

Abstract
γ-Aminobutyric acid type A receptors (GABAARs) are the major sites of fast inhibitory neurotransmission in the brain, and the numbers of these receptors at the cell surface can determine the strength of GABAergic neurotransmission. Chronic changes in neuronal activity lead to an adaptive modulation in the efficacy of GABAergic synaptic inhibition, brought about in part by changes in the number of synaptic GABAARs, a mechanism known as homeostatic synaptic plasticity. Reduction in the number of GABAARs in response to prolonged neuronal activity blockade is dependent on the ubiquitin-proteasome system. The underlying biochemical pathways linking chronic activity blockade to proteasome-dependent degradation of GABAARs are unknown. Here, we show that chronic blockade of L-type voltage-gated calcium channels (VGCCs) with nifedipine decreases the number of GABAARs at synaptic sites but not the overall number of inhibitory synapses. In parallel, blockade of L-type VGCCs decreases the amplitude but not the frequency of miniature inhibitory postsynaptic currents or expression of the glutamic acid decarboxylase GAD65. We further reveal that the activation of L-type VGCCs regulates the turnover of newly translated GABAAR subunits in a mechanism dependent upon the activity of the proteasome and thus regulates GABAAR insertion into the plasma membrane. Together, these observations suggest that activation of L-type VGCCs can regulate the abundance of synaptic GABAARs and the efficacy of synaptic inhibition, revealing a potential mechanism underlying the homeostatic adaptation of fast GABAergic inhibition to prolonged changes in activity.
Introduction
γ-Aminobutyric acid type A receptors (GABAARs),2 the major sites of action for both benzodiazepines and barbiturates, are Cl−-selective ligand-gated ion channels that can be assembled from seven subunit classes (α1–6, β1–3, γ1–3, δ, ϵ, π, and θ), providing the structural basis for extensive heterogeneity of GABAAR structure (1–3). A combination of molecular, biochemical, and genetic approaches suggests that, in the brain, the majority of benzodiazepine receptor subtypes are composed of α, β, and γ2 subunits (1). γ2-containing receptors are highly enriched at synaptic sites in neurons and are responsible for mediating phasic inhibition (4–6). In contrast, receptors composed of α, β, and δ subunits are believed to form a specialized population of extrasynaptic receptors that mediate tonic inhibition (7). GABAARs are assembled within the endoplasmic reticulum (ER) and then transported to the plasma membrane for insertion, whereas misfolded or unassembled receptor subunits are rapidly targeted for ER-associated degradation (5, 6, 8, 9), a process that can be modulated by neuronal activity (9). The number of GABAARs on the neuronal cell surface is a critical determinant for the efficacy of synaptic inhibition and, at steady state, is determined by the rates of receptor insertion and removal from the plasma membrane (10, 11). Cell-surface GABAARs are dynamic entities that exhibit rapid rates of constitutive endocytosis, with internalized receptors being subject to rapid recycling or lysosomal degradation (5, 6, 10). Phosphorylation of the intracellular domains between transmembrane domains 3 and 4 of the GABAAR β and γ subunits by serine/threonine and tyrosine kinases has been shown to alter receptor function either by a direct effect on receptor properties, such as the probability of channel opening or desensitization, or by regulating trafficking of the receptor to and from the cell surface (11, 12).
The activity of neurons in neural circuits is highly regulated, and when firing frequency either falls below or rises above normal physiological levels, compensatory mechanisms come into play to restore normal activity, a process known as homeostatic plasticity (13, 14). Homeostatic synaptic scaling is one homeostatic mechanism that involves uniform adjustments in the strength of all synapses in response to changes in activity while maintaining the individual weights between synapses (15, 16). GABA regulates the excitability of neural circuits, and a number of studies in cultures of dissociated neurons have provided evidence that chronic changes in neuronal activity can lead to homeostatic scaling of GABAergic synaptic strength (9, 17–19), which appears to be dependent on brain-derived neurotrophic factor and tumor necrosis factor α (19, 20). There is now increasing evidence that homeostatic regulation of GABAergic synaptic strength is achieved by modulating the numbers of postsynaptic GABAARs (9, 17, 19). Furthermore, presynaptic changes underlying activity-dependent scaling of inhibitory synaptic strength have been reported in which the levels of the GABA-synthesizing enzyme GAD65, the GABA transporter, and presynaptic GABA content are modulated by activity (17, 18, 21). Currently, little is known about the mechanisms underlying the modulation of synaptic GABAAR abundance in response to chronic changes in neuronal activity. However, it has been recently established that prolonged alteration in neuronal activity modulates the ubiquitination and proteasomal degradation of GABAARs in the ER, resulting in changes in the number of receptors at synaptic sites and the efficacy of synaptic inhibition (9).
A number of important questions remain. For instance, how do chronic changes in activity translate into the homeostatic regulation of GABAAR abundance at synaptic sites? In this study, we have begun to investigate the role of Ca2+ influx through L-type voltage-gated calcium channels (VGCCs) in the modulation of activity-dependent expression of GABAARs and the efficacy of synaptic inhibition.
EXPERIMENTAL PROCEDURES
Antibodies
Rabbit anti-β3 IgG polyclonal antibodies have been described previously (22, 23). Rabbit anti-green fluorescent protein IgG, mouse anti-synapsin IgG, and rabbit anti-GAD65 antibodies were purchased from Synaptic Systems. Peroxidase-conjugated and fluorescent dye-conjugated IgG secondary antibodies were from Jackson ImmunoResearch Laboratories. Fluorescently labeled α-bungarotoxin (α-Bgt) was purchased from Invitrogen.
