with other reagents? To address this question, we characterized the conformation of MdfA-A147C after modification by various cysteine-reactive compounds. We chose six structurally diverse sulfhydryl reagents that do not resemble known MdfA substrates (Fig. 5A). None of the reagents significantly affected the TPP binding activity of the nonreactive Cys-less MdfA (Fig. 5B). Similarly, although the PK-produced fragment CF2 of Cys-less MdfA is slightly more stable than that of MdfA-A147C, no further stabilization was induced by the free sulfhydryl reagents compared with TPP- or the Cm-induced CF2 stabilization (Fig. 5C, compare lanes 4–9 with lanes 2 and 3). Next, the reagents were covalently bound to MdfA-A147C (Fig. 5D), and various effects of the modifications on TPP binding were clearly evident (Fig. 5E). NEM induced a Cm-like stimulation of [3H]TPP binding, demonstrating that such a conformational response to modifications at position 147 is not unique to mBBr, despite the structural difference between mBBr and NEM. In contrast, four of the other reagents (AMS, MTSES, MTSEA, and MTSMT) inhibited TPP binding, whereas the smallest reagent, IAA, did not have any effect. The results also clearly show that Cm did not stimulate TPP binding to any of the modified proteins (Fig. 5E). This is not surprising, in light of the suggestion that residue 147 is important for Cm recognition by MdfA and that modifications at this position by mutagenesis (6) or chemically (this study) might disrupt Cm recognition by the transporter.
The stimulatory effect of Cm and covalently bound mBBr or NEM on TPP binding suggests an allosteric mechanism, in agreement with the notion that residue 147 is not localized at the TPP-binding site. Therefore, the observed inhibitory effect of certain modifications (AMS, MTSES, MTSEA, and MTSMT, ∼50–80% decrease in TPP binding), at this position, on TPP binding to MdfA is somewhat puzzling (Fig. 5E). Previous studies demonstrated that positively charged substrates, such as TPP, form electrostatic interactions with the negatively charged Glu26 of MdfA (14, 15), pinpointing a probable binding site for TPP. In the structural model (Fig. 1), Glu26 and Ala147 are located relatively close to each other on opposite faces of the putative substrate recognition pocket, suggesting that the Cm and TPP-binding sites might not be very far apart. Accordingly, it is possible that certain modifications partially hinder TPP accessibility and thus prevent detecting possible conformational effects that might have been triggered by the modifications. Therefore, we investigated this question by limited proteolysis.
Fig. 5F shows the results of limited proteolysis of various adducts of MdfA-A147C, which can be classified into three groups. NEM and IAA had a moderate substrate-like stabilizing effect on CF2; MTSMT and MTSEA stabilized CF2 more significantly; MTSES and AMS had no effect on CF2 (Fig. 5F, lane 2 in each panel). Notably, unlike mBBr, which exhibited a strong TPP-like effect (Fig. 4C, lane 6), the effects of NEM, IAA, MTSMT, and MTSEA on proteolysis were quantitatively more similar to that of Cm (Fig. 5F, left panel, lane 3, compare with lane 2 in every the other panel). The addition of Cm could hardly further stabilize CF2 in these samples, in agreement with the diminished effect of Cm on TPP binding to these adducts (Fig. 5E). In conclusion, with the exception of AMS and MTSES, various dissimilar compounds are able to induce Cm-like conformational changes in MdfA when attached covalently to residue A147C. These results suggest that the Mdr recognition pocket of MdfA is conformationally sensitive to the presence of various compounds, such that once a molecule is bound, be it a real substrate or a synthetic covalent attachment, it might induce a substrate-like conformational change in the transporter. Therefore, the Cm-like conformational change in MdfA appears to be induced promiscuously.