Neuronal Cell Culture and Transfections
Cultures of hippocampal neurons were prepared from embryonic day 18 rats (9, 22, 24). Dissociated embryonic day 18 rat cortical neurons were transfected with 3 μg of plasmid DNA/2 × 106 neurons using the rat neuron Nucleofector™ kit (Lonza). 60-mm dishes were seeded with 0.4 × 106 neurons and used in experiments after 18–21 days in vitro (DIV).
Biotinylation
Hippocampal neurons were chilled on ice for 5 min and then washed twice with phosphate-buffered saline (PBS) containing 1 mm CaCl2 and 0.5 mm MgCl2 (PBS-CM) at 4 °C. Cells were incubated for 15 min at 4 °C in 1 mg/ml sulfo-N-hydroxysuccinimide (NHS)-biotin (Pierce) dissolved in PBS-CM. To quench unreacted biotin, neurons were washed three times (10 min each wash at 4 °C) with PBS-CM + 75 mm glycine and then washed twice with PBS and lysed in radioimmune precipitation assay (RIPA) buffer (50 mm Tris (pH 8), 150 mm NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, 2 mm EDTA, and mammalian protease inhibitor mixture (Sigma)). Protein concentrations were determined using the micro BCA protein assay kit (Pierce), and equal amounts of solubilized protein were added to UltraLink-immobilized NeutrAvidin biotin-binding protein (Pierce) for 2 h at 4 °C. Avidin beads were washed twice for 15 min at 4 °C with high salt (500 mm NaCl) RIPA buffer, followed by a 15-min wash at 4 °C with RIPA buffer (150 mm NaCl). Precipitated biotinylated proteins and total proteins were resolved by SDS-PAGE, and β3 was detected by immunoblotting with rabbit anti-β3 IgG antibodies (9, 22, 25), followed by peroxidase-conjugated anti-rabbit IgG antibodies and detection with ECL. Blots were imaged using the Fujifilm LAS-3000 imaging system, and bands were quantified with Fujifilm Multi Gauge software.
Immunocytochemistry
Neurons expressing pHβ3 (where pH is pHluorin; 18–21 DIV) were fixed in 4% paraformaldehyde, stained without membrane permeabilization with rabbit anti-green fluorescent protein IgG antibodies, and then permeabilized with 0.1% Triton X-100 for 4 min. Neurons were labeled with anti-synapsin IgG antibodies, visualized by confocal microscopy, and analyzed using MetaMorph imaging software (Molecular Devices). To quantify the fluorescence intensity of cell-surface pHβ3 synaptic staining, images of neurons were thresholded to a point at which dendrites were outlined. Synapsin staining was thresholded to a set value and kept constant for control and test neurons. Next, a 25-μm section along a given proximal dendrite was selected, and a 1-bit binary image (exclusive) was made of the synapsin staining in the outlined dendrite. We then subtracted all pHβ3 staining that did not co-localize with the binarized synapsin staining. As a result, only BBSβ3 staining that co-localized with synapsin remained, and the average fluorescence intensity of these pHβ3 puncta was determined (9). Data were analyzed from 10–12 neurons for each condition from at least two to three different cultures. To quantify the number of pHβ3 synapses, thresholds were set and kept constant for control and test neurons. Receptor clusters were defined as being 0.5–2 μm in length and ∼2–3-fold more intense than background diffuse fluorescence and were co-localized with synapsin staining (9, 26). pHβ3 synaptic puncta were counted from 1-bit binary masks. Data were analyzed from 10–12 neurons for each condition (25 μm/dendrite/cell). Analyses were all performed blind to experimental condition.
GABAAR BBSβ3 Insertion Assay
Hippocampal neurons (18–21 DIV) expressing BBSβ3 were first labeled with 10 μg/ml unlabeled α-Bgt for 15 min at 15 °C to block existing cell-surface receptors. The neurons were then washed three times with PBS at 15 °C, followed by a 5-min incubation at 37 °C with 1 μg/ml Alexa 594-conjugated α-Bgt (9). All incubations were performed in the presence of 200 μm tubocurarine (Sigma) to block α-Bgt binding to endogenous acetylcholine receptors (25, 27–29). Cells were fixed in 4% paraformaldehyde. Confocal images were collected using a ×60 objective lens acquired with Olympus FluoView Version 1.5 software, and the same image acquisition settings for BBSβ3 with or without nifedipine were used. These images were analyzed using MetaMorph imaging software. A three-dimensional reconstruction of an imaged neuron was made from a series of Z sections, and then the average fluorescence intensity of Alexa 594-conjugated α-Bgt staining was measured along 30 μm of two proximal dendrites/neuron after subtraction of background fluorescence.
Metabolic Labeling and Immunoprecipitation
Hippocampal neurons were incubated in methionine-free Dulbecco's modified Eagle's medium for 15 min and then labeled with 500 μCi/ml [35S]methionine (PerkinElmer Life Sciences) for 30 min. Neurons were washed and incubated in complete Neurobasal medium with an excess of unlabeled methionine (100-fold) for an additional 0–4 h. Neurons were lysed in 1% SDS and 25 mm Tris (pH 7.4), and lysates were diluted 10-fold with RIPA buffer lacking SDS (50 mm Tris (pH 8), 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 4 mm EDTA, and mammalian protease inhibitor mixture). GABAAR β3 subunits were immunoprecipitated with rabbit anti-β3 IgG antibodies from equal amounts of solubilized protein as detailed previously (9, 10, 22, 30). Precipitated material was then subjected to SDS-PAGE, and β3 band intensities were determined by phosphorimage spectrometry (Bio-Rad).
Electrophysiological Recordings
Patch-clamp recordings in the whole cell mode were used to measure the properties of miniature inhibitory postsynaptic currents (mIPSCs) in cultured hippocampal neurons. mIPSCs were isolated by the inclusion of tetrodotoxin (TTX; 0.5 μm), d-2-amino-5-phosphopentanoic acid (20 μm), and 6,7-dinitroquinoxaline-2,3-dione (20 μm) to block action potentials, N-methyl-d-aspartic acid (NMDA), and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kainate receptors, respectively, as detailed previously (9). The cell membrane potential was held at −70 mV. A mini analysis program (Synaptosoft, Leonia, NJ) was used to analyze the spontaneous synaptic events. Statistical comparisons of the amplitude and frequency of mIPSCs were made using Student's t test. The threshold for detection of mIPSC amplitude was 15 pA.
RESULTS
Blocking Ca2+ Influx through L-type VGCCs Reduces the Expression Levels of GABAARs
The synaptic expression levels of GABAARs are dynamically regulated in response to chronic changes in neuronal activity (9, 17, 19). Furthermore, bidirectional changes in neuronal activity modulate the ubiquitin-dependent proteasomal degradation of GABAARs (9). Given that L-type VGCCs in neurons couple membrane depolarization to numerous processes, including gene expression (31) and changes in synaptic efficacy (32, 33), we speculated that Ca2+ influx through L-type VGCCs may play a role in regulating activity-dependent expression of GABAARs. To determine the effects of blocking Ca2+ influx through L-type VGCCs on GABAAR expression, we used a class of compounds known as dihydropyridines, which block L-type VGCCs (34). Cultured hippocampal neurons (18–21 DIV) were incubated for 24 h with or without 10 μm nifedipine. We chose a time point of 24 h given that homeostatic scaling of GABAergic synaptic strength occurs over hours to days (9, 17, 19). Cell-surface proteins were labeled with Sulfo-NHS-biotin and isolated using immobilized avidin, and precipitated cell-surface proteins were then resolved by SDS-PAGE. GABAAR β3 subunits were detected by immunoblotting with rabbit anti-β3 IgG antibodies. Nifedipine reduced the cell-surface expression and the total pool of GABAAR β3 subunits by 46 ± 7.3 and 45 ± 3.9%, respectively, compared with control levels (Fig. 1A).
FIGURE 1.

Blockade of L-type VGCCs reduces expression of GABAARs. A, hippocampal neurons (18–21 DIV) were treated with 10 μm nifedipine (Nif) for 24 h and then biotinylated with Sulfo-NHS-biotin and lysed in RIPA buffer. Cell-surface proteins were isolated with immobilized avidin. Immunoblots show the total and cell-surface levels of GABAAR β3 subunits as indicated. Graphs represent quantification of band intensities of cell-surface and total β3 subunits normalized to controls (Ctrl). Data represent the mean ± S.E. percentage of control values. *, significantly different from the control (p < 0.01; t test; n = 4). B, nimodipine reduced the expression of the cell-surface and total levels of GABAAR β3 subunits. Hippocampal neurons were incubated with 15 μm nimodipine for 24 h and then biotinylated with Sulfo-NHS-biotin and lysed in RIPA buffer. Immunoblots show the cell-surface and total levels of GABAAR β3 subunits as indicated. Graphs represent quantification of band intensities, and data represent the mean ± S.E. percentage of control values. *, significantly different from the control (p < 0.05; t test; n = 3).
We also determined the effects of another member of the dihydropyridine class of compounds on GABAAR expression levels, nimodipine, which is cell-impermeable, unlike nifedipine. These experiments revealed that a 24-h treatment with nimodipine (15 μm) reduced the cell-surface and total levels of GABAAR β3 subunits by 42 ± 10.8 and 41 ± 6.2%, respectively, compared with control levels (Fig. 1B).
To determine whether GABAARs localized at synaptic sites were also regulated by Ca2+ influx through L-type VGCCs, we used a construct containing superecliptic pHluorin encoded within the N terminus of the GABAAR β3 subunit (pHβ3) in our experiments (9, 26, 29). Embryonic day 18 hippocampal neurons were nucleofected with pHβ3 and cultured for 18–21 DIV. Neurons were then incubated with nifedipine (10 μm) for 24 h and fixed in 4% paraformaldehyde. To label only the cell-surface population of pHβ3, rabbit anti-green fluorescent protein IgG antibodies were used on non-permeabilized neurons, and synaptic sites were labeled with mouse IgG antibodies to the presynaptic marker synapsin-1 following permeabilization (Fig. 2A). We chose to stain synapsin because the levels of this synaptic marker are unaltered by chronic changes in neuronal activity (17). Images were collected using confocal microscopy, and based on their morphology, only pyramidal cells were chosen. These studies revealed that nifedipine reduced the levels of pHβ3-containing GABAARs at synaptic sites by 47.2 ± 6.15% compared with control levels (Fig. 2B). It should be noted that the nifedipine-induced reduction in the expression levels of total and synaptic GABAAR β3 subunits is approximately equal to the reduction observed following chronic blockade of neuronal activity with TTX (9).