Conformational Effects of Cysteine Modifications at Other Locations in MdfA
To test whether the proposed promiscuity in inducing conformational changes is restricted to modifications in a particular site in the recognition pocket of MdfA (position 147), we examined two other single cysteine mutants: (i) MdfA-V335C, in which the engineered cysteine is inside the putative substrate binding pocket, according to previous genetic and biochemical data (6, 16) and the structural model; and (ii) MdfA-G410C, containing a cysteine in the nonessential C terminus of MdfA (17), outside of the putative binding pocket (Fig. 1). Initially we examined whether the mutant proteins and their covalent adducts are functional. Direct [3H]TPP binding experiments showed that both mutants could bind TPP to various extents and were stimulated by Cm (Fig. 6A). Modification of the C-terminal residue G410C by NEM or MTSEA did not affect TPP binding or its stimulation by Cm, consistent with its proposed location in a nonessential part of the protein outside of the Mdr recognition pocket. Notably, however, mBBr covalently bound at residue G410C inhibited TPP binding to some extent, most likely because it is a substrate (Figs. 3E and 4D) and is somehow able to enter the recognition pocket even if attached to the C-terminal tail of MdfA. Experimental attempts to test this hypothesis are underway. In contrast to MdfA-G410C, modification of MdfA-V335C by either mBBr or MTSEA had a strong inhibitory effect on TPP binding, whereas NEM had a small stimulatory effect. Thus, it appears that modifying V335C affects the interactions of MdfA with TPP, in agreement with the proposed role of residue Val335 in substrate binding (6, 16). Interestingly, however, none of the modifications in residue V335C prevented the Cm-induced stimulation. Thus, unlike Ala147, residue Val335 does not appear to be critical for the interaction with Cm or for the conformational response to Cm, suggesting that Val335 represents another face of the drug-binding pocket.
FIGURE 6.

Effects of sulfhydryl reagents covalently bound to MdfA via residue V335C or G410C. MdfA constructs harboring a single cysteine at position 335 (V335C) or 410 (G410C) were modified by NEM, MTSEA, or mBBr or were left unmodified. The proteins were then purified and assayed as follows. A, [3H]TPP binding without or with Cm or excess of cold TPP, as indicated. B and C, proteolysis of unmodified or modified V335C-MdfA (B) and G410C-MdfA (C) by PK in the absence or presence of Cm or TPP (see the legend to Fig. 4). 10-min time points indicate the fast part of the cleavage kinetics and are shown as controls.
We then investigated how the substrates and the chemical modifications influence the conformation of MdfA-G410C and MdfA-V335C by utilizing the limited proteolysis assay. Almost all of the modified MdfA mutants responded to Cm and TPP, as judged by the stabilization of CF2 against proteolysis by PK (Fig. 6, B and C, lanes 3 and 4). A notable exception is the MTSEA-modified MdfA-V335C, which was impaired in TPP binding according to both the direct binding assay (Fig. 6A, left panel) and the limited proteolysis assay (Fig. 6B, third panel from the left, lane 4). This adduct, however, did respond to Cm, as shown both in the TPP binding assay where Cm stimulated binding (Fig. 6A, left panel) and in the limited proteolysis assay where Cm stabilized the CF2 fragment of MTSEA-modified MdfA-V335C (Fig. 6B, third panel from the left, lane 3). Additionally, we observed that modification of residue V335C by either mBBr or NEM affected the cleavage of MdfA by PK in a substrate-like manner, by stabilizing CF2 (Fig. 6B compare lane 2 in all panels), whereas modifying residue G410C did not have this effect (Fig. 6C, compare lane 2 in all panels). Thus, certain reagents attached to the putative recognition pocket (position 335) trigger a substrate-like conformational effect, whereas modifying a site outside the pocket (position 410) did not influence the conformation of MdfA. Overall, although the V335C site differs from A147C in its response to Cm binding, in both sites modification leads to a conformational change. Another property shared by these positions is their location in the substrate-binding pocket, as inferred from previous experiments showing that: (i) mutations in these positions modulate the activity of MdfA toward certain drugs and (ii) substrate binding protects MdfA-A147C and MdfA-V335C against modification by sulfhydryl reagents (6, 16).