FIGURE 2.

Blockade of L-type VGCCs reduces the synaptic expression of β3-containing GABAARs. A, images of hippocampal neurons (18–21 DIV) expressing GABAAR pHβ3 treated with or without nifedipine (Nif; 10 μm) for 24 h as indicated. Neurons were fixed, and non-permeabilized cells were stained with rabbit anti-green fluorescent protein IgG antibodies and Rhodamine Red-X-conjugated anti-rabbit IgG antibodies to label cell-surface pHβ3 subunits (shown in red). pHluorin fluorescence is not shown. Cells were then permeabilized and incubated with mouse anti-synapsin IgG antibodies and Cy5-conjugated anti-mouse IgG antibodies to label synaptic sites (shown in green). The boxed areas in the left panels are magnified in the right panels. Scale bars = 10 μm. B, quantification of the fluorescence intensity of pHβ3 at synaptic sites. Data represent the mean ± S.E. percentage of control (Ctrl) values. *, significantly different from the control (p < 0.001; t test; n = 10–12 neurons in two independent experiments). C, quantification of the number of synaptic sites containing GABAAR pHβ3 subunits (no significant difference; n = 10–12 neurons in two independent experiments).
The number of GABAergic inhibitory synapses in cultured neurons is modulated by activity blockade (9, 17). Therefore, we determined whether blocking Ca2+ influx had any effect on the number of synaptic sites containing GABAARs. We used the synaptic marker synapsin-1 to label presynaptic sites in neurons expressing pHβ3 following treatment with nifedipine for 24 h. We observed no change in the number of synaptic sites containing pHβ3 (Fig. 2C). Collectively, these data suggest that Ca2+ influx through L-type VGCCs modulates the total pool and the synaptic expression levels of GABAARs but not the number of GABAergic synapses.
Blocking Ca2+ Influx through L-type VGCCs Reduces the Efficacy of Synaptic Inhibition
Given the robust effects of blocking L-type VGCCs on the synaptic accumulation of GABAAR β3 subunits, the effect of nifedipine on the efficacy of synaptic inhibition was determined. Analysis of mIPSCs in cultured hippocampal neurons (18–21 DIV) was performed following nifedipine treatment (10 μm for 24 h) using patch-clamp recording in the whole cell mode as detailed previously (9). mIPSCs were recorded from cultured hippocampal pyramidal neurons in the presence of TTX (500 nm) and in the presence of d-2-amino-5-phosphonopentanoate and 6,7-dinitroquinoxaline-2,3-dione to inhibit neuronal depolarization and ionotropic glutamate receptor activation, respectively. mIPSC amplitudes and frequencies were compared between control neurons and those treated with nifedipine (Fig. 3, A–D). The data revealed that mIPSC amplitude was reduced from 57.6 ± 3.6 pA (n = 16) in control neurons to 38.3 ± 1.9 pA (n = 16) in nifedipine-treated neurons, which represents a 33.5% decrease (Fig. 3, C and D). Also, nifedipine shifted the entire distribution of amplitudes to the left, toward smaller values (Fig. 3B), suggesting that there is a uniform reduction in synaptic inhibition. Nifedipine treatment did not significantly alter mIPSC frequency (control, 1.68 ± 0.17 Hz (n = 16); nifedipine-treated, 1.35 ± 0.23 Hz (n = 16)). Overall, the above data suggest that the efficacy of synaptic inhibition can be modulated by prolonged blockade of Ca2+ influx through L-type VGCCs.
FIGURE 3.

Blockade of L-type VGCCs reduces GABAergic synaptic transmission. A and B, representative mIPSC traces (A) and cumulative distribution of mIPSC amplitude (B) from control and nifedipine-treated (10 μm, 24 h) hippocampal neurons (18–21 DIV). The threshold for mIPSC detection was 15 pA. C, scatter plot of mIPSC amplitudes from control and nifedipine-treated neurons. The mean amplitude in each neuron was the average of all the mIPSC events above 15 pA. D, bar graph of the average (mean ± S.E.) mIPSC amplitude and frequency in control (n = 16) and nifedipine-treated (n = 16) neurons. *, significantly different from the control (p < 0.001; t test).
Previous reports have shown that chronic changes in neuronal activity regulate the expression levels of GAD65 (17, 18), the main enzyme involved in GABA synthesis, leading to reduced levels of GABA and decreased synaptic inhibition (18). We therefore tested whether Ca2+ influx through L-type VGCCs had any effect on GAD65 expression levels. We treated hippocampal neurons with 10 μm nifedipine for 24 h and determined the expression levels of GAD65 by Western blotting. We observed no change in the expression levels of GAD65 (Fig. 4A) but found reduced steady-state levels of GAD65 when neurons were treated with 1 μm TTX for 24 h (Fig. 4B), in agreement with previous studies (17, 18). These data suggest that Ca2+ influx through L-type VGCCs does not regulate GAD65 expression levels and consequently suggest that GABA synthesis is unaffected under these conditions.
FIGURE 4.

GAD65 expression is unaltered by L-type VGCC blockade. A, hippocampal neurons (18–21 DIV) were treated with 10 μm nifedipine (Nif) for 24 h and then lysed in RIPA buffer. Western blotting was performed with rabbit anti-GAD65 IgG antibodies. The graph represents quantification of band intensities, and data represent the mean ± S.E. of control (Ctrl) values. No significant difference from the control was observed (t test; n = 4). B, neuronal activity blockade with TTX reduced GAD65 expression. Hippocampal neurons were treated with 1 μm TTX for 24 h and then lysed in RIPA buffer. Western blotting was performed with rabbit anti-GAD65 IgG antibodies. The graph represents quantification of band intensities, and data represent the mean ± S.E. percentage of control values. *, significantly different from the control (p < 0.05; t test; n = 3).
Ca2+ Influx through L-type VGCCs Regulates the Proteasome-dependent Turnover and Membrane Insertion of GABAARs
Given that the steady-state levels of GABAARs were reduced by nifedipine, we tested whether Ca2+ influx through L-type VGCCs directly influenced the turnover of newly synthesized GABAAR β3 subunits within the ER/secretory pathway. To assess this, we used metabolic labeling with [35S]methionine in pulse-chase experiments. Hippocampal neurons were treated with nifedipine for 24 h and then labeled with [35S]methionine for 30 min and chased for 4 h (Fig. 5A). Therefore, at the time point we chose (4 h post-labeling), β3-containing GABAARs would not have yet reached the cell surface because they take up to 6 h to reach this compartment from the ER (8). Following lysis of neurons, GABAAR β3 subunits were immunoprecipitated with anti-β3 IgG antibodies and resolved by SDS-PAGE. At 4 h, a 26.7 ± 1.94% decrease in β3 subunits was observed in neurons previously treated with nifedipine, whereas in control neurons at 4 h, there was no significant degradation (Fig. 5A). These data implicate Ca2+ influx through L-type VGCCs in modulating ER-associated degradation of de novo synthesized GABAAR β3 subunits.
FIGURE 5.
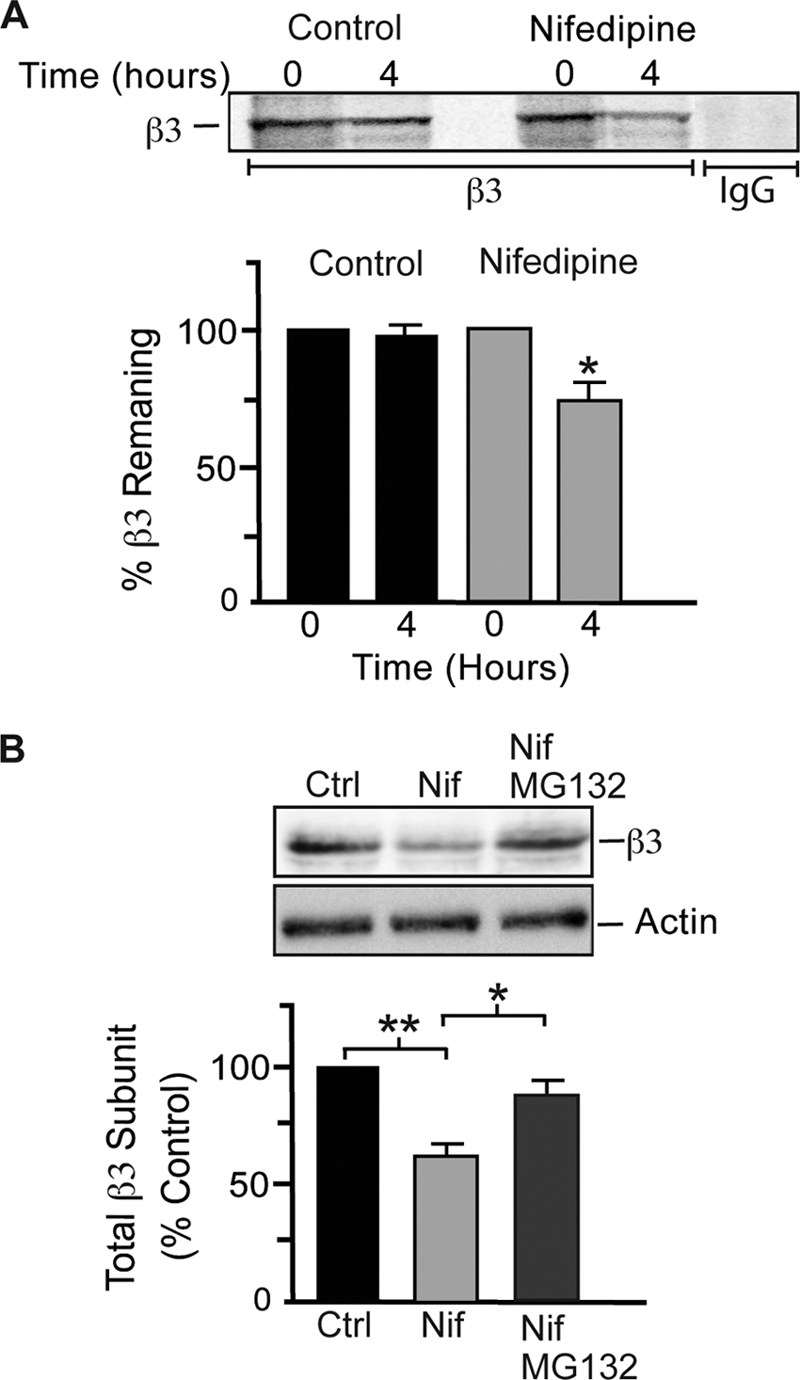
Blockade of L-type VGCCs increases the turnover of GABAARs. A, hippocampal neurons (18–21 DIV) were treated with 10 μm nifedipine for 24 h, followed by a pulse chase with [35S]methionine. Neurons were lysed in 1% SDS and diluted in RIPA buffer, and GABAAR β3 subunits were immunoprecipitated with anti-β3 IgG antibodies or nonspecific IgG antibodies (as indicated) and subjected to SDS-PAGE. Band intensities were quantified by phosphorimage spectrometry, and data represent the mean ± S.E. percentage of levels at time 0. *, significantly different from 0 h (p < 0.05; t test; n = 3). B, inhibition of proteasome activity blocked the effects of nifedipine on GABAAR β3 expression. Hippocampal neurons (18–21 DIV) were treated with or without 10 μm nifedipine for 24 h, and 10 μm MG132 was added for the last 8 h of the nifedipine incubation as indicated. Neurons were lysed, and Western blots were probed with anti-β3 IgG antibodies. Data represent the mean ± S.E. percentage of control (Ctrl) values. *, significantly different (p < 0.05); **, significantly different (p < 0.01; one-way analysis of variance and Bonferroni post-test; n = 3).