In conclusion, the promiscuous induction of conformational changes is not restricted to modifications at position 147. We propose that the Mdr recognition pocket of MdfA is generally conformationally sensitive and that this may explain how many dissimilar substrates are able to induce similar conformational changes upon binding, a prerequisite for Mdr transport.
DISCUSSION
In this study, we utilized purified, detergent-solubilized MdfA to investigate how this secondary transporter responds conformationally to substrate binding. The results show that conformational changes in MdfA are generated promiscuously: (i) dissimilar substrates induce similar conformational changes in MdfA and (ii) reagents that are not chemically related to MdfA substrates induce substrate-like effects, provided that they are forced to bind at the putative Mdr recognition pocket of the transporter. A possible explanation for these phenomena is that MdfA has a sensitive conformational switch in the pocket that can be triggered by either substrate binding or chemical modifications.
Mdr transporters from various families were shown to contain multiple drug interaction sites, and these sites are often allosterically linked (18, 19). In MdfA, binding of Cm was shown to increase the binding affinity for TPP (9). In theory, such stimulation of binding could result either from direct interaction between simultaneously bound substrates or from substrate-induced conformational changes that influence the interaction of the transporter with a second substrate. Our studies show that a Cm-like stimulatory effect on TPP binding affinity can also be exerted by two Cm-unrelated compounds, mBBr and NEM, but only when covalently attached to MdfA-A147C. Considering the structural differences between these compounds and between them and Cm, a conformational change is a possible explanation for this effect, and this notion gains further support from proteolysis trials. In addition, these results suggest that Ala147 constitutes part of the Cm interaction site in MdfA, in agreement with previous genetic and biochemical studies (6). Indeed, the location of Ala147 in the three-dimensional structural model of MdfA (Fig. 1) suggests that it lines the putative Mdr recognition pocket.
Mdr transporters belong to structurally diverse families of transport proteins. This implies that perhaps the Mdr transport function evolved multiple times during evolution (20). Intriguingly, however, Mdr transporters from different families share similar characteristics (1). For example, the mechanism underlying Mdr binding is thought to be universal, mainly based on hydrophobic interaction with substrates (1, 3, 21). Additionally, transporters from different families were shown to extract substrates not only from the aqueous environment (22–24) but also directly from the membrane (18, 25, 26). Thus, Mdr transporters from various families cope with common challenges by adopting similar strategies (4). Better understanding of how Mdr transporters respond conformationally to dissimilar substrates in a transport-competent manner would add mechanistically important clues to how dissimilar compounds are actively exported by such transporters. For example, in a P-glycoprotein mutant that is defective in biogenesis, it has been reported that mutations in the binding site improve protein maturation and that this effect can also be achieved by substrates (27–32). Thus, binding site modifications also have substrate-mimicking effects in this ATP-binding cassette Mdr transporter. However, the question of whether promiscuously triggered conformational responses represent a phenomenon common to Mdr transporters in general remains to be investigated.
The two largest families of Mdr transporters (major facilitator superfamily and ATP-binding cassette) consist mainly substrate-specific transporters. This implies that the molecular and structural mechanisms underlying Mdr and substrate-specific transporters might be similar. One property likely to be shared by both Mdr and substrate-specific transporters is their ability to conformationally respond to substrate binding (5, 6, 33) for efficient coupling. This feature suggests that the multispecificity of Mdr transporters is reflected in their ability not only to bind dissimilar substrates but also to generate transport-competent conformational responses to these compounds. This question was partially addressed in this study.
Utilizing direct and indirect measurements of substrate binding and limited proteolysis, we could detect a substrate-induced change in MdfA, which leads to a conformation having high affinity for TPP and resistance to cleavage by PK. In principle, substrates can protect against proteolysis by directly masking potential cleavage sites. However, we favor the possibility that the effects described here are conferred by induced conformational changes for the following reasons: (i) different substrates that bind to different sites have similar effects on the proteolytic profile (Fig. 4A); (ii) modifications in two different locations of the putative Mdr recognition pocket of MdfA had a similar stabilizing effect against PK (Figs. 4C, 5F, and 6); and (iii) modification of a single cysteine can either have a stabilizing effect or not, depending on the attached compound (Figs. 5E and 6).