We next tested whether proteasome activity was involved in the increased turnover of GABAARs following blockade of L-type VGCCs in cultured hippocampal neurons. We treated neurons with nifedipine (10 μm) for 24 h and added the proteasome inhibitor MG132 (10 μm) during the last 8 h of the nifedipine treatment. These experiments revealed that the effects of nifedipine on the steady-state levels of GABAAR β3 subunits were blocked by MG132 (Fig. 5B). Nifedipine decreased the expression levels of β3 subunits by 38 ± 4.6% compared with control levels, whereas the addition of MG132 brought β3 subunit levels up to 85 ± 5.77% of control levels (Fig. 5B). Together, these data suggest that Ca2+ influx through L-type VGCCs can directly regulate the turnover of newly synthesized GABAARs and that proteasome activity is required for this process.
As activity-dependent L-type Ca2+ influx modulates the turnover of GABAARs, we speculated that this may influence the rate of insertion of receptors into the plasma membrane. To test this, we used a construct that encodes a bungarotoxin-binding site and pHluorin tag within the N terminus of the GABAAR β3 subunit (BBSβ3) in an insertion assay as described previously (9, 29). Hippocampal neurons (18–21 DIV) expressing BBSβ3 were incubated with 10 μm nifedipine for 24 h. Neurons were then incubated with excess unlabeled α-Bgt at 15 °C to block existing receptors and then incubated at 37 °C with Alexa 594-conjugated α-Bgt for a number of time points (0–10 min) to label newly inserted BBSβ3 at the plasma membrane (Fig. 6A). Excess unlabeled α-Bgt blocked accumulation of Alexa 594-conjugated α-Bgt staining on the plasma membrane, and α-tubocurarine (200 μm) was added to block α-Bgt binding to endogenous acetylcholine receptors. At different time points, neurons were fixed and imaged by confocal microscopy. The insertion of BBSβ3 increased linearly over a 10-min time period, which was reduced by nifedipine treatment (Fig. 6B). We therefore used a time point of 5 min to assess the influence of Ca2+ influx on the insertion of BBSβ3 and normalized data to those seen in untreated neurons, which were assigned a value of 100% (Fig. 6C). These experiments revealed that blocking Ca2+ influx with nifedipine significantly reduced the insertion of BBSβ3-containing GABAARs into the membrane by 52.7 ± 5% compared with control levels (Fig. 6C).
FIGURE 6.

Insertion of GABAARs is modulated by Ca2+ influx through L-type VGCCs. A, images of pHluorin fluorescence and Alexa 594-conjugated α-Bgt staining at 5 min of insertion in the presence or absence of nifedipine (Nif; 10 μm) as indicated. Hippocampal neurons expressing BBSβ3 (18–21 DIV) were treated with or without nifedipine (10 μm) for 24 h. Neurons were then incubated with 10 μg/ml unlabeled α-Bgt to block existing cell-surface BBSβ3, washed, incubated for 5 min with 1 μg/ml Alexa 594-conjugated α-Bgt to label newly inserted BBSβ3, and fixed. The boxed areas in the upper right-hand corners show pHluorin fluorescence. The rectangles in the left panels (Alexa 594-conjugated α-Bgt staining) are enlarged in the right panels. B, increase in BBSβ3 insertion over time with or without nifedipine (10 μm). The assay was performed as described for A. C, graph showing quantification of Alexa 594-conjugated α-Bgt fluorescence intensity at 5 min of insertion for control (Ctrl) and nifedipine (10 μm)-treated neurons. Data represent the mean ± S.E. percentage of control BBSβ3 values. *, significantly different from the control (p < 0.01; t test; n = six to eight neurons from two independent cultures).
DISCUSSION
In this study, we have shown that prolonged blockade of Ca2+ influx through L-type VGCCs leads to reduced total and synaptic expression of GABAARs and diminished GABAergic synaptic transmission without affecting the levels of GAD65. Reduced GABAAR expression following L-type VGCC blockade is a result of increased turnover of de novo synthesized GABAARs, which is dependent on the activity of the proteasome. In addition, chronic blockade of Ca2+ influx reduces the insertion of GABAARs into the neuronal membrane. Overall, prolonged reduction in L-type Ca2+ influx decreases the numbers of synaptic GABAARs and reduces synaptic inhibition, highlighting a possible role for L-type VGCCs in the activity-dependent scaling of GABAergic synaptic strength.
The number of GABAARs is a critical factor in determining the strength of GABAergic synaptic inhibition (35). It has been previously reported that prolonged changes in neuronal activity lead to an adaptive modulation of the numbers of GABAARs at synaptic sites and the efficacy of synaptic inhibition (9, 17, 19, 36). Therefore, a critical expression locus of activity-dependent scaling of GABAergic synaptic strength is the postsynaptic change in GABAAR numbers. Furthermore, the direct ubiquitination of GABAARs and the ubiquitin-proteasome system play a critical role in the activity-dependent modulation of the abundance of these receptors at synaptic sites (9). In this study, we have shown that blocking Ca2+ influx through L-type VGCCs in cultured hippocampal neurons mimics the effects of TTX on GABAAR turnover, accumulation at synaptic sites, and efficacy of synaptic inhibition (9). These observations suggest that activity-dependent scaling of GABAergic synaptic strength is mediated in part by Ca2+ influx through L-type VGCCs. Interestingly, a recent report has shown that the increase in AMPA receptor number brought about by activity blockade with TTX is mediated by somatic Ca2+ influx and that TTX prevents these action potential-triggered Ca2+ transients in the cell soma (37). In addition, the effects of TTX on AMPA receptor accumulation could be partially mimicked by blocking L-type VGCCs, but the authors speculate that other VGCCs (T- and R-type) contribute to this phenomenon (37). Furthermore, Ca2+ influx through L-type VGCCs is critical for activity-dependent changes in the composition of AMPA receptors (33). Thus, these studies highlight the importance of L-type Ca2+ influx in the homeostatic scaling of glutamatergic synaptic strength.
In addition to the changes in abundance of GABAARs, a number of presynaptic factors also determine GABAergic synaptic strength. We did not observe a change in GAD65 expression or the frequency of mIPSCs when L-type Ca2+ influx was blocked. This is in stark contrast to the presynaptic changes that have been reported when neuronal activity was blocked with TTX. Activity blockade in cultured neurons with TTX induces down-regulation of GAD65 and vesicular inhibitory amino acid transporter expression (17–19, 21), reduces the number of GABAergic synapses (9, 17), and results in a smaller mIPSC frequency (9, 17). Thus, Ca2+ influx through L-type VGCCs may be responsible for only mediating activity-dependent postsynaptic changes in GABAergic synaptic strength. However, it appears that other mechanisms regulate presynaptic factors governing the homeostatic scaling of GABAergic synaptic strength. Perhaps activity-dependent changes in the expression of GAD65 are mediated by Ca2+ influx through NMDA receptors and not L-type VGCCs, as one report has shown that NMDA receptor activation dominates Ca2+ influx in interneurons (38) and that chronic blockade of NMDA receptors reduces GAD67 expression (39).
This study has begun to delineate the biochemical pathways underlying the scaling of the strength of GABAergic synaptic neurotransmission. However, there are a number of unanswered questions. For instance, we have revealed that Ca2+ influx can regulate the turnover of GABAARs, but the exact processes leading to the modulation of receptor turnover and insertion still remain to be determined but could possibly involve Ca2+-dependent phosphorylation of GABAAR subunits and subsequent modulation of receptor ER export/degradation.
In conclusion, this study has demonstrated the importance of L-type Ca2+ influx in regulating the abundance of GABAARs at synaptic sites and the efficacy of synaptic inhibition. Furthermore, this work emphasizes the importance of Ca2+ signaling in mediating activity-dependent scaling of GABAergic synaptic strength.
This work was supported, in whole or in part, by National Institutes of Health Grants NS047478, NS048045, NS051195, NS056359, and NS054900 from NINDS (to S. J. M.) and by a fellowship from the American Society for Epilepsy (to R. S. S.).
- GABAAR
- γ-aminobutyric acid type A receptor
- ER
- endoplasmic reticulum
- VGCC
- voltage-gated calcium channel
- α-Bgt
- α-bungarotoxin
- DIV
- days in vitro
- PBS
- phosphate-buffered saline
- NHS
- N-hydroxysuccinimide
- RIPA
- radioimmune precipitation assay
- pH
- pHluorin
- BBS
- bungarotoxin-binding site
- mIPSC
- miniature inhibitory postsynaptic current
- TTX
- tetrodotoxin
- NMDA
- N-methyl-d-aspartic acid
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid.