The precise role of the observed conformational change remains to be shown. Nevertheless, this change is probably of functional relevance because it is induced by various substrates of the transporter and not by compounds that are not substrates (unless covalently attached at the substrate-binding site). Substrate-induced conformational changes are critical for the transport mechanism, as shown for well characterized transporters (34, 35), and our results suggest that with MdfA, dissimilar substrates induce similar conformational changes and thus might share the same transport mechanism.
The observation that even nonsubstrate compounds can trigger a substrate-like conformational change when forced to bind to the putative Mdr recognition pocket of the transporter is intriguing. We propose that this phenomenon reflects an inherent property of Mdr transporters, which evolved in such a manner that enables them to respond to many structurally unrelated stimuli in the pocket. This property represents an additional level of multispecificity in MdfA (36) and suggests that functional conformational changes can be triggered promiscuously.
Acknowledgments
We thank Marija Jankovic and Philip Hu for help in the initial characterization of modified MdfA-A147C.
This work was supported in part by the Israel Cancer Research Foundation, the Minerva Foundation, and the Israel Science Foundation.
- Mdr
- multidrug
- PMSF
- phenylmethanesulfonyl fluoride
- DDM
- β-dodecyl maltopyranoside
- Mal-PEG
- maleimide-polyethyleneglycol
- Cm
- chloramphenicol
- PK
- proteinase K
- mBBr
- monobromobimane
- NEM
- N-ethylmaleimide
- AMS
- 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid
- IAA
- iodoaceteadime
- MTSEA
- 2-aminoethyl methanethiosulfonate
- MTSES
- 2-sulfonatoethyl methanethiosulfonate
- MTSMT
- (trimethylammonium)methyl methanethiosulfonate
- TPP
- tetraphenylphosphonium.
REFERENCES
- 1.Higgins C. F. (2007)Nature 446,749–757 [DOI] [PubMed] [Google Scholar]
- 2.Saier M. H., Jr., Paulsen I. T. (2001)Semin. Cell Dev. Biol. 12,205–213 [DOI] [PubMed] [Google Scholar]
- 3.Fluman N., Bibi E. (2009)Biochim. Biophys. Acta 1794,738–747 [DOI] [PubMed] [Google Scholar]
- 4.Venter H., Shahi S., Balakrishnan L., Velamakanni S., Bapna A., Woebking B., van Veen H. W. (2005)Biochem. Soc. Trans. 33,1008–1011 [DOI] [PubMed] [Google Scholar]
- 5.Ambudkar S. V., Lelong I. H., Zhang J., Cardarelli C. O., Gottesman M. M., Pastan I. (1992)Proc. Natl. Acad. Sci. U.S.A. 89,8472–8476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adler J., Bibi E. (2004)J. Biol. Chem. 279,8957–8965 [DOI] [PubMed] [Google Scholar]
- 7.Sigal N., Cohen-Karni D., Siemion S., Bibi E. (2006)J. Mol. Microbiol. Biotechnol. 11,308–317 [DOI] [PubMed] [Google Scholar]
- 8.Saier M. H., Jr., Beatty J. T., Goffeau A., Harley K. T., Heijne W. H., Huang S. C., Jack D. L., Jähn P. S., Lew K., Liu J., Pao S. S., Paulsen I. T., Tseng T. T., Virk P. S. (1999)J. Mol. Microbiol. Biotechnol. 1,257–279 [PubMed] [Google Scholar]
- 9.Lewinson O., Bibi E. (2001)Biochemistry 40,12612–12618 [DOI] [PubMed] [Google Scholar]