REFERENCES
- 1.Rudolph U., Möhler H. (2004) Annu. Rev. Pharmacol. Toxicol. 44, 475–498 [DOI] [PubMed] [Google Scholar]
- 2.Rudolph U., Möhler H. (2006) Curr. Opin. Pharmacol. 6, 18–23 [DOI] [PubMed] [Google Scholar]
- 3.Sieghart W., Sperk G. (2002) Curr. Top. Med. Chem. 2, 795–816 [DOI] [PubMed] [Google Scholar]
- 4.Essrich C., Lorez M., Benson J. A., Fritschy J. M., Lüscher B. (1998) Nat. Neurosci. 1, 563–571 [DOI] [PubMed] [Google Scholar]
- 5.Kittler J. T., Moss S. J. (2003) Curr. Opin. Neurobio.l 13, 341–347 [DOI] [PubMed] [Google Scholar]
- 6.Lüscher B., Keller C. A. (2004) Pharmacol. Ther. 102, 195–221 [DOI] [PubMed] [Google Scholar]
- 7.Farrant M., Nusser Z. (2005) Nat. Rev. Neurosci. 6, 215–229 [DOI] [PubMed] [Google Scholar]
- 8.Gorrie G. H., Vallis Y., Stephenson A., Whitfield J., Browning B., Smart T. G., Moss S. J. (1997) J. Neurosci. 17, 6587–6596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saliba R. S., Michels G., Jacob T. C., Pangalos M. N., Moss S. J. (2007) J. Neurosci. 27, 13341–13351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kittler J. T., Thomas P., Tretter V., Bogdanov Y. D., Haucke V., Smart T. G., Moss S. J. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 12736–12741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacob T. C., Moss S. J., Jurd R. (2008) Nat. Rev. Neurosci. 9, 331–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moss S. J., Smart T. G. (2001) Nat. Rev. Neurosci. 2, 240–250 [DOI] [PubMed] [Google Scholar]
- 13.Turrigiano G. G., Nelson S. B. (2004) Nat. Rev. Neurosci. 5, 97–107 [DOI] [PubMed] [Google Scholar]
- 14.Burrone J., Murthy V. N. (2003) Curr. Opin. Neurobiol. 13, 560–567 [DOI] [PubMed] [Google Scholar]
- 15.Turrigiano G. G., Leslie K. R., Desai N. S., Rutherford L. C., Nelson S. B. (1998) Nature 391, 892–896 [DOI] [PubMed] [Google Scholar]
- 16.Turrigiano G. G. (2008) Cell 135, 422–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kilman V., van Rossum M. C., Turrigiano G. G. (2002) J. Neurosci. 22, 1328–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hartman K. N., Pal S. K., Burrone J., Murthy V. N. (2006) Nat. Neurosci. 9, 642–649 [DOI] [PubMed] [Google Scholar]
- 19.Swanwick C. C., Murthy N. R., Kapur J. (2006) Mol. Cell. Neurosci. 31, 481–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stellwagen D., Malenka R. C. (2006) Nature 440, 1054–1059 [DOI] [PubMed] [Google Scholar]
- 21.De Gois S., Schäfer M. K., Defamie N., Chen C., Ricci A., Weihe E., Varoqui H., Erickson J. D. (2005) J. Neurosci. 25, 7121–7133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jovanovic J. N., Thomas P., Kittler J. T., Smart T. G., Moss S. J. (2004) J. Neurosci. 24, 522–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brandon N. J., Jovanovic J. N., Colledge M., Kittler J. T., Brandon J. M., Scott J. D., Moss S. J. (2003) Mol. Cell. Neurosci. 22, 87–97 [DOI] [PubMed] [Google Scholar]
- 24.Kittler J. T., Delmas P., Jovanovic J. N., Brown D. A., Smart T. G., Moss S. J. (2000) J. Neurosci. 20, 7972–7977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saliba R. S., Pangalos M., Moss S. J. (2008) J. Biol. Chem. 283, 18538–18544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacob T. C., Bogdanov Y. D., Magnus C., Saliba R. S., Kittler J. T., Haydon P. G., Moss S. J. (2005) J. Neurosci. 25, 10469–10478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pedersen S. E., Cohen J. B. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 2785–2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sekine-Aizawa Y., Huganir R. L. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 17114–17119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bogdanov Y., Michels G., Armstrong-Gold C., Haydon P. G., Lindstrom J., Pangalos M., Moss S. J. (2006) EMBO J. 25, 4381–4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brandon N. J., Delmas P., Kittler J. T., McDonald B. J., Sieghart W., Brown D. A., Smart T. G., Moss S. J. (2000) J. Biol. Chem. 275, 38856–38862 [DOI] [PubMed] [Google Scholar]
- 31.West A. E., Griffith E. C., Greenberg M. E. (2002) Nat. Rev. Neurosci. 3, 921–931 [DOI] [PubMed] [Google Scholar]
- 32.Grover L. M., Teyler T. J. (1990) Nature 347, 477–479 [DOI] [PubMed] [Google Scholar]
- 33.Thiagarajan T. C., Lindskog M., Tsien R. W. (2005) Neuron 47, 725–737 [DOI] [PubMed] [Google Scholar]
- 34.Tsien R. W., Tsien R. Y. (1990) Annu. Rev. Cell Biol. 6, 715–760 [DOI] [PubMed] [Google Scholar]
- 35.Nusser Z., Cull-Candy S., Farrant M. (1997) Neuron 19, 697–709 [DOI] [PubMed] [Google Scholar]
- 36.Nusser Z., Hájos N., Somogyi P., Mody I. (1998) Nature 395, 172–177 [DOI] [PubMed] [Google Scholar]
- 37.Ibata K., Sun Q., Turrigiano G. G. (2008) Neuron 57, 819–826 [DOI] [PubMed] [Google Scholar]
- 38.Goldberg J. H., Yuste R., Tamas G. (2003) J. Physiol. 551, 67–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kinney J. W., Davis C. N., Tabarean I., Conti B., Bartfai T., Behrens M. M. (2006) J. Neurosci. 26, 1604–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]