- 10.Kubista M., Sjoback R., Eriksson S., Albinsson B. (1994)Analyst 119,417–419 [Google Scholar]
- 11.Sigal N., Vardy E., Molshanski-Mor S., Eitan A., Pilpel Y., Schuldiner S., Bibi E. (2005)Biochemistry 44,14870–14880 [DOI] [PubMed] [Google Scholar]
- 12.Kosower E. M., Kosower N. S. (1995)Methods Enzymol. 251,133–148 [DOI] [PubMed] [Google Scholar]
- 13.Dunham T. D., Farrens D. L. (1999)J. Biol. Chem. 274,1683–1690 [DOI] [PubMed] [Google Scholar]
- 14.Adler J., Lewinson O., Bibi E. (2004)Biochemistry 43,518–525 [DOI] [PubMed] [Google Scholar]
- 15.Edgar R., Bibi E. (1999)EMBO J. 18,822–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adler J., Bibi E. (2005)J. Biol. Chem. 280,2721–2729 [DOI] [PubMed] [Google Scholar]
- 17.Adler J., Bibi E. (2002)J. Bacteriol. 184,3313–3320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitchell B. A., Paulsen I. T., Brown M. H., Skurray R. A. (1999)J. Biol. Chem. 274,3541–3548 [DOI] [PubMed] [Google Scholar]
- 19.Loo T. W., Bartlett M. C., Clarke D. M. (2003)J. Biol. Chem. 278,39706–39710 [DOI] [PubMed] [Google Scholar]
- 20.Neyfakh A. A. (2002)Mol. Microbiol. 44,1123–1130 [DOI] [PubMed] [Google Scholar]
- 21.Vazquez-Laslop N., Zheleznova E. E., Markham P. N., Brennan R. G., Neyfakh A. A. (2000)Biochem. Soc. Trans. 28,517–520 [PubMed] [Google Scholar]
- 22.Nagano K., Nikaido H. (2009)Proc. Natl. Acad. Sci. U.S.A. 106,5854–5858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cole S. P., Deeley R. G. (2006)Trends. Pharmacol. Sci. 27,438–446 [DOI] [PubMed] [Google Scholar]
- 24.Mao Q., Deeley R. G., Cole S. P. (2000)J. Biol. Chem. 275,34166–34172 [DOI] [PubMed] [Google Scholar]
- 25.Bolhuis H., van Veen H. W., Brands J. R., Putman M., Poolman B., Driessen A. J., Konings W. N. (1996)J. Biol. Chem. 271,24123–24128 [DOI] [PubMed] [Google Scholar]
- 26.Bolhuis H., van Veen H. W., Molenaar D., Poolman B., Driessen A. J., Konings W. N. (1996)EMBO J. 15,4239–4245 [PMC free article] [PubMed] [Google Scholar]
- 27.Loo T. W., Clarke D. M. (1997)J. Biol. Chem. 272,709–712 [DOI] [PubMed] [Google Scholar]
- 28.Loo T. W., Clarke D. M. (1998)J. Biol. Chem. 273,14671–14674 [DOI] [PubMed] [Google Scholar]
- 29.Loo T. W., Bartlett M. C., Clarke D. M. (2006)J. Biol. Chem. 281,29436–29440 [DOI] [PubMed] [Google Scholar]
- 30.Loo T. W., Bartlett M. C., Clarke D. M. (2007)J. Biol. Chem. 282,32043–32052 [DOI] [PubMed] [Google Scholar]
- 31.Loo T. W., Bartlett M. C., Clarke D. M. (2008)J. Biol. Chem. 283,24860–24870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loo T. W., Bartlett M. C., Clarke D. M. (2009)J. Biol. Chem. 284,24074–24087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smirnova I., Kasho V., Choe J. Y., Altenbach C., Hubbell W. L., Kaback H. R. (2007)Proc. Natl. Acad. Sci. U.S.A. 104,16504–16509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hollenstein K., Dawson R. J., Locher K. P. (2007)Curr. Opin. Struct. Biol. 17,412–418 [DOI] [PubMed] [Google Scholar]
- 35.Toyoshima C. (2008)Arch. Biochem. Biophys. 476,3–11 [DOI] [PubMed] [Google Scholar]
- 36.Edgar R., Bibi E. (1997)J. Bacteriol. 179,2274–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]